

Abstract
Emerging evidence demonstrated that particulate matter 2.5 (PM2.5) exposure served as an important risk factor of cardiovascular diseases. Some studies also reported that COX-2/mPGES-1/PGE2 cascade played a pathogenic role in vascular injury. However, the relationship between the PM2.5 exposure and the activation of COX-2/mPGES-1/PGE2 cascade in endothelial cells is still unknown. In the present study, mouse aorta endothelial cells were exposed to PM2.5. Strikingly, following the PM2.5 treatment, we observed dose- and time-dependent upregulation of COX-2 at both protein and mRNA levels as determined by Western blotting and qRT-PCR, respectively. However, COX-1 mRNA expression was not affected by PM2.5 treatment. Next, we examined mPGES-1 expression. As expected, mPGES-1 protein was markedly increased by PM2.5 exposure in line with a significant increment of PGE2 release in medium. At the same time, we observed a dose-dependent upregulation of another two PGE2 synthases of mPGES-2 and cPGES determined by qRT-PCR. Inhibition of COX-2 by using a specific COX-2 inhibitor NS-398 markedly blocked cell apoptosis, inflammation, and PGE2 secretion. Taken together, these results suggested that PM2.5 could activate inflammatory axis of COX-2/PGES/PGE2 in vascular endothelial cells to promote cell apoptosis and inflammatory response.
Keywords: PM2.5, endothelial cells, COX-2, apoptosis, inflammation
Introduction
It is known that ambient air pollution has become an important insult of many human diseases in the world, including respiratory and cardiovascular diseases [1]. According to the World Health Organization (WHO) report, of the 17.5 million cardiovascular diseases (CVD)-related deaths in 2012, 3.7 million deaths was associated with ambient air pollution, accounting for 6.7% of total deaths worldwide. Air pollutant is a complex mixture containing gases, particulate matter (PM), semi-volatile and volatile organics, and other pollutants. The United State Environmental Protection Agency (USEPA) defined PM as “a complex mixture of extremely small particles and gases which includes acids, organic chemicals, metals, soils and dust”. According to aerodynamic diameter, PM is classified into four categories: total suspended particulate (TSP≤100 μm, PM100); coarse particulate matter (TSP≤10 μm, PM10); fine particulate matter (TSP≤2.5 μm, PM2.5), and ultrafine particles (TSP≤0.1 μm, PM0.1). Particulate matter comes mainly from human activities and natural phenomena (e.g. wildfires, volcanoes, and land dust) [2]. In both, human-generated sources including emissions from industrial production, biomass burning, and vehicle exhaust are the main contributors [3]. PM contains many toxic heavy metals, including chromium, lead, arsenic, cadmium, and nickel which are harmful to human health [4]. The smaller aerodynamic diameter, the larger adsorption area, and the higher risk to human health [5]. Compared with PM10, PM2.5 have smaller size and lighter quality, which can enter into respiratory tract and vascular system, leading to the pulmonary and systemic inflammation and oxidative stress, activating coagulation system, disturbing autonomic nerve and vascular endothelium function [6-9].
Epidemiological studies have revealed both short- and long-term exposure to high concentration of PM2.5 significantly increased morbidity and mortality [10]. In cardiovascular system, exposure to PM2.5 was associated with a variety of cardiovascular diseases, including myocardial infarction [11,12], arrhythmias [13,14], heart failure exacerbations [15], coronary heart disease [16,17], arterial wall thickening and atherosclerosis [18,19]. Recently, the pathogenic mechanisms involved in the CVD related to PM2.5 were chiefly documented as: (1) Oxidative stress [20-22]; (2) Pulmonary and systemic inflammatory response [23,24]; (3)Endothelial Dysfunction [25-28]; (4) Autonomic Dysfunctions [29-31]; (5) Abnormal activation of coagulation [32,33]; and (6) Genotoxic Effects [34,35].
As mentioned above, inflammatory response served as an important contributor in PM2.5-related cardiovascular diseases. A recent study reported that cyclooxygenase-2 (COX-2) expression was upregulated in mice myocardial tissue after co-exposure to SO2, NO2, and PM2.5 [24]. COX-2 is the dominant source of prostaglandin (PG), including prostaglandin E2 (PGE2) which is mainly synthesized by prostaglandin E synthase (PGES) [36]. In the past decade, three forms of PGE synthases have been found and characterized: including microsomal prostaglandin E synthase-1 (mPGES-1), microsomal prostaglandin E synthase-2 (mPGES-2) and cytosolic PGES (cPGES). mPGES-1 is the best-characterized PGE synthase [37,38]. Many studies have demonstrated that COX-2/mPGES-1/PGE2 cascade activation is involved in cardiovascular disease, including neointimal hyperplasia after vascular injury, aortic aneurysm, and atherosclerosis [39-42]. Thus, we hypothesized that PM2.5 could activate inflammatory cascade of COX-2/mPGES-1/PGE2 in endothelial cells (ECs) to cause the cell injury and inflammation. To prove this hypothesis, mouse aorta endothelial cells were exposed to PM2.5 and the activation and the role of this cascade were examined.
