

Abstract
MicroRNAs (miRNAs) act as tumor regulators in T-cell acute lymphoblastic leukemia (T-ALL). However, the molecular mechanisms by which miRNA-139 (miR-139) regulates T-ALL remain unclear. In this study, we found that miR-139 was lowly expressed whereas C-X-C chemokine receptor type 4 (CXCR4) was highly expressed in T-ALL cell lines and patient samples. The T-ALL patients simultaneously with high levels of CXCR4 and low expression of miR-139 possessed poor prognosis. Moreover, the introduction of miR-139 inhibited T-ALL cell proliferation and invasion in vitro and suppressed tumor growth and lung metastasis in vivo. CXCR4 was identified as a direct target of miR-139. The suppressive effects of miR-139 were mimicked and counteracted by CXCR4 depletion and overexpression, respectively. Overall, the miR-139/CXCR4 axis plays an important role in T-ALL carcinogenesis.
Keywords: miR-139, CXCR4, T-ALL
Introduction
Acute lymphoblastic leukemia (ALL) is a hematologic malignancy arising from hematopoietic precursors of lymphoid cells and is the most common type of leukemia. T-cell acute lymphoblastic leukemia (T-ALL) accounts for 15% and 25% of pediatric and adult ALL cases, respectively [1]. T-ALL originates from the thymus and spreads fatally to other organs. Despite great improvement in T-ALL therapy by multi-agent combination chemotherapy, the overall survival rate is still about 60%-70% in children and only 30%-40% in adults [2,3]. Thus, identifying the factors and potential mechanisms contributing to T-ALL and securing advanced treatments are necessary and urgent.
microRNAs (miRNAs) are small noncoding RNAs that negatively regulate the gene expression by mRNA degradation or translational repression [4]. The discovery of miRNAs facilitates a new understanding of carcinogenesis, particularly leukemogenesis [5-7]. Dysregulated miRNA disrupts the hematopoietic system and causes leukemia. Several miRNAs, such as miR-203 [8], miRNA-19 [9], miRNA-193b-3p [10], and miRNA-142-3p [11], are involved in T-ALL development and progression. Some studies have demonstrated the aberrant expression and function as well as regulatory network of miR-139 in leukemia [12,13]. However, the biological functions of miR-139 in T-ALL are still unclear.
C-X-C chemokine receptor type 4 (CXCR4) is a common chemokine receptor that is highly expressed in many human cancers, such as breast cancer, ovarian cancer, melanoma, and prostate cancer [14]. The C-X-C motif ligand (CXCL) 12/CXCR4 axis is regulated by various oncogenic pathways and positively correlates with metastasis and poor prognosis; hence, targeting CXCR4 could be an effective strategy for cancer therapy [15,16]. Previous reports highlighted the essential role of CXCL12/CXCR4 in acute myeloid leukemia and B-cell acute lymphoblastic leukemia [17,18]. Recent studies have illuminated the importance of CXCL12/CXCR4 signaling in the maintenance and progression of T-ALL [19,20]. Nevertheless, the function and regulatory mechanism of CXCR4 in T-ALL remain unknown.
In this study, we investigated the expression and function link between miR-139 and CXCR4 and their biological roles in T-ALL. We demonstrated that miR-139 reduces T-ALL cell proliferation and invasion in vitro by directly targeting CXCR4. MiR-139 overexpression or CXCR4 depletion impaired T-ALL growth and lung metastasis in vivo. The inverse correlation between miR-139 and CXCR4 was also identified in T-ALL cells and patient samples. The patients with high expression of miR-139 and low levels of CXCR4 had long survival time. Overall, the results suggest that miR-139/CXCR4 axis is a promising therapeutic target for T-ALL.
Materials and methods
Patients
Forty patients without radiotherapy and chemotherapy signed written consent were enrolled in this study. The patients from 2007 to 2009 were diagnosed basing on routine morphological evaluation, immunophenotyping and cytochemical smears according to the WHO criteria and then followed up to 2014 or until death. This study was approved by the Ethics Committee of the First Affiliated Hospital of Henan University of Science and Technology.
Bone marrow (BM) sample collection and isolation of T cells from BM
BM samples were harvested for mononuclear cells isolation from all the patients and healthy donors by using Ficoll-Paque PLUS (Amersham Biosciences, Uppsala, Sweden) density-gradient centrifugation. Normal T cells were isolated from mononuclear cells of the healthy donors by MACS depletion (Miltenyi Biotec, Bergisch Gladbach, Germany).
Cell lines
Human T-ALL cell lines (HPB-ALL, TALL-1, KOPTK1, Jurkat, CCRF-CEM, and Molt16) and HEK293T cell line were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). T-ALL cell lines were cultured in Roswell Park Memorial Institute 1640 (RPMI1640; Gibco, BRL, Grand Island, USA) supplemented with 10% fetal bovine serum (FBS; Gibco). HEK293T cells were grown in Dulbecco’s Modified Eagle’s Medium (Gibco) containing 10% FBS. All cells were incubated in a humidified incubator with 5% CO2 at 37°C. Cells from passages 2-4 were used for experiments.
