

Abstract
The ZDSD rat is a new obese-diabetic rat model that expresses type 2 diabetes in the presence of an intact leptin pathway. During a long pre-diabetic state, the animals exhibit most of the features of metabolic syndrome including obesity, hyperlipidemia, hypertension, insulin resistance and decreased glucose disposal. The animals used in these studies were either allowed to become spontaneously diabetic at 16-30 weeks of age, or diabetes was induced with a diabetogenic diet. In the presence of either spontaneous or diet-induced diabetes, they develop progressive albuminuria as well as increases in other urinary markers of impaired renal function (kidney injury molecule-1 (KIM-1), β2-microglobulin, clusterin and cystatin C). Typical morphological changes of nephropathy, such as glomerular capillary basement membrane thickening and podocyte effacement, accompany these marker increases. Lisinopril (ACEi) treatment (30 mg/kg/day via the diet) dramatically reduced diabetes-induced albuminuria by 85%, independent of the duration of diabetes or the initial albumin excretion. These results position the ZDSD rat as a relevant model of diabetic nephropathy that can be treated with clinically effective compounds.
Keywords: Diabetes, kidney, nephropathy, renal, rat
Introduction
The physiologic environment of chronic hyperglycemia with obesity is associated with a variety of co-morbidities including cardiovascular disease, atherosclerosis, retinopathy, neuropathy and nephropathy. Prolonged exposure to hyperglycemia is now recognized as a major factor in the pathogenesis of diabetic complications in type 1 and type 2 diabetic patients [1,2]. Chronic hyperglycemia results in accumulation of reactive oxygen species, pro-inflammatory cytokines and advanced glycation end-products (AGE) that ultimately induce end organ damage in vasculature, heart and kidneys [3]. Diabetic nephropathy (DN) is one of the most serious complications of diabetic microvascular changes [4], and is the most common cause of chronic kidney injury leading to end-stage renal disease (ESRD). Chronic hyperglycemia induces hemodynamic as well as metabolic changes in the diabetic kidney [5]. Oxidative stress resulting from hyperglycemia is associated with endothelial dysfunction and the development of hypertension [6]. Hypertension is identified in approximately 50% of patients with type 2 diabetes and metabolic syndrome; the addition of hypertension accelerates the development of DN and ESRD [7]. The prevalence of chronic kidney disease is much higher in patients identified as pre-diabetic or diabetic compared with normoglycemic patients [8]. Insulin resistance, as a precursor to hyperglycemia, is also causative in DN due to the induction of vasoconstriction, sodium retention and arterial hypertension [9].
DN is characterized by albuminuria, increases in serum creatinine and a decreased glomerular filtration rate (GFR). Histological changes include glomerular mesangial expansion, glomerular basement membrane (GBM) thickening, excessive extracellular matrix proteins and fibrosis [7-9]. Severe diabetes is not necessary for the development of DN. Indeed, it has been shown that pre-diabetic patients with impaired glucose tolerance and insulin resistance, before onset of severe hyperglycemia, exhibit the same prevalence for development of chronic kidney disease as overtly diabetic patients [8]. Treatment options for diabetic nephropathy are directed toward lowering blood pressure and improving glycemic control [10]. Angiotensin II receptor blockers (ARB) and angiotensin II converting enzyme inhibitors (ACEi) have been the mainstay to stop or slow progression to ESRD [10-13].
Pre-clinical studies in DN are often carried out in diabetic rodents. The most common model is the STZ rat which employs a normoglycemic rat induced to become diabetic through injection of streptozotocin. This model has been used extensively to evaluate the mechanisms and potential interventions for DN [7,14-16]. The rapid induction of type 1 diabetes in rats after a single dose of STZ (>45 mg/kg) increases blood glucose levels to more than 500 mg/dL within 48 hours [17], and is associated with rapidly developing renal damage. Streptozotocin can also be described as a direct renal toxin; it causes tubular and glomerular hypertrophy with mesangial expansion and thus exerts renal damage in addition to that caused by its induction of beta cell apoptosis and hyperglycemia [17]. The development of renal damage following STZ is likely dramatically different from the development of DN in patients with pre-diabetes/metabolic syndrome who slowly progress to type 2 diabetes.
Rat models with defective leptin receptors, such as the ZDF and ZSF1, have been characterized and used to evaluate compounds to treat DN [18-29]. However, although the ZDF rat does have kidney changes that appear to be specifically associated with the diabetic condition [23], it also has kidney hydronephrosis [30] that makes it less than an ideal model for DN. The ZSF1 rat has been used effectively for the study of DN, but it has the leptin receptor defects from both the ZDF and the SHHF rats [24] which, when combined, cause the obesity in this model. Although these established models have, and likely will continue, to be used in DN studies, the defects in the morphology of the kidney shown by the ZDF, and the leptin receptor defects present in both models are problematic for a translational model.
Mouse models have also been diligently sought as models featuring all of the characteristics of human DN. A recent review by Betz and Conway states that “no model exhibits with all of the features of human DN” [31]. As a consequence, the quest for ideal DN models continues.
The ZDSD/Pco (ZDSD) rat is a new model of obesity, metabolic syndrome and diabetes [32]. The model was developed by crossing a homozygous lean Zucker diabetic fatty (ZDF) male rat with a sub strain of the Crl:CD (SD) rat, selectively bred for high fat diet induced obesity [33,34]. The standard Crl:CD (SD) rat is a sub strain of SD rats that is significantly heavier and more obese than other lines of SD rats; a percentage of these rats is very susceptible to developing obesity, when fed high fat diets [33,34]. The original design was to combine the defect in β-cell gene transcription that is found in lean and obese ZDF rats [35] with the obesity of the Crl:CD (SD) model, to produce an obese diabetic model that preserves the critical leptin pathway. The animals were fed regular rodent chow (Purina 5008) during the 12 years of the model development process. The offspring from the initial crosses were screened and selected for obesity, the propensity to become diabetic and the expression of the other characteristics of metabolic syndrome. This model has been selectively inbred for >30 generations. The ZDSD rat has been shown to develop microvascular and macrovascular complications of diabetes in a fashion similar to that occurring in human diabetes [36]. Spontaneous development of diabetic complications such as impaired wound healing [37], osteoporosis [38,39], decreased nerve conduction velocity [36] and hypertension [40] have also been identified in the model.