Materials and methods
Materials
Mouse aorta endothelial cells (MAECs) were obtained from ATCC. Prostaglandin E2 (PGE2) EIA kit, COX-2 (catalog no. 160106) and mPGES-1 (catalog no. 160140) antibodies were purchased from Cayman Chemicals (Ann Arbor, MI). BAX (catalog no. 50599-2) and Bcl2 (catalog no. 12789-1) antibodies were from Proteintech. Trizol was obtained from Takara. Dulbecco’s modified Eagle’s medium (DMEM) and Fetal bovine serum were obtained from Gibco. Cell culture dishes were obtained from Corning.COX-2 inhibitor NS-398 (catalog no. s1772) was from Beyotime (Shanghai, China). PM2.5 was kindly provided by Dr. Zhengdong Zhang from School of Public Health, Nanjing Medical University, Nanjing, China, collected from Wuhan China.
Cell culture and PM2.5 exposure
MAECs were cultured in DMEM supplemented with 10% FBS, and 1% penicillin/streptomycin in a 5% CO2 atmosphere at 37°C. All cell exposure experiments were performed at 80-90% cell confluence with viability ≥ 90% determined by the trypan blue staining. The cells were digested with 0.25% trypsin and sub-cultured into 6-wells plates or 12-wells plates for 24 h with DMEM supplemented 10% FBS. Then using serum-free medium instead of previous culture medium, and the cells were incubated with freshly dispersed PM2.5 at final concentrations of 25, 50, 100 μg/ml for 12, 24 h at 37°C with or without a pretreatment of NS-398 (COX-2 inhibitor).
Western blotting analysis
After the exposure to PM2.5, MAECs were collected and lysed using RIPA buffer supplemented with protease and phosphatase inhibitors at 4°C for 30 min. Whole cell lysates were collected using centrifugation at 12,000 g for 15 min at 4°C, and protein concentration was determined with BCA Protein Assay Kit (Beyotime). Then proteins were subjected to 10% or 5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane by wet electrophoretic transfer at 300 mA for 60 min. The PVDF membranes were then blocked in 5% Skim milk at room temperature for 1 h and then incubated with the primary antibodies diluted (1:1000) in 5% milk for overnight at 4°C, and then incubated with the secondary antibody diluted (1:3000) at room temperature for 2 h. After completion of incubation; the membranes were washed with TBST for five times. Immuno-reactive bands were detected with ECL reagents using Gel imager (Bio-Rad). Densities of target protein bands were determined with Quantity One 4.4.1 (Bio-Rad). The internal control GAPDH was used to normalize loading variations.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted with TRIZOL according to the protocol of the manufacturer. Oligonucleotides were designed by using Primer 5 software and the sequences are β-actin: 5’-GCTCTGGCTCCTAGCACCAT-3’ (sense) and 5’-GCCACCGATC CACACAGAGT-3’ (antisense); COX-1: 5’-CATTGCACATCCATCCACTC-3’ (sense) and 5’-CCAAAGCGGACACAGACAC-3’ (antisense); COX-2: 5’-AGGACTCTGCTCACGAAGGA-3’ (sense) and 5’-TGACATGGATTGGAACAGCA-3’ (antisense); mPGES-2: 5’-GCTGGGGCTGTACCACAC-3’ (sense) and 5’-GATTCACCTCCACCACCTGA-3’ (antisense); cPGES: 5’-GGTAGAGACCGCCGGAGT-3’ (sense) and 5’-TCGTACCACTTTGCAGAAGCA-3’ (antisense); Mouse IL-6: 5’-GCTTAGGCATAACGCACT-3’ (sense) and 5’-GGAAATCGTGGAAATGAG-3’ (antisense); MouseIL-1β: 5’-AGGCTCCGAGATGAACAA-3’ (sense) and 5’-GAAGGCATTAGAAACAGTCC-3’ (antisense); Mouse TNF-α: 5’-CTGTGAAGGGAATGGGTGTT-3’ (sense) and 5’-CAGGGAAGAATCTGGAAAGGT-3’ (antisense); Mouse MCP-1: 5’-ACCACAGTCCATGCCATCAC (sense) and 5’-TTGAGGTGGTTGTGGAAAAG-3’ (antisense); Mouse ICAM-1: 5’-CGCTTCCGCTACCATCAAC-3’ (sense) and 5’-GGCGGCTCAGTATCTCCTC-3’ (antisense); Mouse VCAM-1: 5’-GGAGAGACAAAGCAGAAGTGG-3’ (sense) and 5’-AACACAAGCGTGGATTTGG (antisense); Real-time PCR was carried out using SYBR Green (Vazyme). The PCR cycle was as follows: initial denaturant at 95°C for 5 min, followed by 40 cycles of denaturant at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 30 s. The amount of target genes was analyzed using the 2^-ΔCt method following the normalization through β-actin.
Apoptosis assay
MAECs were treated and stained with FITC annexin V and propidium iodide according to the instruction of manufacturer. The stained cells were analyzed by using a BD FACSCalibur flow cytometer (Bedford, MA) and the data analysis was performed with FlowJo software.
EIA assay
Cell culture medium was collected after PM2.5 exposure with or without a pretreatment of NS-398 (COX-2 inhibitor), and the PGE2 levels were determined using an EIA kit (Cayman Chemical Company) according to the manufacturer’s instructions.