Establishment of CCRF-CEM-luciferase (luc) cell line
CCRF-CEM-luc cell line was established as previously described [21,22]. Lentivirus pLV-luc was purchased from Inovogen Biotechnology (Delhi, India) and infected with CCRF-CEM cells. After 16 days of screening with puromycin (200 μg/ml; Sigma, St. Louis, MO, USA), a single clone with stable luciferase expression was obtained and named CCRF-CEM-luc cell line.
Cell transfection and infection
MiR-139 mimics and its negative control RNA (miR-NC) were synthesized by Qiagen (Hilden, Germany). Plasmid pcDNA3.1-CXCR4 was obtained from Biovector (Beijing, China). Transfection was performed by electroporation using Amaxa NucleofectorTM Device (Lonza, Germany) following the manufacturer’s instructions. CCRF-CEM cells were transfected with 100 nM miR-139 mimics or miR-NC or co-transfected with 100 nM miR-139 mimics and 2 μg of CXCR4-expressing plasmid in six-well plates for in vitro experiments. Lentivirus pGCsilence (pGCsi), pGCsi-miR-139, pLKO.1, and pLKO.1-shCXCR4 were purchased from GenePharma (Shanghai, China) and infected with CCRF-CEM-luc cells. All constructs were confirmed by DNA sequencing. After 12 days of screening with puromycin (5 μg/ml; Sigma), stable clones were generated and harvested for in vivo assays.
Dual-luciferase reporter assay
For the dual-luciferase report assay, the wild type (WT) 3’-UTR of CXCR4 and a variant containing mutations in the putative miR-139 binding sites were inserted downstream of the firefly luciferase gene in the pGL3 vector (Promega, Madison, WI, USA). The primers used to amplify the WT and mutant (MUT) 3’-UTRs are listed as follows. WT 3’-UTR, forward: 5’-ATACTCGAGAGTCAACATGCCTGCCCCAAAC A-3’ and reverse: 5’-CACGCGGCCGCCTAGACAGACAAGGAAAGTTTAATGG-3’. MUT 3’-UTR, forward: 5’-ATTACGACATGTATCAATGCATAGGGAAGGAA-3’ and reverse: 5’-AAGGGTCGTGGCTCCCATGCTCCACGTGAAA-3’. All constructs were confirmed by DNA sequencing. CCRF-CEM cells were co-transfected with reporter constructs, an internal control vector (pGL3), and a synthetic miR-139 mimic. After 48 h of transfection, luciferase activity, normalized to the activity of Renilla, was determined using the Dual-Luciferase Reporter Assay System (Promega) and a luminometer (Glomax 20/20; Promega) following the manufacturer’s protocol.
RNA extraction and quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from the cells by using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. To quantify the miR-139 expression, complementary DNA (cDNA) was synthesized with miScript Reverse Transcription Kit (Qiagen) and then amplified using SYBR Premix Ex Taq™ (TaKaRa, Otsu, Shiga, Japan). U6 was used as an internal control. To quantify the mRNA levels of CXCR4, cDNA was generated using the Reverse Transcription Kit (Promega). qPCR assay was performed using a standard IQTM SYBR Green Supermix kit (Bio-Rad, Berkeley, USA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous control. The used primer pairs were miR-139, 5’-TCTACAGTGCACGTGTCTCCAG-3’; U6 forward 5’-CTCGCTT CGGCAGCAC A-3’, U6 reverse 5’-AACGCTTCACGAATTTGCGT-3’; CXCR4 forward 5’-GTAGA GCGAGTGTTGCCATG-3’, CXCR4 reverse 5’-TTGAAATGGACGTTTTCATCC-3’; and GAPDH forward 5’-ACACCCACTCCTCCACCTTT-3’, GAPDH reverse 5’-TTA CTCCTTGGAGGCCATGT-3’. Gene expression was measured in triplicate, quantified using the 2-ΔΔCT method, and normalized to a control. All qPCR assays were performed on an Applied Biosystems 7500 system (Applied Biosystems, Warrington, UK).
Cell stimulation
Cell stimulation was carried out as previously described [22]. After being serum- starved for 5 h at 37°C, cells were stimulated with 100 ng/ml of CXCL12 for various time periods. After stimulation, cells were harvested for analysis.
Cell proliferation assay
Cell proliferation was determined by Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). CCRF-CEM cells were seeded into 96-well plates at a density of 1 × 103 cells per well (n = 5 for each time point) in a final volume of 100 μl. The cells were cultured for 1, 2, 3, or 4 days after transfection with miR-NC or miR-139 mimics or miR-139 mimics + CXCR4-expressing plasmid. In accordance with the manufacturer’s instructions for CCK-8, CCK-8 solution (10 μl) was added to each well, and the absorbance at 450 nm was measured after incubation for 3 h to calculate the number of viable cells.