Obesity and metabolic syndrome are clear predictors of chronic kidney disease largely due to the potentiation of chronic inflammation by insulin resistance [41]. In addition, the lipoprotein abnormalities and increased hemodynamics and vascular dysfunction associated with metabolic syndrome have all been implicated as causative for the development of renal disease [42,43]. This cluster of metabolic dysregulations is exhibited by the ZDSD rat, positioning the model as a unique tool for the study of DN.
In validation of the ZDSD rat as a novel tool for the study of DN, we have characterized the spontaneous development of DN in the ZDSD rat and determined the efficacy of the ACEi, Lisinopril, in reducing albuminuria.
Materials and methods
Spontaneous development of renal dysfunction in untreated ZDSD rats
Male ZDSD/Pco rats (n=16) were obtained from the PreClinOmics, A Crown Bioscience company (now Crown Bioscience-Indiana), colony. They were maintained on Purina 5008 regular rodent chow from weaning and throughout the study duration. Beginning at 10 weeks of age, blood samples (fed) were collected from the tail vein for assessment of blood glucose, blood urea nitrogen (BUN) and creatinine. Twenty-four-hour urine samples were collected at room temperature and without additives. Blood and urine samples were obtained in animals every 2-4 weeks until 30 weeks of age. Animals were considered diabetic when morning blood glucose (fed) reached ≥250 mg/dL. Urine volume and body weight were recorded. Urine was assayed for cystatin C, clusterin, urinary kidney injury molecule-1 (KIM-1) and β2-microglobulin using Luminex multiplex kits RKTX1-37K (clusterin, KIM-1) and RKTX2-37K (albumin, β2-microglobulin, cystatin C). Kidneys were fixed in 10% buffered formalin and stained with periodic acid Schiff (PAS) and H&E for pathological assessment of untreated 33 week old animals. Age-matched SD rats (n=10) were included for comparison.
Kidneys from selected diabetic animals at 33 weeks of age, as described above, were also prepared for light microscopy. Representative sections were selected for microscopy.
Electron microscopy
Control Crl:CD (SD) (n=2) and ZDSD male rats that were spontaneously diabetic for 12 (n=2) and 16.5 (n=2) weeks were perfused with 4% paraformaldehyde then fixed in 2% paraformaldehyde and 2% glutaraldehyde in 0.1M phosphate buffer. After fixation the specimens were rinsed with phosphate buffered saline (PBS) followed by post fixation with 1% osmium tetroxide in phosphate buffer for one hour. After rinsing with PBS, the tissue specimens were dehydrated through a series of graded ethyl alcohols from 70 to 100%. After dehydration, the specimens were infiltrated with 2 changes of 100% propylene oxide and a 50:50 mixture of propylene oxide and the embedding resin (Embed 812, Electron Microscopy Sciences, Hatfield, PA) overnight. The next day the specimens were transferred to fresh 100% embedding media for a minimum of 2 hours, then embedded in a fresh change of 100% embedding media. Following polymerization overnight at 60°C, the blocks were ready to be sectioned. Thin sections were cut (70-80 nm), stained with uranyl acetate and lead citrate, then viewed on a Tecnai BioTwin (FEI, Hillsboro, OR) with digital images taken with an AMT (Advanced Microscope Techniques, Danvers, MA) CCD camera. Multiple measurements were taken (144 to 162 per group) using the software on the Tecnai of three peripheral capillary loops in each of three glomeruli from each animal. Thickness values that were more than 2.5 SD from the mean were considered outliers and not used leaving 141 to 158 measurements per group.
Effect of Lisinopril on renal function
Male ZDSD rats, were maintained on Purina 5008 chow ad lib until 18 weeks of age. A diabetogenic diet (Purina 5SCA) was initiated and continued for 3 weeks to accelerate the development of hyperglycemia. Animals were then maintained for the duration of the study on Purina 5008 regular rodent chow. In this study, treatment was started 5, 9 and 13 weeks after animals became hyperglycemic (ages 29, 33 and 37 weeks; respectively). In each age group, diabetic rats were sorted into untreated (Purina 5008 chow, n=12) and treated (5008 chow admixed with Lisinopril 250 ppm, n=13) based on body weight and fed glucose level. Treatment was continued for 4 weeks. Blood and 24-hour urine samples were collected before and after 4 weeks of treatment. Urine was collected at room temperature and without preservatives. Fed glucose was measured in whole blood by StatStrip (Xpress II, Novo Biomedical); HbA1c (whole blood), creatinine (serum) and BUN (serum) were assayed in serum using a AU480 clinical analyzer (Beckman Coulter, Brea, CA); albumin and creatinine were measured in urine by MSD (MesoScale Discovery, Rockville, MD) and AU480 analyzer, respectively. Estimated glomerular filtration (eGFR) was calculated using the following formula: Urine creatinine (mg/dL) × urine volume (mls/min)/serum creatinine (mg/dL).
Statistical analysis
All data are presented as Mean ± SEM. Statistical analysis was performed using Prism for Windows (version 6.07 GraphPad, San Diego, CA). With regard to data presented on the spontaneous development of DN in ZDSD rats, a two-way ANOVA was conducted to compare the effect of strain on body weight, blood glucose, urine volume, urine albumin, urine KIM-1, urine clusterin, urine cystatin C and urine β2-microglobulin. Post-hoc comparisons between strains were made using Sidak’s multiple comparison tests and are indicated on graphs. With regard to the effects of Lisinopril, means were compared using one-way ANOVA/pooled t-test or paired t-tests. Significant effects indicated on graphs (*P<0.05).
Results
Spontaneous development of renal dysfunction in untreated ZDSD
ZDSD rats were significantly heavier when compared to age-matched SD rats at 10 weeks of age (380.5 ± 5.0 vs. 326.4 ± 3.6 g) through 26 weeks of age (556.8 ± 7.6 vs. 498.5 ± 6.9 g). ZDSD weight gain leveled at about 20 weeks and subsequently decreased. Body weight in 28-30 week old animals was not significantly different when compared to control SD animals (Figure 1A).