Statistical analysis
All results obtained were expressed as means ± standard deviation (SD) of three independent experiments. Data were analyzed using analysis of variance (ANOVA) statistical analysis followed by a post-hoc t test or unpaired Student’s t-test. Statistical significance was set at P < 0.05.
Results
PM2.5 exposure upregulated COX-2 expression in mouse aorta endothelial cells
COX-2 has a known role in mediating inflammatory response in many diseases including cardiovascular complications. However, whether PM2.5 could affect COX-2 expression in vascular endothelial cells was not reported. In the present study, we treated mouse aortic endothelial cells with PM2.5 and found that PM2.5 at the concentration of 25 μg/ml and 50 μg/ml for 24 h moderately but significantly enhanced COX-2 protein expression (Figure 1A, 1B). Further increment of PM2.5 concentration to 100 μg/ml led to a more robust induction of COX-2 protein expression by 3 folds (Figure 1A, 1B). Then we examined COX-2 mRNA expression in endothelial cells following treatment with PM2.5. Similarly, COX-2 mRNA level was dose-dependently enhanced as compared with the control group (Figure 1C). To observe whether PM2.5 has a time-dependent effect on regulating COX-2 expression, we treated endothelial cells with PM2.5 at different time points (0 h, 12 h, and 24 h). As shown by Figure 2A, 2B, PM2.5 time-dependently upregulated COX-2 protein expression. For the mRNA regulation, 12-h PM2.5 exposure led to a maximal COX-2 induction in the cells (Figure 2C).
Figure 1.
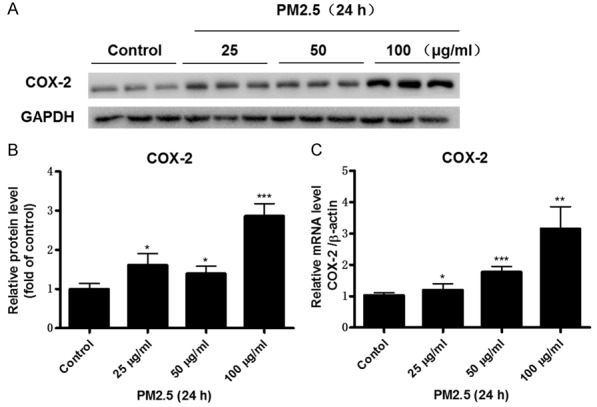
PM2.5 dose-dependently induced COX-2 expression in endothelial cells. Mouse aorta endothelial cells were treated with different doses of PM2.5 (0, 25, 50, 100 μg/ml) for 24 h. A. Western blotting analysis of COX-2. B. Densitometric analysis of COX-2 Western blots. C. COX-2 mRNA expression in endothelial cells was determined by qRT-PCR. The values represent the means ± SD (n=6 in each group). *p<0.05 vs. Control group. **p<0.01 vs. Control group. ***p<0.001 vs. Control group.
Figure 2.
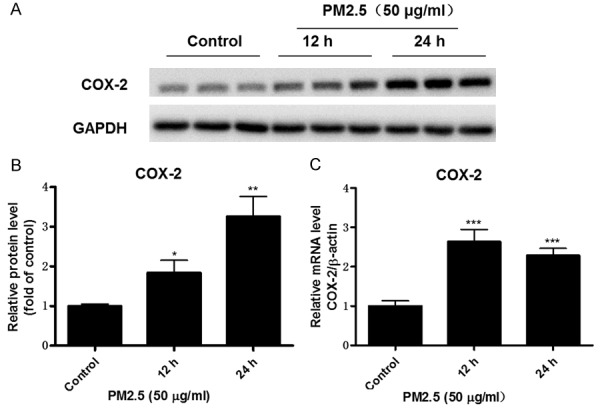
PM2.5 time-dependently induced COX-2 expression in endothelial cells. Mouse aorta endothelial cells were treated with PM2.5 (50 μg/ml) at different time points (0, 12, and 24 h). A. Western blotting analysis of COX-2. B. Densitometric analysis of COX-2 Western blots. C. COX-2 mRNA expression in endothelial cells was determined by qRT-PCR. The values represent the means ± SD (n=6 in each group). *p<0.05 vs. Control group. **p<0.01 vs. Control group. ***p<0.001 vs. Control group.
In general, COX-1 served as an enzyme for the generation of baseline prostaglandins and could not be regulated under many pathological conditions. As expected, PM2.5 at a dose of 50 μg/ml did not affect the mRNA expression of COX-1 in a time course study (Figure 3). These results demonstrated that PM2.5 has a potent and selective role in upregulating COX-2 expression in vascular endothelial cells.
Figure 3.
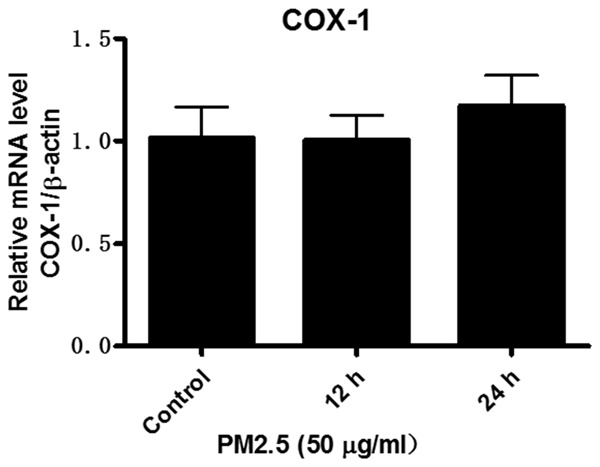
PM2.5 exposure did not change COX-1 expression in endothelial cells. Mouse aorta endothelial cells were treated with PM2.5 (50 μg/ml) at different time points (0, 12, and 24 h). COX-1 mRNA expression in endothelial cells was determined by qRT-PCR. The values represent the means ± SD (n=6 in each group).