Colony formation assay
The colony-forming activity of CCRF-CEM cells was assessed by using MethoCult H4434 (Stem Cell Technologies, Vancouver, BC, Canada) in accordance with the manufacturer’s instructions. After 24 h of transfection with RNA oligonucleotides, cells were resuspended at 1 × 104 cells/ml of MethoCult methylcellulose-based medium. All conditions were performed in triplicate at 37°C and 5% CO2, and colonies were counted after 14 days after staining with 1% crystal violet (Sigma).
Cell cycle analysis
CCRF-CEM cells were seeded into 12-well plates. After 48 h of transfection with RNA oligonucleotides, the cells were harvested, resuspended in PBS, and fixed with ice-cold 70% ethanol and treated with 1 mg/ml RNase at 4°C overnight. Intracellular DNA was labeled with 10 μl of propidium iodide (PI, 50 μg/ml; Sigma) at 37°C for 30 min and then analyzed using a BD FACSCalibur flow cytometer (BD Technologies, Carlsbad, CA, USA). The cell proportions in the G1, S, and G2/M phases were calculated using ModFit software (Verity Software House Inc., Topsham, ME, USA).
Apoptosis determination
Apoptosis assay was performed using Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit I (BD Bioscience, San Diego, CA, USA) following the manufacturer’s protocol. CCRF-CEM cells were treated with 10 μg/ml of Vincristine (VCR) before transfection. After 48 h of transfection with RNA oligonucleotides, cells were harvested, centrifuged, and resuspended in 100 μl of FITC-binding buffer. Approximately 5 μl of ready-to-use Annexin V-FITC (BD Bioscience) and 5 μl of PI were added to the mixture. Cells were incubated for 30 min in the dark. Annexin V-FITC and PI fluorescence were assessed by BD FACSCalibur flow cytometer (BD Technologies) and analyzed by CellQuest software (BD Bioscience).
Migration and invasion assays
An 8-μm Transwell insert (Costar, Dallas, TX, USA) was used for the migration assay. A total of 5 × 103 cells were suspended in the serum-free medium and seeded into the upper chamber of the Transwell, and RPMI 1640 medium containing 100 ng/ml CXCL12 was added to the lower chamber. For invasion assays, 1 × 104 cells were suspended in serum-free medium and added to the upper chamber of 8-μm pore-size Transwells precoated with 200 μl of Matrigel (BD Bioscience) at a concentration of 200 μg/ml. The medium with 100 ng/ml CXCL12 was added to the lower chamber as a chemoattractant. After 24 h of incubation, the filters were fixed in methanol and stained with 4’-6-diamidino-2-phanylindole (DAPI; Sigma). After cells that failed to migrate or invade through the pores were carefully removed, five random fields were counted per chamber under an inverted florescent microscope (Carl Zeiss, Berlin, Germany).
Western blotting
Protein was extracted, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto nitrocellulose membranes (Millipore, Bedford, MA, USA). The membranes were blocked in Tris-buffered saline with 0.2 % of Tween 20 (TBST) containing 5% non-fat milk. Western blotting was performed with primary antibodies targeting CXCR4 (Abcam, Cambridge, UK) and β-actin (Abnova, Taiwan, China), followed by the secondary antibody conjugated with horseradish peroxidase (Sigma). Immunoreactivity was visualized by enhanced chemiluminescence kit (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in accordance with the manufacturer’s instruction.
Terminal transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL assay was performed as previously described [20]. Tissue specimens were fixed with 10% formalin overnight, embedded with paraffin, non-serially sectioned (4 μm), and mounted onto poly-L-lysine-coated slides. After deparaffinization in xylene and rehydration in a graded series of ethanol solutions, the sections were rinsed with PBS and incubated with FITC-labeled terminal deoxynucleotidyl transferase nucleotide mix (Roche, Mannheim, BW, Germany) at 37°C for 60 min according to the manufacturer’s protocol. Subsequently, the sections were washed twice in PBS and counterstained with 10 mg/ml DAPI. TUNEL-positive cells were imaged, mounted using a fluorescent microscope (Carl-Zeiss), and ultimately expressed as a percentage of the total cells determined by DAPI staining.
In vivo tumor growth, metastasis, and apoptosis assays
Animal experiments were formally approved by the Institutional Committee for Animal Research and performed in conformity with the national guidelines for the care and use of laboratory animals. For tumor growth assays, six-week-old female mice with severe combined immune deficiency (SCID; Institute of Zoology, Chinese Academy of Sciences, Beijing, China) received subcutaneous injections of 1 × 106 CCRF-CEM-luc cells with stably expressing miR-139 or shCXCR4 or their controls (n = 6 mice/group) through the hind flank. At 10, 20, 30, 40, and 50 days after inoculation, tumor volumes were monitored and calculated as follows: tumor volume = width2 × length/2. All mice were sacrificed at 50 days post-inoculation, and tumors were removed and weighed. For apoptosis assay, tumor tissues were sectioned and stained with TUNEL kits. TUNEL-positive cells were examined and calculated under a fluorescence microscope (Carl Zeiss). Mice were injected with 5 × 106 CCRF-CEM-luc cells with a stable expression of miR-139 or shCXCR4 or their controls through the tail vein to establish the tumor metastasis model. Mice were sacrificed at 5 weeks post-injection, and lungs were removed and embedded in paraffin. Three non-sequential sections per animal were obtained. And sections were stained with hematoxylin/eosin (Maixin Biotech) and analyzed for metastasis by light microscopy (Carl Zeiss). The total number of metastases per lung section was determined and averaged.