Figure 1.
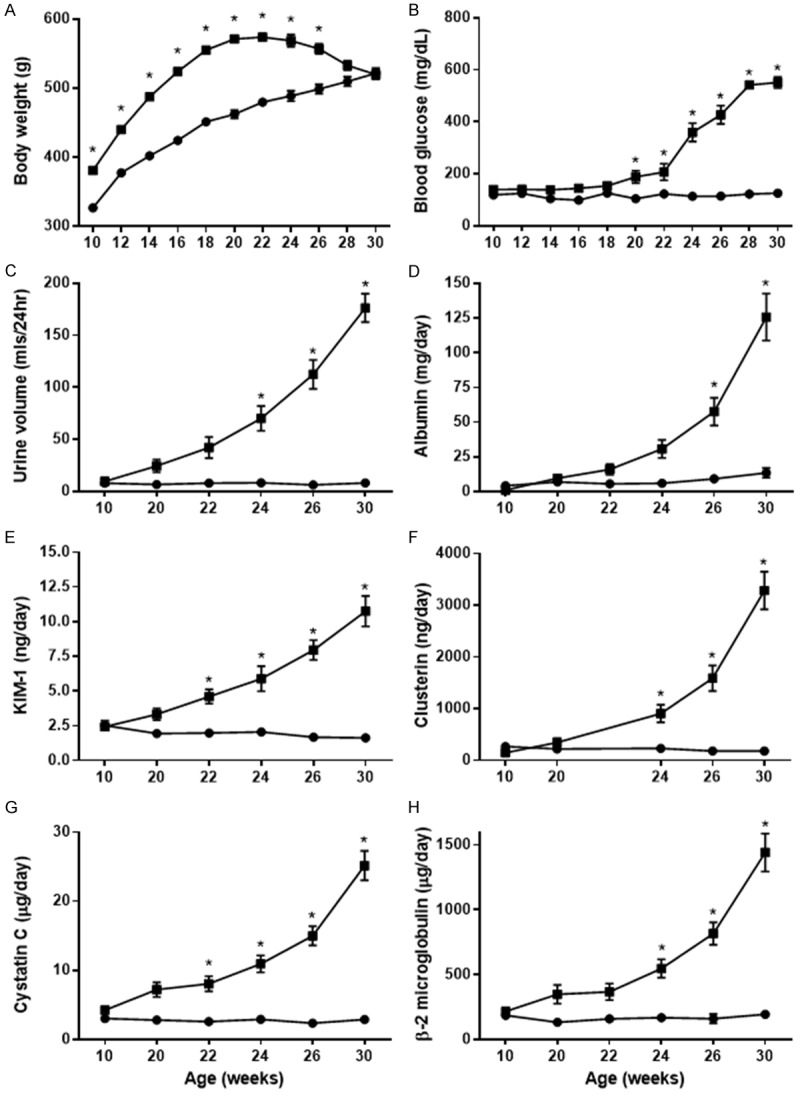
Weight (A), blood glucose (B) and urinary markers (C-H) of renal injury in ZDSD rats compared to age-matched SD rats. ZDSD rats (■, 10-30 weeks of age) are significantly heavier (A) and have significantly higher blood glucose levels (B) compared to age-matched SD rats (●). Renal injury is evident in ZDSD rats compared to SD rats as evidenced by significantly higher urine volume, albumin excretion and excretion of renal injury biomarkers (C-H) (two-way ANOVA, Sidak’s; *, P<0.05).
Hyperglycemia developed spontaneously in ZDSD rats compared to SD rats and although glucose levels tended to run higher in animals as young as 10 weeks of age (118.8 ± 2.9 vs. 139.4 ± 2.1 mg/dL) they do not become statistically significant until 20 weeks of age. Glucose levels remained quite steady in SD animals as they age, while there was a progressive increase in glucose in aging ZDSD rats. Glucose levels were significantly higher compared to SD rats from 20 weeks of age, and reached 550.9 ± 21.2 mg/dL at 30 weeks (Figure 1B).
Urine volume (mls/day) was not significantly different in ZDSD rats compared to age-matched SD rats at 10 weeks of age. However, as blood glucose increased, urine volume became significantly different starting at 24 weeks when compared to age-matched SD rats (Figure 1C).
Albumin excretion was comparable to that of SD rats in animals up to 20 weeks of age (6.9 ± 1.4 vs. 9.5 ± 2.7 mg/day for SD and ZDSD rats, respectively). Urinary albumin excretion became significantly different, compared to SD rats, at 26 weeks reaching 125.7 ± 16.9 mg/day in 30 week old animals (Figure 1D).
In addition to increased albumin excretion, age-related increases in the excretion of early urinary markers of renal injury (β2-microglobulin, KIM-1, clusterin and cystatin C) were noted in ZDSD animals (Figure 1E-H). Figure 1G demonstrates significantly higher excretion of cystatin C in the urine of ZDSD animals compared to SD animals starting at 22 weeks of age (8.1 ± 0.1 vs. 2.6 ± 0.3 µg/day, respectively). Figure 1G illustrates that cystatin C levels in urine of SD animals remained steady throughout the study period; however, levels in ZDSD rats increased rapidly compared to baseline values over the 20-week observation period and were significantly higher compared to SD rats at each of 22 to 30 weeks. In 30 weeks old animals, cystatin C was increased by 9 fold over that of SD rats (25.1 ± 2.1 vs. 2.9 ± 0.2 µg/day, respectively). A similar pattern was apparent for KIM-1, clusterin and β2-microglobulin; significant differences in these markers first appeared in 22 to 24 week old animals (Figure 1E, 1F and 1H). A seven-fold increase in KIM-1 (10.8 ± 1.1 vs. 1.6 ± 0.1 ng/day) and a seven-fold increase in β2-microglobulin (1441.9 ± 146.5 vs. 194.1 ± 22.5 ng/day) were prominent in 30 week old rats.