PM2.5 upregulated mPGES-1 expression in mouse aorta endothelial cells
mPGES-1 served as an inducible PGE2-generating enzyme under many pathological conditions. Thus, we examined mPGES-1 expression via Western blotting and observed a robust upregulation by more than 4 folds following PM2.5 exposure for 12 h at a dose of 50 μg/ml (Figure 4A, 4B). This result demonstrated that PM2.5 is a potent stimulant of mPGES-1 in vascular endothelial cells.
Figure 4.
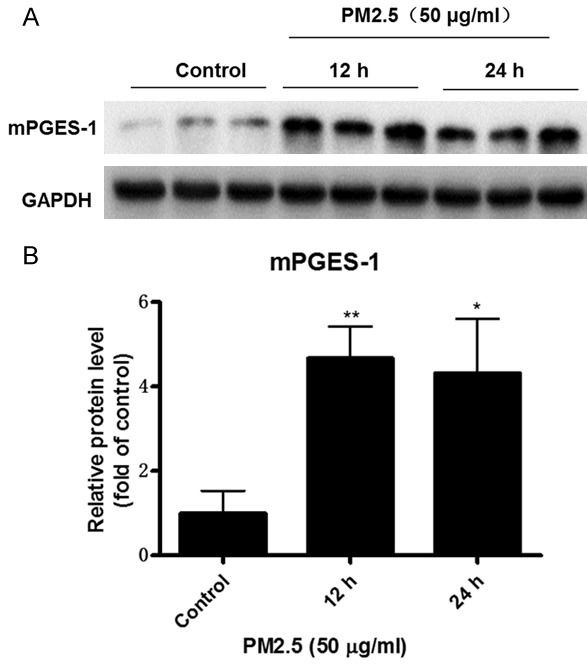
PM2.5 induced mPGES-1 expression in endothelial cells. Mouse aorta endothelial cells were treated with PM2.5 (50 μg/ml) at different time points (0, 12, and 24 h). A. Western blotting analysis of mPGES-1. B. Densitometric analysis of mPGES-1 Western blots. The values represent the means ± SD (n=6 in each group). *p<0.05 vs. Control group. **p<0.01 vs. Control group.
PM2.5 upregulated mPGES-2 and cPGES expression in mouse aorta endothelial cells
In general, among three PGE synthases, mPGES-1 was proven as an inducible enzyme and responsible for the production of induced PGE2. However, mPGES-2 and cPGES were thought to be mainly responsible for the maintenance of baseline PGE2. In the present study, we also examined the mPGES-2 and cPGES expressions in endothelial cells exposed to PM2.5 at different doses (25 μg/ml, 50 μg/ml, and 100 μg/ml) for 24 h. As shown by the data, PM2.5 dose-dependently increased the expressions of mPGES-2 and cPGES as determined by qRT-PCR (Figure 5A, 5B). These results demonstrated that both mPGES-2 and cPGES could be upregulated by PM2.5 in vascular endothelial cells.
Figure 5.
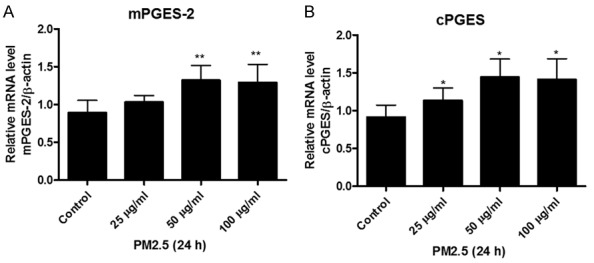
PM2.5 upregulated the mRNA expressions of mPGES-2 and cPGES in endothelial cells. Mouse aorta endothelial cells were treated with different doses of PM2.5 (0, 25, 50, 100 μg/ml) for 24 h. A. mPGES-2 mRNA expression in endothelial cells was determined by qRT-PCR. B. cPGES mRNA expression in endothelial cells was determined by qRT-PCR. The values represent the means ± SD (n=6 in each group). *p<0.05 vs. Control group. **p<0.01 vs. Control group.
Inhibiting COX-2 significantly blocked PM2.5-induced PGE2 production
In agreement with the upregulation of COX-2 and PGE2 synthases, we observed a marked increase of PGE2 production following PM2.5 treatment (Figure 6). Strikingly, such an induction of PGE2 was abolished by a specific COX-2 inhibitor NS-398 (Figure 6).
Figure 6.