Bioluminescence imaging and quantification
Bioluminescence imaging was conducted as previously described [22]. Six-week-old female SCID mice were subcutaneously injected with 1 × 106 CCRF-CEM-luc cells infected with control lentivirus or lentivirus expressing miR-139 or shCXCR4. Tumor growth was assessed by in vivo luciferase imaging of the xenografts at 15 days after treatment. Mice were intraperitoneally injected with D-luciferin (Promega) at a dose of 150 mg/kg per mouse and anesthetized during image acquisition with the Xenogen IVIS imaging system. Signals in the defined regions of interest were quantified as luminescence radiance (photons/s/cm2/sr) by Living Image software (Xenogen Corporation, Berkeley, CA, USA).
Statistical analysis
Data are expressed as means ± standard deviation (SD) from three independent experiments. Data were analyzed by Student’s t-test and analysis of variance (ANOVA). Survival curves were plotted using the Kaplan-Meier method and the survival rate was analyzed by Log-rank test. Pearson correlation analysis was conducted to assess the statistical significance between cases with high or low levels of miR-139 or CXCR4. All P values were two-sided and obtained using SPSS 13.0 software package (SPSS Inc., IL, USA). P < 0.05 was considered statistically significant.
Results
Reduced miR-139 expression and increased CXCR4 levels in T-ALL cells
We initially found that miR-139 levels were significantly lower in T-ALL cells (HPB-ALL, TALL-1, KOPTK1, Jurkat, CCRF-CEM, and Molt16) than that in normal T cells (Figure 1A). qPCR and Western blot analyses showed that CXCR4 expression was enhanced in T-ALL cell lines compared with normal T cells (Figure 1B and 1C). Moreover, CXCR4 expression was inversely correlated with miR-139 level in T-ALL cell lines (Figure 1D). The CCRF-CEM cell line with the lowest level of miR-139 and the highest level of CXCR4 was selected for further studies. These results suggest that miR-139 and CXCR4 play key roles in T-ALL development.
Figure 1.

Expression levels of miR-139 and CXCR4 in T-ALL cell lines. (A and B) qPCR assays were conducted to assess miR-139 expression (A) and CXCR4 mRNA levels (B) in T-ALL cell lines (HPB-ALL, TALL-1, KOPTK1, Jurkat, CCRF-CEM, and Molt16) and normal T cells. U6 and GAPDH were used as the internal controls, respectively. (C) Protein levels of CXCR4 in T-ALL cell lines and normal T cells were determined by Western blot. β-actin was used as the endogenous control. (D) Inverse correlation between miR-139 and CXCR4 levels in T-ALL cell lines. All data are shown as mean ± SD of three separate experiments. *P < 0.05, **P < 0.01 vs. normal T cell group.
MiR-139 directly targets CXCR4 in T-ALL cells
The targets of miR-139 were predicted using three algorithms, namely, TargetScan, PicTar, and miRBase (Figure 2A). Figure 2B shows a complementary sequence of miR-139 to the 3’-UTR of CXCR4 mRNA. Dual-luciferase reporter assay revealed that miR-139 decreased the activity of the luciferase reporter fused to the 3’-UTR-WT of CXCR4 but did not inhibit that of the reporter fused to the MUT version (Figure 2C). Moreover, the introduction of miR-139 reduced the mRNA and protein levels of CXCR4 in CCRF-CEM cells (Figure 2D and 2E). These data demonstrate that miR-139 directly targets CXCR4 in T-ALL cells.
Figure 2.

Identification of CXCR4 as a direct target of miR-139. (A) Putative target genes of miR-139 were predicted using TargetScan, PicTar, and miRBase software. (B) Predicted binding sites of miR-139 in the MUT and WT 3’-UTR of CXCR4. (C) Dual-luciferase reporter assays were performed 24 h after co-transfection of CCRF-CEM cells with miR-NC or miR-139 mimics and a pGL3 construct containing the WT or MUT 3’-UTR of CXCR4. Data were normalized to those from cells co-transfected with miR-NC and pGL3 plasmid. mRNA (D) and protein (E) levels of CXCR4 in CCRF-CEM cells transfected with miR-NC or miR-139 mimics were detected by qPCR and Western blot assays. GAPDH and β-actin were used as internal controls. All data are shown as mean ± SD of three separate experiments. **P < 0.01 vs. miR-NC group. WT: wild-type; MUT: mutant.