Fixed kidneys from 33 weeks old ZDSD rats were examined for morphological evidence of renal disease. Figure 2 represents a typical pattern of tubule, glomerular and interstitial changes that developed in ZDSD rats as a result of long-standing diabetes. Histopathology scores (0-5) were assigned for glomerulopathy with mesangial expansion and capsule thickening, tubular dilation and degeneration, protein casts and inflammation. Scores averaged 2.5-3.0 for each assessment for ZDSD kidneys (Figure 2A and 2B).
Figure 2.

Light microscopic pictures of diabetic kidney pathology. The upper panel (A) shows a glomerulus with a nodule in the lower right quadrant of the picture. This glomerulus also demonstrates mesangial expansion (A). The lower panel illustrates sclerosis in two glomeruli and other pathological changes (B).
Basement membrane thickening was apparent in electron micrographs of glomeruli in ZDSD rats when compared to control rats (≈320 nm vs. 163 nm, respectively) (Figure 3A and 3B). Podocyte effacement was also evident on the convex surface (left side) of the diabetic glomerularcapillary (Figure 3B) when compared to regular distribution of podocyte foot processes in Figure 3A. Figure 3C demonstrated significant increases in thickness of the glomerular capillary basement membranes at 12 and 16.5 weeks of diabetes.
Figure 3.

Electron micrographs of glomerular capillary walls. The picture on the left (A) is from a control kidney while the one on the right (B) is from a ZDSD rat that was diabetic for 12 weeks. The micrographs are aligned with podocytes on the left, and capillary endothelium on the right of the basement membrane. There is a clear thickening of the basement membrane in the diabetic ZDSD when compared to control capillary of age-matched SD rat. (C) represents the quantification of the basement membrane thickness between CD control and ZDSD at 12 weeks and 16.5 weeks of diabetes (155, 158 and 141 measurements, respectively; 6 glomeruli from 2 animals, one-way ANOVA followed by Dunnett’s multiple comparisons test; *P<0.05).
Effect of Lisinopril on renal function in diabetic ZDSD
Body weight
Based on average feed intake and body weight, Lisinopril was delivered at approximately 30 mg/kg/day over the 4-week period. Lisinopril had no significant effect on body weight in diabetic ZDSD rats following 4 weeks of treatment (Table 1).
Table 1.
Data before and after vehicle and Lisinopril treatment
| Vehicle | Lisinopril | |||
|---|---|---|---|---|
|
|
||||
| Measurement | Baseline | Termination | Baseline | Termination |
| Body Weight (g) | 496.0 ± 6.7 | 468.2 ± 6.9† | 500.1 ± 6.4 | 452.6 ± 6.9† |
| Blood Glucose (mg/dL) | 563.2 ± 15.2 | 572.0 ± 15.7 | 563.2 ± 17.7 | 718.3 ± 16.6*,† |
| HbA1c (%) | 10.2 ± 0.21 | 10.9 ± 0.20† | 10.4 ± 0.2 | 12.3 ± 0.3† |
| Urinary Volume (ml/day) | 171.4 ± 18.4 | 224.3 ± 9.3† | 139.5 ± 18.9 | 222.5 ± 17.9*,† |
| Urinary Albumin (mg/day) | 47.3 ± 16.8 | 145.8 ± 39.4† | 43.7 ± 12.2 | 4.6 ± 1.1*,† |
| eGFR (ml/min) | 5.07 ± 0.20 | 5.72 ± 0.24† | 4.63 ± 0.33 | 3.93 ± 0.19*,† |
| Serum Creatinine (mg/dL) | 0.44 ± 0.01 | 0.41 ± 0.01 | 0.43 ± 0.01 | 0.49 ± 0.02*,† |
| Serum BUN (mg/dL) | 18.8 ± 0.6 | 22.3 ± 0.7† | 18.9 ± 0.6 | 37.6 ± 1.2*,† |
Significantly different from vehicle at termination, t-test P<0.05.
Significantly different from corresponding baseline data, paired t-test P<0.05.
Glucose
Baseline blood glucose in diabetic ZDSD rats was not different between the two groups (Table 1). Following vehicle treatment, blood glucose did not change significantly, but administration of Lisinopril elicited a significant increase compared to baseline and vehicle treatment (Table 1). Although hyperglycemia was accompanied by elevated HbA1c values from baseline with both vehicle and Lisinopril (4 weeks of treatment), Lisinopril treatment resulted in a significant increase in HbA1c compared to vehicle treated animals at 4 weeks (Table 1).
Urine volume
Baseline urine volume in diabetic ZDSD rats was not significantly different between the groups at the beginning and the end of treatment; however, urine volume increased significantly in both groups as diabetes progressed. No significant effect of Lisinopril compared to vehicle was noted (Table 1).
Albumin excretion
Baseline albumin excretion averaged 45.5 ± 10.0 mg/day in diabetic ZDSD rats and there were no significant differences between treatment groups at baseline (Table 1). Albuminuria increased significantly after administration of vehicle for 4 weeks while administration of Lisinopril significantly reduced the albuminuria compared to vehicle treatment at 4 weeks and reduced albumin compared to its baseline value by 89% (Table 1).
Lisinopril prevented the progressive rise in albumin excretion as diabetes developed, independent of the duration of diabetes at the start of treatment (-89.2 ± 3.3, -81.8 ± 7.5 and -90.9 ± 1.9%), and lowered the levels compared to baseline values in animals that were diabetic for 5, 9 and 13 weeks, respectively (Figure 4).
Figure 4.

Effect of Lisinopril on urinary albumin excretion in animals with the duration of diabetes. Lisinopril treatment elicited a significant decrease in urinary albumin excretion when compared to vehicle treatment. When stratified according to the duration of diabetes before initiation of treatment, the normalization of albumin excretion by Lisinopril treatment is consistent. All paired groups of animals are significantly different (paired t-test; *P<0.05).