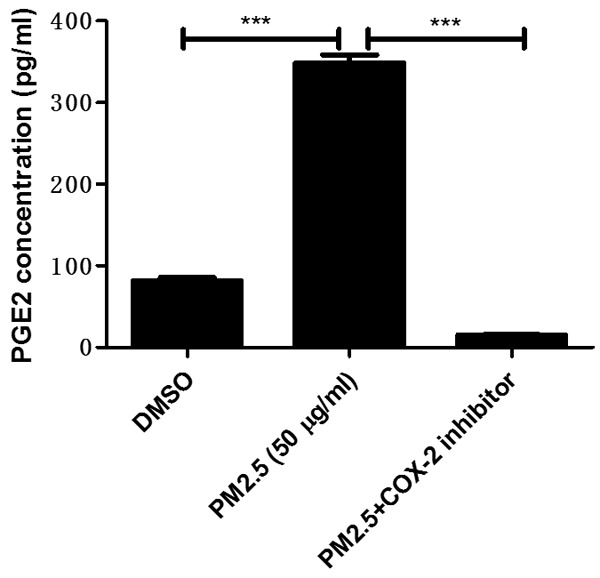
COX-2 inhibitor blocked PM2.5-induced PGE2 release in endothelial cells. Mouse aorta endothelial cells were pretreated with COX-2 inhibitor NS-398 (10 μM) for 1 hour and then were treated with PM2.5 (50 μg/ml) for 24 h. PGE2 levels in the medium were examined by EIA assay. The values represent the means ± SD (n=6 in each group). ***p<0.001 vs. DMSO or PM2.5 group.
Inhibiting COX-2 significantly blocked PM2.5-induced MAEC apoptosis
To further evaluate the effect of COX-2 on PM2.5-induced MAEC injury, we detected the apoptotic response in MAECs after the PM2.5 administration. The results demonstrated that PM2.5 could induce the apoptosis in MAECs which was completely blocked by inhibiting COX-2 (Figure 7A). Meantime, the increment of BAX and the decrement of Bcl2 were largely normalized following COX-2 inhibition (Figure 7B, 7C). These results suggested a detrimental role of COX-2 in mediating PM-2.5-induced endothelial cell apoptosis.
Figure 7.
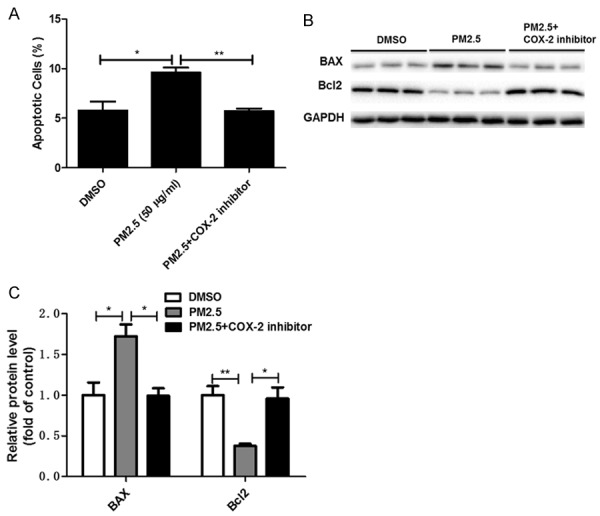
COX-2 inhibitor blocked PM2.5-induced apoptosis in endothelial cells. Mouse aorta endothelial cells were pretreated with COX-2 inhibitor NS-398 (10 μM ) for 1 hour and then were treated with PM2.5 (50 μg/ml) for 24 h. A. Analysis of endothelial cell apoptosis. B. Western blotting analysis of BAX and Bcl2. C. Densitometric analysis of BAX and Bcl2 Western blots. The values represent the means ± SD (n=6 in each group). *p<0.05 vs. DMSO or PM2.5 group. **p<0.01 vs. DMSO or PM2.5 group. ***p<0.001 vs. DMSO or PM2.5 group.
Inhibiting COX-2 significantly attenuated PM2.5-induced inflammation in MAECs
Inflammation plays an important role in the onset and progression of cardiovascular diseases. COX-2/PGES/PGE2 pathway is known as a potent inflammatory cascade. Thus, we examined the inflammatory markers including IL-6, IL-1β, TNF-α, MCP-1, ICAM-1, and VCAM-1 in MAECs (Figure 8A-F). As expected, PM2.5 significantly enhanced the expressions of these inflammatory markers, which was markedly ameliorated by the treatment of a specific COX-2 inhibitor. These data indicated that COX-2 mediated an inflammatory response induced by PM2.5 in endothelial cells.
Figure 8.