MiR-139 inhibits T-ALL cell proliferation and apoptosis resistance by targeting CXCR4
We investigated whether CXCR4 is a functional target of miR-139. CCRF-CEM cells were treated with miR-139 mimic or miR-NC or miR-139 mimic + CXCR4-expressing plasmid in the presence of 100 ng/ml CXCL12. Cell viability significantly decreased in the miR-139-transfected cells compared with the miR-NC-treated cells, which was markedly attenuated by CXCR4 overexpression (Figure 3A). MiR-139 reduced the colony formation of CCRF-CEM cells, and the ectopic expression of CXCR4 rescued the inhibitory effects of miR-139 (Figure 3B). The increase in G1 phase and decrease in G2/M phases by miR-139 were counteracted by CXCR4 restoration (Figure 3C). As shown in Figure 3D, miR-139 augmented the percentage of apoptotic CCRF-CEM cells pretreated with 10 μg/ml VCR, which was neutralized by CXCR4 overexpression. These results indicate that miR-139 inhibits CXCR4-elicited proliferation and apoptosis resistance in T-ALL cells.
Figure 3.
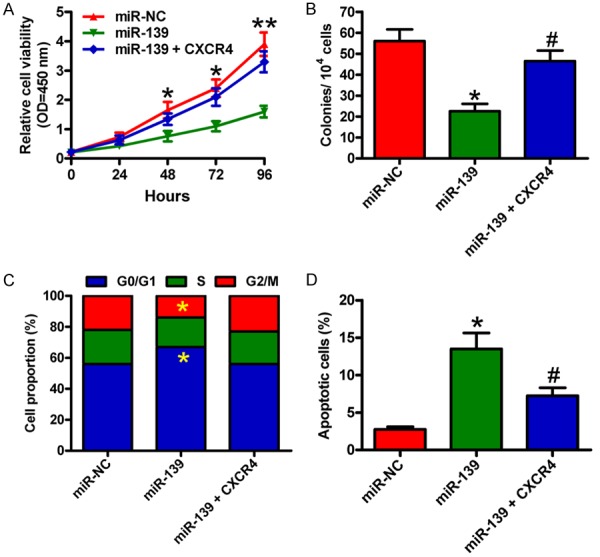
MiR-139 reduced the proliferation and apoptosis resistance of T-ALL cells by targeting CXCR4. CCRF-CEM cells were transfected with miR-NC or miR-139 mimics or miR-139 mimics + CXCR4-expressing plasmid. A. CCK-8 assay was carried out to determine cell viability. B. Quantitative analysis of colony formation of CCRF-CEM cells. C. Flow cytometry analysis of CCRF-CEM cell fraction in G1, S, and G2/M phases after 48 h of transfection. D. CCRF-CEM cells subjected to VCR pretreatment were transfected with miR-NC or miR-139 mimics or miR-139 mimics + CXCR4-expressing plasmid. Apoptosis was measured by flow cytometry. All data are shown as mean ± SD of three separate experiments. *P < 0.05, **P < 0.01 vs. miR-NC group; #P < 0.05 vs. miR-139 group.
MiR-139 reduces the migration and invasion of T-ALL cells by downregulating CXCR4
Whether miR-139 suppresses the migration and invasion of T-ALL cells by targeting CXCR4 was evaluated by Transwell assays. As expected, miR-139 inhibited the migration of CCRF-CEM cells, whereas CXCR4 overexpression partially counteracted the decrease (Figure 4A and 4B). Similarly, the invasion of CCRF-CEM cells was reduced by miR-139, and the inhibitory effect was attenuated by CXCR4 restoration (Figure 4C and 4D). These results demonstrate that miR-139 potently suppresses the CXCR4-mediated migration and invasion of T-ALL cells.
Figure 4.

MiR-139 inhibited the CXCR4-mediated migration and invasion of T-ALL cells. CCRF-CEM cells were transfected with miR-NC or miR-139 mimics or miR-139 mimics + CXCR4-expressing plasmid. (A and B) Migration assay was performed for CCRF-CEM cells. Images were captured after 24 h of transfection (A), and the number of migrated cells was calculated (B). (C and D) Cell invasion was detected using Transwell chambers (C), and the number of invaded cells was quantified (D). All data are shown as the mean ± SD of three separate experiments. Scale: 10 μm. *P < 0.05 vs. miR-NC group; #P < 0.05 vs. miR-139 group.
MiR-139 overexpression or CXCR4 depletion retards tumorigenesis and metastasis of T-ALL in vivo
A xenograft or metastasis mouse model was established by the subcutaneous or intravenous injection of CCRF-CEM-luc cells stably expressing a miR-139 precursor or shCXCR4 or their vector controls. Compared with the control groups, the miR-139 overexpression or CXCR4 knockdown group exhibited significant reductions in tumor growth (Figure 5A), volume (Figure 5B), and weight (Figure 5C). Apoptotic cells were much more in the tumors of the ectopic miR-139 expression or CXCR4-silencing group than that in the tumors derived from the control groups (Figure 5D). MiR-139 restoration or CXCR4 depletion also effectively retarded lung metastases in vivo (Figure 5E). These results indicate that the suppression of T-ALL growth and lung metastasis by miR-139 is mediated by CXCR4 reduction in vivo.