Glomerular filtration
The estimated glomerular filtration rate (eGFR) was calculated from urine and serum creatinine values. At baseline, eGFR averaged 4.8 ± 0.2 mls/min in diabetic ZDSD rats and was not different among treatment groups. Following 4 weeks of treatment, eGFR increased significantly in the vehicle treated animals; in contrast, eGFR in Lisinopril treated animals decreased significantly and was significantly lower following treatment with Lisinopril compared to vehicle (Table 1).
Serum creatinine
Baseline serum creatinine averaged 0.43 ± 0.004 mg/dL in diabetic ZDSD rats and there were no significant differences among treatment groups. Serum creatinine did not change with the progression of more severe diabetes in the vehicle group, but serum creatinine in Lisinopril treated rats increased significantly compared to baseline values and to vehicle treated animals after 4 weeks of treatment (Table 1).
Serum BUN
Baseline serum BUN averaged 18.9 ± 0.41 mg/dL and there were no differences among treatment groups. Compared to vehicle, administration of Lisinopril elicited a significant increase in BUN following the 4-week treatment (Table 1).
Discussion
Diabetic nephropathy (DN) is regarded as the leading cause of ERSD and is estimated to occur in 20-40% of diabetic patients [44]. The cluster of conditions known as metabolic syndrome carries with it a number of risk factors for the development of DN including obesity, hypertension and insulin resistance. Obesity has been shown to be a predictor for development of DN, and high fat feeding has been shown to increase body fat and induce renal injury in obese mice [45] and in humans, where reduction in obesity (via gastric bypass) produced remission in DN [46]. Hypertension occurs in 50% of patients with type 2 diabetes and contributes to the development of DN through a number of mechanisms associated with arterial damage and hemodynamics [47]. In addition, increased levels of inflammatory mediators and over production of reactive oxygen species (ROS) have been identified in diabetic animals and patients. Inflammation and ROS have been shown to be major contributors to the initiation and progression of DN [48-50]. Indeed, current therapeutic approaches for DN are centered on the control of blood pressure, reduction of hyperglycemia and life-style changes to reduce or eliminate obesity [51,52]. Albuminuria represents a biomarker of generalized endothelial dysfunction within the kidney and is predictive of the existence of endothelial dysfunction within the cardiovascular system [53]. Albuminuria has been identified as a risk factor for both renal and cardiovascular morbidity and mortality in diabetic [54,55] and in non-diabetic patients [56]. These risk factors have all been identified as contributory to the DN that spontaneously develops in the ZDSD rat.
In current clinical practice, DN presents in two stages: microalbuminuria (30-300 mg/day) and macroalbuminuria (>300 mg/day). Early DN is defined as the presence of microalbuminuria with a normal or mildly decreased eGFR (>60 ml/min/1.73 m2) [57]. While the diagnosis of nephropathy in diabetic patients is focused on the presence of albuminuria, it has become clear that significant changes in renal architecture and function have been shown in patients with normal albuminuria [58,59]. Declining renal function in the absence of albuminuria has been shown to occur in 25% of diabetic patients [60,61]. Histological examination of renal biopsies from diabetic patients with impaired renal function in the absence of albuminuria indicated vascular and tubulo-interstitial damage [62]. Thus, while concurrent monitoring of albuminuria and eGFR are the mainstay of diagnoses and evaluation of treatment for DN, earlier detection is paramount to preventing or slowing the progression to ESRD in diabetic patients. Early tubular injury has been identified as a major contributor to early DN and is predictive of progression to ESRD [63-65]. Indeed, tubular damage markers are elevated in the urine of diabetic patients [66]. Tubular damage markers such as KIM-1 and β2-microglobulin were elevated in the urine of diabetic patients before the onset of microalbuminuria and, as such, represent sensitive markers for the early detection of DN [64,67-72]. High urinary KIM-1, like albumin, was associated with increased long term risk. Clusterin is upregulated in renal tissues following insult and is associated with the process of cellular repair. Increases in urinary clusterin have been observed in a wide variety of renal diseases including glomerulonephritis and renal tubular injury [73,74]. Clusterin was found to be elevated by 2 fold in the urine of diabetic patients compared to normal volunteers [75]. Increased urinary [76,77] and serum [78] cystatin C are recognized as markers of renal tubular dysfunction. Although not yet routinely used in clinical practice, measurements of serum cystatin C may be preferred over serum creatinine in estimation of GFR especially when renal function is not stable, as occurs in early DN [79]. Cystatin C was elevated by 4 fold in the urine of diabetic patients compared to normal volunteers [65]. It is evident from the biomarker profile in human diabetic patients that glomerular and tubular injury occurs in the setting of hyperglycemia. In a similar fashion, biomarkers for glomerular (albumin) and tubular injury (KIM-1, clusterin, cystatin C and β2-microglobulin) in the setting of hyperglycemia are reflected in the spontaneous development of DN in the ZDSD rat shown in this investigation.
The interruption of an upregulated renin-angiotensin-aldosterone system (RAAS) remains the cornerstone of renoprotective strategies for diabetic patients. This is accomplished with monotherapy using angiotensin converting enzyme inhibitors (ACEi), angiotensin II receptor blockers (ARB), direct renin inhibitors (aliskiren), or dual therapy with ACEi/ARB. The ACEi Lisinopril is a commonly used first line anti-hypertensive agent and has been shown to reduce blood pressure and reduce albuminuria in hypertensive and normotensive diabetic patients [80-82].
It has been suggested that in early DN, chronic hyperglycemia enhances glucose transport to the proximal tubule which elicits hyperfiltration, increased glomerular pressure and subsequent mesangial expansion with proteinuria [83,84]. This hyperfiltration has been documented in the early stages of DN in humans [85] and in hypertensive rats made diabetic with STZ [86]. A reversal of the hyperfiltration is a predominant benefit with ACEi therapy and is thought to be the result of preferential dilation of the efferent arteriole, and a decrease in glomerular capillary pressure, with improvement in renal blood flow. Concomitant with this reduction in GFR, increases in BUN and serum creatinine have been noted in patients with renal insufficiency [87]. Similar to human DN, and as illustrated by high eGFR in untreated ZDSD rats, hyperfiltration is also a key feature of DN in this model.