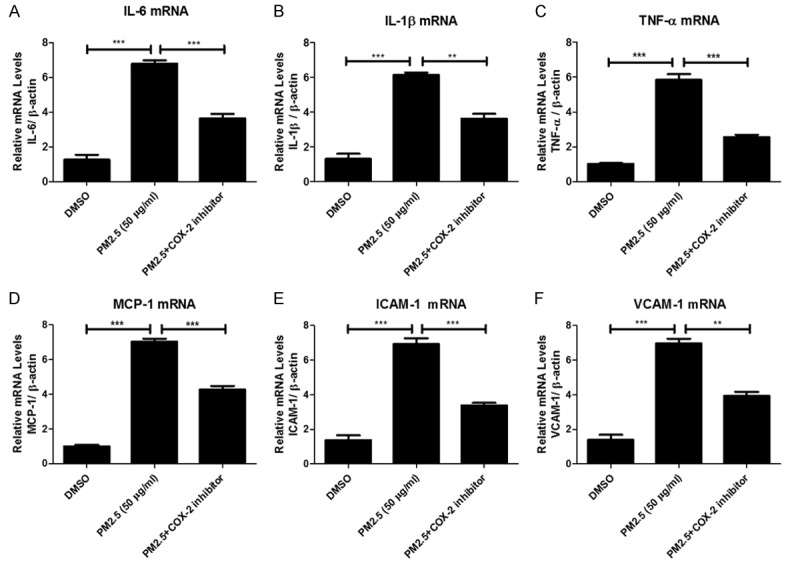
COX-2 inhibitor blocked PM2.5-induced inflammatory response in endothelial cells. Mouse aorta endothelial cells were pretreated with COX-2 inhibitor NS-398 (10 μM) for 1 hour and then were treated with PM2.5 (50 μg/ml) for 24 h. IL-6, IL-1β, TNF-α, MCP-1, ICAM-1 and VCAM-1 mRNA expression in endothelial cells were determined by qRT-PCR. The values represent the means ± SD (n=4 in each group). **p<0.01 vs. DMSO or PM2.5 group. ***p<0.001 vs. DMSO or PM2.5 group.
Discussion
Accumulating evidence demonstrated that PM2.5 inhalation is associated with the increased risk of CVD, as well as the mortality and morbidity. A recent research proved that episodic exposure to PM2.5 was associated with elevated circulating endothelial microparticles indicating the endothelial cell apoptosis and endothelial injury, and enhanced inflammatory cytokines indicating a systemic inflammation [43]. Although systemic inflammation and endothelial dysfunction induced by PM2.5 have been observed and recognized by the researchers, the exact mechanism mediating such effects remains unclear. In consideration of a known role of COX-2/PGES/PGE2 axis in mediating inflammatory response, we performed experiments to define the effect of PM2.5 on regulating endothelial COX-2/PGES/PGE2, as well as the role of this axis in mediating the inflammation and endothelial cell apoptosis.
COX-2 is responsible for the formation of five major prostanoids. Emerging evidence demonstrated that COX-2 was upregulated in inflamed vascular regions and played a pathogenic role in the atherosclerosis, aneurysm, and balloon-induced artery injury [44-46]. In the present study, we found that COX-2 was robustly upregulated in response to the PM2.5 exposure. Considering the known role of COX-2 contributing to the inflammation and inflammatory vascular diseases, we could speculate that COX-2 induction in endothelial cells might contribute to the PM2.5-associated local and systemic inflammation and vascular injury. To prove this hypothesis, we treated vascular endothelial cells with a specific COX-2 inhibitor prior to the PM2.5 treatment. As expected, inhibition of COX-2 not only entirely blocked cell apoptosis but also attenuated inflammatory response, suggesting a potent role of COX-2 in mediating PM2.5-induced cell apoptosis and inflammation.
mPGES-1 is the best characterized PGE2 synthase and has an established role in mediating inflammation [36]. Some groups reported that the deficiency of mPGES-1 could prevent the formation of atherosclerosis, neointimal hyperplasia after vascular injury and the formation of aortic aneurysm induced by angiotensin II [39,41,42], indicating a detrimental role of mPGES-1 in mediating the inflammatory vascular diseases. In this study, we found PM2.5 strikingly enhanced the expression of mPGES-1 in line with the COX-2 induction and PGE2 release in vascular endothelial cells, suggesting that mPGES-1 might contribute to the PM2.5-related vascular inflammatory injury coupling with COX-2.
In the current study, we also observed the induction of mPGES-2 and cPGES. Although these two enzymes have been thought as the contributor of baseline PGE2 generation, recent studies from KO animals did not support this notion. The mPGES-2 is a Golgi membrane-associated protein. Its maturation relies on the spontaneous cleavage of the N-terminal hydrophobic domain [47]. Systemic mPGES-2 deletion in mice did not reduce the basal PGE2 levels in different organs [48]. The cPGES is abundantly expressed in the cytosol of various types of cells [49] .Similar as the findings from systemic mPGES-2 KO mice, deletion of cPGES did not lower the PGE2 levels in various organs [50]. These reports suggested that although both mPGES-2 and cPGES were upregulated by PM2.5 at mRNA levels, they were unlikely involved in the PGE2 production in this experimental setting.
In summary, using mouse aortic endothelial cells, we observed a striking effect of PM2.5 on activating COX-2/mPGES-1/PGE2 axis which might serve as the pathogenic mechanism in PM2.5-induced endothelial cell apoptosis and inflammation. Thus, targeting COX-2/mPGES-1/PGE2 axis might be an effective strategy in preventing PM2.5-associated cardiovascular complications.
Acknowledgements
This work was supported by scientific grants fromNational Natural Science Foundation of China (Nos. 81670647, 81600352, 81530023, 81570616, 81470928, 81325004, and 81270797), the Natural Science Foundation of Jiangsu Province (No. BK20160137, BK20141079, BL2014007), and the Nanjing Medical University (2015NJMUZD053).
Disclosure of conflict of interest
None.