Figure 5.

MiR-139 overexpression or CXCR4 knockdown suppressed tumor growth and metastasis and promoted T-ALL cell apoptosis in vivo. SCID mice were injected subcutaneously or intravenously with CCRF-CEM-luc cells that were infected with a control lentivirus (Lenti-pGCsi or Lenti-pLKO.1) or a recombinant lentivirus expressing a miR-139 precursor (Lenti-pGCsi-miR-139) or shCXCR4 (Lenti-shCXCR4). A. In vivo luciferase image for the detection of xenograft growth. B. Tumor volume was measured and calculated every 10 days. C. Tumor weight was measured after 50 days of implantation. D. TUNEL assay was conducted to detect the percentage of apoptotic cells. E. Numbers of metastatic foci in the lungs from various groups at 5 weeks after tail vein injection. All data are shown as mean ± SD of three separate experiments. *P < 0.05, **P < 0.01 vs. Lenti-pGCsi or Lenti-pLKO.1 group.
MiR-139 and CXCR4 expression levels are highly associated with the prognosis of T-ALL
Clinically, we found that miR-139 expression was markedly downregulated in T-ALL samples compared with those from healthy donors (Figure 6A). This finding is consistent with the results among T-ALL cells. By contrast, qPCR analysis showed that CXCR4 expression was higher in T-ALL samples than in specimens from healthy donors (Figure 6B). Intriguingly, CXCR4 level inversely correlated with miR-139 level in T-ALL patients (Figure 6C). Moreover, the five-year overall survival rate of patients with high miR-139 levels and low CXCR4 expression was higher than that of patients with low miR-139 levels and high CXCR4 expression (Figure 6D). These findings suggest that miR-139 expression is negatively associated with CXCR4 level in T-ALL specimens and that the combination of miR-139 and CXCR4 is a prognostic indicator for T-ALL patients.
Figure 6.

MiR-139 and CXCR4 levels are highly associated with the prognosis of T-ALL patients. A. qPCR analysis of miR-139 levels in T-ALL samples compared with those from healthy donors (HD) (n = 20). U6 was used as the internal control. B. mRNA levels of CXCR4 in T-ALL and HD specimens were measured by qPCR assays (n = 20). GAPDH was used as the endogenous control. C. Correlation between CXCR4 level and miR-139 expression in T-ALL specimens (n = 20). D. Five-year overall survival rate of T-ALL patients with miR-139 low expression and CXCR4 high level (n = 29) and the patients with miR-139 high expression and CXCR4 low level (n = 11). All data are shown as mean ± SD of three separate experiments. *P < 0.05 vs. HD group; **P < 0.01 vs. HD group. HD: healthy donors.
Discussion
MiRNA and gene expression profiles have provided valuable insights into the molecular mechanisms of leukemia [7]. In this study, we found that miR-139 is downregulated in T-ALL cells and samples, negatively associating with CXCR4 expression. Integrative bioinformatics prediction and Dual-luciferase reporter assay indicated that CXCR4 is a direct target of miR-139. Functionally, the inhibitory effects of miR-139 on T-ALL cell proliferation, apoptosis resistance, and motility were reversed by CXCR4 restoration in vitro. MiR-139 overexpression and CXCR4 depletion significantly reduced the malignant characteristics of T-ALL in vivo. The patients with high miR-139 levels and low CXCR4 expression has higher five-year overall survival rate than the patients with low miR-139 level and high CXCR4 expression. Overall, these results suggest that miR-139 acts as a tumor suppressor in T-ALL by targeting CXCR4.
Reportedly, miR-139 is highly silenced in numerous tumors [13,23-26]. However, the expression of miR-139 in T-ALL has rarely been identified. Herein, we demonstrated that miR-139 was lowly expressed in T-ALL cell lines and patient samples. The elevated miR-139 expression is associated with a favorable outcome in a cohort of 165 pediatric patients with AML [13]. In this study, we found that the five-year overall survival rate of patients with high miR-139 was obviously higher than that of patients with miR-139 low expression. Therefore, miR-139 may contribute to the prognostic assessment of T-ALL patients. Alemdehy et al. [27] evaluated the inhibitory effect of miR-139 in leukemic transformation. Consistently, our results showed that miR-139 significantly inhibited CCRF-CEM cell proliferation, migration, and invasion. These findings indicate that miR-139 acts a suppressor in T-ALL carcinogenesis.