Large increases in creatinine are seen clinically in some patients [88], but these are relatively rare and even these large increases are not typically considered problematic unless other clinical signs of worsening nephropathy are present. In a clinical study [88] of 13,166 cases, 31 had increases of serum creatinine from <1.2 to >2.5 mg/dL. Although smaller increases are more common, they are also not typically considered to be an indication of worsening nephropathy but rather considered to be indications of drug efficacy [87]. Nevertheless, elevated creatinine levels are issues with ACEi and ARB treatments [89] and require monitoring to identify potentially worsening nephropathy.
The ZDSD rat presents with a progressive hyperglycemia and albuminuria consistent with DN in human disease. Also, similar to DN in patients, urinary biomarkers used clinically to assess glomerular and tubular injury were prominently elevated in the model prior to the development of overt diabetes or albuminuria. Histologic lesions that are also identified in human DN including basement membrane thickening and mesangial expansion were evident with chronic hyperglycemia in ZDSD rats. In addition, this model responded to ACEi treatment in a similar fashion to that of diabetic patients with nephropathy. Similar to clinical experience, Lisinopril reduced hyperfiltration, increased serum BUN and creatinine and prevented albuminuria. The changes in intra-renal hemodynamics elicited by ACEi therapy could also explain the rises in blood glucose and associated increase in HbA1c seen in the paper since a decrease in glomerular filtration will likely also result in a decrease in glucose excretion in these untreated rats. This may not be seen clinically since this phenomenon would not be observed with reasonably well treated patients.
Conclusion
Obesity and metabolic syndrome are clear predictors of chronic kidney disease, largely due to the potentiation of chronic inflammation by insulin resistance. In addition, the lipoprotein abnormalities, increased hemodynamics and vascular dysfunction associated with metabolic syndrome have all been implicated as causative for renal disease. The ZDSD rat spontaneously develops significant renal disease corresponding to hyperglycemia and hypertension [40], the severity of which correlates with the level of hyperglycemia. Elevations in biomarkers for renal dysfunction (i.e., cystatin C, KIM-1, clusterin, and β2-microglobulin) as well as significant albuminuria and histological analysis have proven the ZDSD rat to exhibit nephropathy that closely mimics that observed in obese, insulin resistant patients.
ACEi treatment, which is the mainstay of clinical treatment for DN, was also effective in significantly reducing albuminuria in the diabetic ZDSD rat. Due to the demonstration of hypertension in this model, it should be considered that this reduction is likely to be at least partially due to a reduction in the hypertension-induced glomerular hyperfiltration [19], although it is known that ACEi can reduce albuminuria at doses that do not effect blood pressure [90]. The increased creatinine and BUN levels resulting from ACEi therapy may increase acceptance of the model, as these are indications of clinical efficacy of ACEi and ARB therapy according to Schoolwerth [87]. Interestingly, while reductions in hyperfiltration were noted in the STZ rat following Lisinopril, the elevations in BUN and creatinine seen both clinically and in the ZDSD rat were not apparent in the STZ model.
This study strongly supports the ZDSD rat as a translational model of diabetic nephropathy with the potential to add value to mechanistic as well as drug discovery efforts.
Acknowledgements
The project was partially funded by a grant from the NIH R44 DK074242 and by internal funds at PreClinOmics, Indianapolis, IN 46268. The authors would like to acknowledge our deceased colleague and co-author, Vincent H Gattone, 2nd from the Department of Anatomy at Indiana University School of Medicine who encouraged the pursuit of the nephropathy in this model and, with the assistance of Caroline Miller, contributed the light and electron microscopy to this paper. The authors would also like to acknowledge the multiple staff at PreClinOmics, now Crown Bioscience Indiana, who made contributions to the development, breeding and monitoring of the ZDSD rat and their valuable assistance in the experiments presented in this paper. Of these, the longest term contributors are Leah Shanahan, Kathryn Coy, Melissa Bass and Julius Mongu. A special acknowledgement goes to Joseph Pesek, past President of PreClinOmics, who encouraged and contributed to the development of the ZDSD model and to the review of the research performed with the model. RGP and KMZ contributed to the design, analysis of data, drafting and review. CVJ reviewed, edited and approved the final document.
Disclosure of conflict of interest
The ZDSD rat was developed, in part, from the ZDF rat and as such RGP receives a small portion of license fees that are paid to Indiana University for sales of the ZDSD rat, he is also a consultant with Crown Bioscience, the distributor of the ZDSD rat. CVJ is an employee and KMZ is a consultant with Crown Bioscience.