References
- 1.Dockery DW, Pope CA 3rd, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG Jr, Speizer FE. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 2.Lee BJ, Kim B, Lee K. Air pollution exposure and cardiovascular disease. Toxicol Res. 2014;30:71–75. doi: 10.5487/TR.2014.30.2.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao J, Xu H, Xu Q, Chen B, Kan H. Fine particulate matter constituents and cardiopulmonary mortality in a heavily polluted Chinese city. Environ Health Perspect. 2012;120:373–378. doi: 10.1289/ehp.1103671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greene NA, Morris VR. Assessment of public health risks associated with atmospheric exposure to PM2.5 in Washington, DC, USA. Int J Environ Res Public Health. 2006;3:86–97. doi: 10.3390/ijerph2006030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Q, Hong X, Wold LE. Cardiovascular effects of ambient particulate air pollution exposure. Circulation. 2010;121:2755–2765. doi: 10.1161/CIRCULATIONAHA.109.893461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franck U, Odeh S, Wiedensohler A, Wehner B, Herbarth O. The effect of particle size on cardiovascular disorders--the smaller the worse. Sci Total Environ. 2011;409:4217–4221. doi: 10.1016/j.scitotenv.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 7.Mills NL, Amin N, Robinson SD, Anand A, Davies J, Patel D, de la Fuente JM, Cassee FR, Boon NA, Macnee W, Millar AM, Donaldson K, Newby DE. Do inhaled carbon nanoparticles translocate directly into the circulation in humans? Am J Respir Crit Care Med. 2006;173:426–431. doi: 10.1164/rccm.200506-865OC. [DOI] [PubMed] [Google Scholar]
- 8.Brook RD, Rajagopalan S, Pope CA 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 9.Dominici F, Peng RD, Bell ML, Pham L, McDermott A, Zeger SL, Samet JM. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA. 2006;295:1127–1134. doi: 10.1001/jama.295.10.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franchini M, Mannucci PM. Thrombogenicity and cardiovascular effects of ambient air pollution. Blood. 2011;118:2405–2412. doi: 10.1182/blood-2011-04-343111. [DOI] [PubMed] [Google Scholar]
- 11.Peters A, Dockery DW, Muller JE, Mittleman MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103:2810–2815. doi: 10.1161/01.cir.103.23.2810. [DOI] [PubMed] [Google Scholar]
- 12.D’Ippoliti D, Forastiere F, Ancona C, Agabiti N, Fusco D, Michelozzi P, Perucci CA. Air pollution and myocardial infarction in Rome: a casecrossover analysis. Epidemiology. 2003;14:528–535. doi: 10.1097/01.ede.0000082046.22919.72. [DOI] [PubMed] [Google Scholar]
- 13.Peters A, Liu E, Verrier RL, Schwartz J, Gold DR, Mittleman M, Baliff J, Oh JA, Allen G, Monahan K, Dockery DW. Air pollution and incidence of cardiac arrhythmia. Epidemiology. 2000;11:11–17. doi: 10.1097/00001648-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Dockery DW, Luttmann-Gibson H, Rich DQ, Link MS, Mittleman MA, Gold DR, Koutrakis P, Schwartz JD, Verrier RL. Association of air pollution with increased incidence of ventricular tachyarrhythmias recorded by implanted cardioverter defibrillators. Environ Health Perspect. 2005;113:670–674. doi: 10.1289/ehp.7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz J, Morris R. Air pollution and hospital admissions for cardiovascular disease in Detroit, Michigan. Am J Epidemiol. 1995;142:23–35. doi: 10.1093/oxfordjournals.aje.a117541. [DOI] [PubMed] [Google Scholar]
- 16.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007;356:447–458. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 17.Madrigano J, Kloog I, Goldberg R, Coull BA, Mittleman MA, Schwartz J. Long-term exposure to PM2.5 and incidence of acute myocardial infarction. Environ Health Perspect. 2013;121:192–196. doi: 10.1289/ehp.1205284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunzli N, Jerrett M, Garcia-Esteban R, Basagana X, Beckermann B, Gilliland F, Medina M, Peters J, Hodis HN, Mack WJ. Ambient air pollution and the progression of atherosclerosis in adults. PLoS One. 2010;5:e9096. doi: 10.1371/journal.pone.0009096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, Fayad ZA, Fuster V, Lippmann M, Chen LC, Rajagopalan S. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294:3003–3010. doi: 10.1001/jama.294.23.3003. [DOI] [PubMed] [Google Scholar]
- 20.Wauters A, Dreyfuss C, Pochet S, Hendrick P, Berkenboom G, van de Borne P, Argacha JF. Acute exposure to diesel exhaust impairs nitric oxide-mediated endothelial vasomotor function by increasing endothelial oxidative stress. Hypertension. 2013;62:352–358. doi: 10.1161/HYPERTENSIONAHA.111.00991. [DOI] [PubMed] [Google Scholar]
- 21.Li R, Kou X, Geng H, Xie J, Tian J, Cai Z, Dong C. Mitochondrial damage: an important mechanism of ambient PM2.5 exposure-induced acute heart injury in rats. J Hazard Mater. 2015;287:392–401. doi: 10.1016/j.jhazmat.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Zou Y, Jin C, Su Y, Li J, Zhu B. Water soluble and insoluble components of urban PM2.5 and their cytotoxic effects on epithelial cells (A549) in vitro. Environ Pollut. 2016;212:627–635. doi: 10.1016/j.envpol.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Q, Chen H, Yang T, Rui W, Liu F, Zhang F, Zhao Y, Ding W. Direct effects of airborne PM2.5 exposure on macrophage polarizations. Biochim Biophys Acta. 2016;1860:2835–2843. doi: 10.1016/j.bbagen.2016.03.033. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Ji X, Ku T, Sang N. Inflammatory response and endothelial dysfunction in the hearts of mice co-exposed to SO2, NO2, and PM2.5. Environ Toxicol. 2015 doi: 10.1002/tox.22200. [DOI] [PubMed] [Google Scholar]