Many studies have revealed that miR-139 targets different effectors, including CXCR4 [25,26]. As an important chemokine receptor, CXCR4 elevation frequently emerges and promotes the growth and metastasis of malignancies [25,26,28-30]. Similarly, our investigation on the intrinsic function of CXCR4 in T-ALL revealed that CXCR4 counteracted the inhibitory effects of miR-139 on the proliferation, apoptosis resistance, and motility of human T-ALL cells in vitro, which is a direct supportive evidence for CXCR4 function. Passaro et al. [19] reported that CXCR4 is highly important for T-ALL propagation in vivo, and their findings are supported by both Notch-induced mouse T-ALL and human T-ALL xenograft models. Consistently, we found that CXCR4 depletion markedly reduced the malignant characteristics of T-ALL in vivo. The upregulation of CXCR4 cell surface expression was also highlighted in diagnostic T-ALL cases [19,20]. We demonstrated that, in opposition to miR-139, CXCR4 was significantly upregulated in T-ALL specimens, and the combination of miR-139 and CXCR4 was confirmed as a potential prognostic indicator in T-ALL patients.
Our study has limitations in deciphering the role of the CXCL12/CXCR4 axis in miR-139-downregulated T-ALL. Previously, Wani et al. [21] illuminated that CXCL12 enhances CXCR4-mediated cell migration and ERK and STAT3 signaling in breast cancer cells. In the present study, we only illuminated the promoting effects of CXCR4 on T-ALL malignancies but did not further identify CXCR4 downstream molecules and their functions.
In summary, miR-139 is downregulated in T-ALL. In vitro and in vivo studies confirmed that miR-139 is a novel suppressor of T-ALL by directly targeting CXCR4. Furthermore, the combination of miR-139 and CXCR4 is a potential prognostic factor in T-ALL patients. Overall, these findings suggest that miR-139 impedes T-ALL growth and metastasis by targeting CXCR4 and highlight the potential role of miR-139/CXCR4 in prognostic evaluation and therapeutic use of T-ALL patients.
Disclosure of conflict of interest
None.
References
- 1.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535–1548. doi: 10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg JM, Silverman LB, Levy DE, Dalton VK, Gelber RD, Lehmann L, Cohen HJ, Sallan SE, Asselin BL. Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber cancer institute acute lymphoblastic leukemia consortium experience. J. Clin. Oncol. 2003;21:3616–3622. doi: 10.1200/JCO.2003.10.116. [DOI] [PubMed] [Google Scholar]
- 3.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, Cheng C, Su X, Rubnitz JE, Basso G, Biondi A, Pui CH, Downing JR, Campana D. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schotte D, Akbari Moqadam F, Lange-Turenhout EA, Chen C, van Ijcken WF, Pieters R, den Boer ML. Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia. 2011;25:1389–1399. doi: 10.1038/leu.2011.105. [DOI] [PubMed] [Google Scholar]
- 6.Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in down syndrome. Blood. 2009;113:2619–2628. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao H, Wang D, Du W, Gu D, Yang R. MicroRNA and leukemia: tiny molecule, great function. Crit Rev Oncol Hematol. 2010;74:149–155. doi: 10.1016/j.critrevonc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Bueno MJ, Perez de Castro I, Gomez de Cedron M, Santos J, Calin GA, Cigudosa JC, Croce CM, Fernandez-Piqueras J, Malumbres M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan AA, Leslie CS, Parker JS, Paddison PJ, Tam W, Ferrando A, Wendel HG. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12:372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mets E, Van der Meulen J, Van Peer G, Boice M, Mestdagh P, Van de Walle I, Lammens T, Goossens S, De Moerloose B, Benoit Y, Van Roy N, Clappier E, Poppe B, Vandesompele J, Wendel HG, Taghon T, Rondou P, Soulier J, Van Vlierberghe P, Speleman F. MicroRNA-193b-3p acts as a tumor suppressor by targeting the MYB oncogene in T-cell acute lymphoblastic leukemia. Leukemia. 2015;29:798–806. doi: 10.1038/leu.2014.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, Hong M, Jiang T, Jiang Q, Lu J, Huang X, Huang B. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (TALL) by targeting glucocorticoid receptor-alpha and cAMP/PKA pathways. Leukemia. 2012;26:769–777. doi: 10.1038/leu.2011.273. [DOI] [PubMed] [Google Scholar]