References
- 1.Gugliucci A. Glycation as the glucose link to diabetic complications. J Am Osteopath Assoc. 2000;100:621–634. [PubMed] [Google Scholar]
- 2.Ospina NS, Montori VM. Review: in type 2 diabetes, GLP-1 agonists plus basal insulin reduce HbA1c without increasing hypoglycemia. Ann Intern Med. 2015;162:JC6. doi: 10.7326/ACPJC-2015-162-2-006. [DOI] [PubMed] [Google Scholar]
- 3.Aronson D. Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol. 2008;45:1–16. doi: 10.1159/000115118. [DOI] [PubMed] [Google Scholar]
- 4.John S. Complication in diabetic nephropathy. Diabetes Metab Syndr. 2016;10:247–249. doi: 10.1016/j.dsx.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Li C, Sun G. Histone acetylation and its modifiers in the pathogenesis of diabetic nephropathy. J Diabetes Res. 2016;2016:4065382. doi: 10.1155/2016/4065382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan Q, Sun D, Li X, Chen G, Zheng Q, Li L, Gu C, Feng B. Association of blood glucose level and hypertension in elderly Chinese subjects: a community based study. BMC Endocr Disord. 2016;16:40. doi: 10.1186/s12902-016-0119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Si X, Li P, Zhang Y, Zhang Y, Lv W, Qi D. Renoprotective effects of olmesartan medoxomil on diabetic nephropathy in streptozotocin-induced diabetes in rats. Biomed Rep. 2014;2:24–28. doi: 10.3892/br.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao QR, Fan LJ, Jiang W, Zhao DF, Wan H, Pan DY, Lin X, Zhang T, Shen J. [Prevalence of chronic kidney disease and its risk factors in subjects with different glucose metabolism status] . Nan Fang Yi Ke Da Xue Xue Bao. 2016;36:697–700. [PubMed] [Google Scholar]
- 9.Lopes HF, Correa-Giannella ML, Consolim-Colombo FM, Egan BM. Visceral adiposity syndrome. Diabetol Metab Syndr. 2016;8:40. doi: 10.1186/s13098-016-0156-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pofi R, Di MF, Gigante A, Rosato E, Isidori AM, Amoroso A, Cianci R, Barbano B. Diabetic nephropathy: focus on current and future therapeutic strategies. Curr Drug Metab. 2016;17:497–502. doi: 10.2174/138920021705160324165553. [DOI] [PubMed] [Google Scholar]
- 11.Tian ML, Shen Y, Sun ZL, Zha Y. Efficacy and safety of combining pentoxifylline with angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker in diabetic nephropathy: a meta-analysis. Int Urol Nephrol. 2015;47:815–822. doi: 10.1007/s11255-015-0968-2. [DOI] [PubMed] [Google Scholar]
- 12.Huang H, Hu L, Lin J, Zhu X, Cui W, Xu W. Effect of fosinopril on chemerin and VEGF expression in diabetic nephropathy rats. Int J Clin Exp Pathol. 2015;8:11470–11474. [PMC free article] [PubMed] [Google Scholar]
- 13.Srivastava A, Adams-Huet B, Vega GL, Toto RD. Effect of losartan and spironolactone on triglyceride-rich lipoproteins in diabetic nephropathy. J Investig Med. 2016;64:1102–1108. doi: 10.1136/jim-2016-000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Trad B, Al-Batayneh K, El-Metwally S, Alhazimi A, Ginawi I, Alaraj M, Alkofahi E, Aljumaili O, Kosba A. Nigella sativa oil and thymoquinone ameliorate albuminuria and renal extracellular matrix accumulation in the experimental diabetic rats. Eur Rev Med Pharmacol Sci. 2016;20:2680–2688. [PubMed] [Google Scholar]
- 15.Zhou Y, Li L, Yu Y, Yang H, Yao H, Zhang Y, Zhang L, Deng C, Zhao L, Zhang Y, Song B. [Impact of glycogen synthase kinase-3beta (GSK-3beta) inhibitor on Wnt and NF-kappaB signal pathways in a rat model of diabetic nephropathy] . Zhonghua Bing Li Xue Za Zhi. 2015;44:783–787. [PubMed] [Google Scholar]
- 16.Taskiran E, Erbas O, Yigitturk G, Meral A, Akar H, Taskiran D. Exogenously administered adenosine attenuates renal damage in streptozotocin-induced diabetic rats. Ren Fail. 2016;38:1276–82. doi: 10.1080/0886022X.2016.1207054. [DOI] [PubMed] [Google Scholar]
- 17.Goyal SN, Reddy NM, Patil KR, Nakhate KT, Ojha S, Patil CR, Agrawal YO. Challenges and issues with streptozotocin-induced diabetes-A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem Biol Interact. 2016;244:49–63. doi: 10.1016/j.cbi.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 18.Castoldi G, di Gioia CR, Bombardi C, Maestroni S, Carletti R, Steckelings UM, Dahlof B, Unger T, Zerbini G, Stella A. Prevention of diabetic nephropathy by compound 21, selective agonist of angiotensin type 2 receptors, in Zucker diabetic fatty rats. Am J Physiol Renal Physiol. 2014;307:F1123–F1131. doi: 10.1152/ajprenal.00247.2014. [DOI] [PubMed] [Google Scholar]
- 19.Chadha GS, Morris ME. Effect of type 2 diabetes mellitus and diabetic nephropathy on IgG pharmacokinetics and subcutaneous bioavailability in the rat. AAPS J. 2015;17:965–975. doi: 10.1208/s12248-015-9771-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominguez JH, Wu P, Hawes JW, Deeg M, Walsh J, Packer SC, Nagase M, Temm C, Goss E, Peterson R. Renal injury: similarities and differences in male and female rats with the metabolic syndrome. Kidney Int. 2006;69:1969–1976. doi: 10.1038/sj.ki.5000406. [DOI] [PubMed] [Google Scholar]
- 21.Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M. Podocyte injury promotes progressive nephropathy in zucker diabetic fatty rats. Lab Invest. 2002;82:25–35. doi: 10.1038/labinvest.3780392. [DOI] [PubMed] [Google Scholar]
- 22.Marques C, Mega C, Goncalves A, Rodrigues-Santos P, Teixeira-Lemos E, Teixeira F, Fontes-Ribeiro C, Reis F, Fernandes R. Sitagliptin prevents inflammation and apoptotic cell death in the kidney of type 2 diabetic animals. Mediators Inflamm. 2014;2014:538737. doi: 10.1155/2014/538737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCarthy KJ, Routh RE, Shaw W, Walsh K, Welbourne TC, Johnson JH. Troglitazone halts diabetic glomerulosclerosis by blockade of mesangial expansion. Kidney Int. 2000;58:2341–2350. doi: 10.1046/j.1523-1755.2000.00418.x. [DOI] [PubMed] [Google Scholar]