- 25.Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015:4. doi: 10.1161/JAHA.115.002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nohria A, Gerhard-Herman M, Creager MA, Hurley S, Mitra D, Ganz P. Role of nitric oxide in the regulation of digital pulse volume amplitude in humans. J Appl Physiol (1985) 2006;101:545–548. doi: 10.1152/japplphysiol.01285.2005. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Goodson JM, Zhang B, Chin MT. Air pollution and adverse cardiac remodeling: clinical effects and basic mechanisms. Front Physiol. 2015;6:162. doi: 10.3389/fphys.2015.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park SK, O’Neill MS, Vokonas PS, Sparrow D, Schwartz J. Effects of air pollution on heart rate variability: the VA normative aging study. Environ Health Perspect. 2005;113:304–309. doi: 10.1289/ehp.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song X, Liu Y, Hu Y, Zhao X, Tian J, Ding G, Wang S. Short-Term exposure to air pollution and cardiac arrhythmia: a meta-analysis and systematic review. Int J Environ Res Public Health. 2016:13. doi: 10.3390/ijerph13070642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pieters N, Plusquin M, Cox B, Kicinski M, Vangronsveld J, Nawrot TS. An epidemiological appraisal of the association between heart rate variability and particulate air pollution: a meta-analysis. Heart. 2012;98:1127–1135. doi: 10.1136/heartjnl-2011-301505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buteau S, Goldberg MS. A structured review of panel studies used to investigate associations between ambient air pollution and heart rate variability. Environ Res. 2016;148:207–247. doi: 10.1016/j.envres.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 32.Baja ES, Schwartz JD, Wellenius GA, Coull BA, Zanobetti A, Vokonas PS, Suh HH. Trafficrelated air pollution and QT interval: modification by diabetes, obesity, and oxidative stress gene polymorphisms in the normative aging study. Environ Health Perspect. 2010;118:840–846. doi: 10.1289/ehp.0901396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viehmann A, Hertel S, Fuks K, Eisele L, Moebus S, Mohlenkamp S, Nonnemacher M, Jakobs H, Erbel R, Jockel KH, Hoffmann B. Long-term residential exposure to urban air pollution, and repeated measures of systemic blood markers of inflammation and coagulation. Occup Environ Med. 2015;72:656–663. doi: 10.1136/oemed-2014-102800. [DOI] [PubMed] [Google Scholar]
- 34.Zanobetti A, Baccarelli A, Schwartz J. Geneair pollution interaction and cardiovascular disease: a review. Prog Cardiovasc Dis. 2011;53:344–352. doi: 10.1016/j.pcad.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung MH, Kim HR, Park YJ, Park DS, Chung KH, Oh SM. Genotoxic effects and oxidative stress induced by organic extracts of particulate matter (PM 10) collected from a subway tunnel in Seoul, Korea. Mutat Res. 2012;749:39–47. doi: 10.1016/j.mrgentox.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mancini JA, Blood K, Guay J, Gordon R, Claveau D, Chan CC, Riendeau D. Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharideinduced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276:4469–4475. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- 38.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 39.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. 2006;103:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3003787. 132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, Grant G, Cheng Y, Lawson JA, Assoian RK, Jones PL, Fitzgerald GA. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011;123:631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Pure E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 43.Pope CA, Bhatnagar A, McCracken J, Abplanalp WT, Conklin DJ, O’Toole TE. Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circ Res. 2016;119:1204–1214. doi: 10.1161/CIRCRESAHA.116.309279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006;26:1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 45.Grosser T, Yu Y, Fitzgerald GA. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 46.Yang HM, Kim HS, Park KW, You HJ, Jeon SI, Youn SW, Kim SH, Oh BH, Lee MM, Park YB, Walsh K. Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal hyperplasia through inhibition of Akt signaling. Circulation. 2004;110:301–308. doi: 10.1161/01.CIR.0000135467.43430.16. [DOI] [PubMed] [Google Scholar]
- 47.Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, Ohmiya Y, Watanabe K, Kudo I. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278:37937–37947. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- 48.Jania LA, Chandrasekharan S, Backlund MG, Foley NA, Snouwaert J, Wang IM, Clark P, Audoly LP, Koller BH. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E2 biosynthesis. Prostaglandins Other Lipid Mediat. 2009;88:73–81. doi: 10.1016/j.prostaglandins.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275:32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- 50.Lovgren AK, Kovarova M, Koller BH. cPGES/p23 is required for glucocorticoid receptor function and embryonic growth but not prostaglandin E2 synthesis. Mol Cell Biol. 2007;27:4416–4430. doi: 10.1128/MCB.02314-06. [DOI] [PMC free article] [PubMed] [Google Scholar]