- 12.Krowiorz K, Ruschmann J, Lai C, Ngom M, Maetzig T, Martins V, Scheffold A, Schneider E, Pochert N, Miller C, Palmqvist L, Staffas A, Mulaw M, Bohl SR, Buske C, Heuser M, Kraus J, O’Neill K, Hansen CL, Petriv OI, Kestler H, Dohner H, Bullinger L, Dohner K, Humphries RK, Rouhi A, Kuchenbauer F. MiR-139-5p is a potent tumor suppressor in adult acute myeloid leukemia. Blood Cancer J. 2016;6:e508. doi: 10.1038/bcj.2016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmrich S, Engeland F, El-Khatib M, Henke K, Obulkasim A, Schoning J, Katsman-Kuipers JE, Michel Zwaan C, Pich A, Stary J, Baruchel A, de Haas V, Reinhardt D, Fornerod M, van den Heuvel-Eibrink MM, Klusmann JH. miR-139-5p controls translation in myeloid leukemia through EIF4G2. Oncogene. 2016;35:1822–1831. doi: 10.1038/onc.2015.247. [DOI] [PubMed] [Google Scholar]
- 14.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 15.Kuo YY, Hou HA, Chen YK, Li LY, Chen PH, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Chou WC, Liu CY, Tang JL, Yao M, Tien HF. The N-terminal CEBPA mutant in acute myeloid leukemia impairs CXCR4 expression. Haematologica. 2014;99:1799–1807. doi: 10.3324/haematol.2014.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 2016;35:816–826. doi: 10.1038/onc.2015.139. [DOI] [PubMed] [Google Scholar]
- 17.Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104:550–557. doi: 10.1182/blood-2004-02-0566. [DOI] [PubMed] [Google Scholar]
- 18.Patel B, Dey A, Castleton AZ, Schwab C, Samuel E, Sivakumaran J, Beaton B, Zareian N, Zhang CY, Rai L, Enver T, Moorman AV, Fielding AK. Mouse xenograft modeling of human adult acute lymphoblastic leukemia provides mechanistic insights into adult LIC biology. Blood. 2014;124:96–105. doi: 10.1182/blood-2014-01-549352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C, Tran Quang C, Ghysdael J. CXCR4 is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell. 2015;27:769–779. doi: 10.1016/j.ccell.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, Morrison SJ, Fooksman DR, Aifantis I, Schwab SR. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell. 2015;27:755–768. doi: 10.1016/j.ccell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao HD, Tang HL, Liu NN, Zhao YL, Liu QQ, Zhu XS, Jia LT, Gao CF, Yang AG, Li JT. Targeting ubiquitin-specific protease 22 suppresses growth and metastasis of anaplastic thyroid carcinoma. Oncotarget. 2016;7:31191–31203. doi: 10.18632/oncotarget.9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JT, Jia LT, Liu NN, Zhu XS, Liu QQ, Wang XL, Yu F, Liu YL, Yang AG, Gao CF. MiRNA-101 inhibits breast cancer growth and metastasis by targeting CX chemokine receptor 7. Oncotarget. 2015;6:30818–30830. doi: 10.18632/oncotarget.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnan K, Steptoe AL, Martin HC, Pattabiraman DR, Nones K, Waddell N, Mariasegaram M, Simpson PT, Lakhani SR, Vlassov A, Grimmond SM, Cloonan N. miR-139-5p is a regulator of metastatic pathways in breast cancer. Rna. 2013;19:1767–1780. doi: 10.1261/rna.042143.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan Q, He M, Deng X, Wu WK, Zhao L, Tang J, Wen G, Sun X, Liu Y. Derepression of c-Fos caused by microRNA-139 down-regulation contributes to the metastasis of human hepatocellular carcinoma. Cell Biochem Funct. 2013;31:319–324. doi: 10.1002/cbf.2902. [DOI] [PubMed] [Google Scholar]
- 25.Bao W, Fu HJ, Xie QS, Wang L, Zhang R, Guo ZY, Zhao J, Meng YL, Ren XL, Wang T, Li Q, Jin BQ, Yao LB, Wang RA, Fan DM, Chen SY, Jia LT, Yang AG. HER2 interacts with CD44 to up-regulate CXCR4 via epigenetic silencing of microRNA-139 in gastric cancer cells. Gastroenterology. 2011;141:2076–2087. e2076. doi: 10.1053/j.gastro.2011.08.050. [DOI] [PubMed] [Google Scholar]
- 26.Luo HN, Wang ZH, Sheng Y, Zhang Q, Yan J, Hou J, Zhu K, Cheng Y, Xu YL, Zhang XH, Xu M, Ren XY. MiR-139 targets CXCR4 and inhibits the proliferation and metastasis of laryngeal squamous carcinoma cells. Med Oncol. 2014;31:789. doi: 10.1007/s12032-013-0789-z. [DOI] [PubMed] [Google Scholar]
- 27.Alemdehy MF, Haanstra JR, de Looper HW, van Strien PM, Verhagen-Oldenampsen J, Caljouw Y, Sanders MA, Hoogenboezem R, de Ru AH, Janssen GM, Smetsers SE, Bierings MB, van Veelen PA, von Lindern M, Touw IP, Erkeland SJ. ICL-induced miR139-3p and miR199a-3p have opposite roles in hematopoietic cell expansion and leukemic transformation. Blood. 2015;125:3937–3948. doi: 10.1182/blood-2014-11-612507. [DOI] [PubMed] [Google Scholar]
- 28.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, Min W, McLendon RE, Rich JN, Bao S. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faber A, Hoermann K, Stern-Straeter J, Schultz DJ, Goessler UR. Functional effects of SDF-1alpha on a CD44(+) CXCR4(+) squamous cell carcinoma cell line as a model for interactions in the cancer stem cell niche. Oncol Rep. 2013;29:579–584. doi: 10.3892/or.2012.2171. [DOI] [PubMed] [Google Scholar]
- 30.Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]