- 24.Tofovic SP, Kusaka H, Kost CK Jr, Bastacky S. Renal function and structure in diabetic, hypertensive, obese ZDFxSHHF-hybrid rats. Ren Fail. 2000;22:387–406. doi: 10.1081/jdi-100100882. [DOI] [PubMed] [Google Scholar]
- 25.Tofovic SP, Salah EM, Dubey RK, Melhem MF, Jackson EK. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic nitric oxide synthase inhibition. J Cardiovasc Pharmacol. 2005;46:25–35. doi: 10.1097/01.fjc.0000162765.89437.ae. [DOI] [PubMed] [Google Scholar]
- 26.Tofovic SP, Salah EM, Jackson EK, Melhem M. Early renal injury induced by caffeine consumption in obese, diabetic ZSF1 rats. Ren Fail. 2007;29:891–902. doi: 10.1080/08860220701569846. [DOI] [PubMed] [Google Scholar]
- 27.Bilan VP, Salah EM, Bastacky S, Jones HB, Mayers RM, Zinker B, Poucher SM, Tofovic SP. Diabetic nephropathy and long-term treatment effects of rosiglitazone and enalapril in obese ZSF1 rats. J Endocrinol. 2011;210:293–308. doi: 10.1530/JOE-11-0122. [DOI] [PubMed] [Google Scholar]
- 28.Rafikova O, Salah EM, Tofovic SP. Renal and metabolic effects of tempol in obese ZSF1 rats--distinct role for superoxide and hydrogen peroxide in diabetic renal injury. Metabolism. 2008;57:1434–1444. doi: 10.1016/j.metabol.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Jia Y, Jackson EK, Tofovic SP. 2-methoxyestradiol and 2-ethoxyestradiol retard the progression of renal disease in aged, obese, diabetic ZSF1 rats. J Cardiovasc Pharmacol. 2007;49:56–63. doi: 10.1097/FJC.0b013e31802cb88e. [DOI] [PubMed] [Google Scholar]
- 30.Vora JP, Zimsen SM, Houghton DC, Anderson S. Evolution of metabolic and renal changes in the ZDF/Drt-fa rat model of type II diabetes. J Am Soc Nephrol. 1996;7:113–117. doi: 10.1681/ASN.V71113. [DOI] [PubMed] [Google Scholar]
- 31.Betz B, Conway BR. An update on the use of animal models in diabetic nephropathy research. Curr Diab Rep. 2016;16:18. doi: 10.1007/s11892-015-0706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterson RG, Jackson CV, Zimmerman K, de WW, Huebert N, Hansen MK. Characterization of the ZDSD rat: a translational model for the study of metabolic syndrome and type 2 diabetes. J Diabetes Res. 2015;2015:487816. doi: 10.1155/2015/487816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol. 1997;273:R725–R730. doi: 10.1152/ajpregu.1997.273.2.R725. [DOI] [PubMed] [Google Scholar]
- 34.Levin BE, Dunn-Meynell AA, McMinn JE, Alperovich M, Cunningham-Bussel A, Chua SC Jr. A new obesity-prone, glucose-intolerant rat strain (F.DIO) Am J Physiol Regul Integr Comp Physiol. 2003;285:R1184–R1191. doi: 10.1152/ajpregu.00267.2003. [DOI] [PubMed] [Google Scholar]
- 35.Griffen SC, Wang J, German MS. A genetic defect in beta-cell gene expression segregates independently from the fa locus in the ZDF rat. Diabetes. 2001;50:63–68. doi: 10.2337/diabetes.50.1.63. [DOI] [PubMed] [Google Scholar]
- 36.Davidson EP, Coppey LJ, Holmes A, Lupachyk S, Dake BL, Oltman CL, Peterson RG, Yorek MA. Characterization of diabetic neuropathy in the zucker diabetic sprague-dawley rat: a new animal model for type 2 diabetes. J Diabetes Res. 2014;2014:714273. doi: 10.1155/2014/714273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suckow MA, Gobbett TA, Peterson RG. Wound healing delay in the ZDSD rat. In Vivo. 2017;31:55–60. doi: 10.21873/invivo.11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Creecy A, Uppuganti S, Merkel AR, O’Neal D, Makowski AJ, Granke M, Voziyan P, Nyman JS. Changes in the fracture resistance of bone with the progression of type 2 diabetes in the ZDSD rat. Calcif Tissue Int. 2016;99:289–301. doi: 10.1007/s00223-016-0149-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reinwald S, Peterson RG, Allen MR, Burr DB. Skeletal changes associated with the onset of type 2 diabetes in the ZDF and ZDSD rodent models. Am J Physiol Endocrinol Metab. 2009;296:E765–E774. doi: 10.1152/ajpendo.90937.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson EK, Mi Z, Tofovic SP, Gillespie DG. Effect of dipeptidyl peptidase 4 inhibition on arterial blood pressure is context dependent. Hypertension. 2015;65:238–249. doi: 10.1161/HYPERTENSIONAHA.114.04631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borgeson E, Sharma K. Obesity, immunomodulation and chronic kidney disease. Curr Opin Pharmacol. 2013;13:618–624. doi: 10.1016/j.coph.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai H, Lu S, Tang X, Lu M, Chen R, Chen Z, Yang P, Liu C, Zhou H, Lu Y, Yuan H. Combined association of serum uric acid and metabolic syndrome with chronic kidney disease in hypertensive patients. Kidney Blood Press Res. 2016;41:413–423. doi: 10.1159/000443443. [DOI] [PubMed] [Google Scholar]
- 43.Logue OC, McGowan JW, George EM, Bidwell GL 3rd. Therapeutic angiogenesis by vascular endothelial growth factor supplementation for treatment of renal disease. Curr Opin Nephrol Hypertens. 2016;25:404–409. doi: 10.1097/MNH.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Si X, Li P, Zhang Y, Zhang Y, Lv W, Qi D. Renoprotective effects of olmesartan medoxomil on diabetic nephropathy in streptozotocin-induced diabetes in rats. Biomed Rep. 2014;2:24–28. doi: 10.3892/br.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu D, Gao B, Li M, Yao L, Wang S, Chen M, Li H, Ma C, Ji A, Li Y. Hydrogen sulfide mitigates kidney injury in high fat diet-induced obese mice. Oxid Med Cell Longev. 2016;2016:2715718. doi: 10.1155/2016/2715718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Di J, Yu H, Han X, Li K, Zhang P. The short-term remission of diabetic nephropathy after Roux-en-Y gastric bypass in Chinese patients of T2DM with obesity. Obes Surg. 2015;25:1263–1270. doi: 10.1007/s11695-015-1666-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pugsley MK. The angiotensin-II (AT-II) receptor blocker olmesartan reduces renal damage in animal models of hypertension and diabetes. Proc West Pharmacol Soc. 2005;48:35–38. [PubMed] [Google Scholar]
- 48.Nishikawa T, Brownlee M, Araki E. Mitochondrial reactive oxygen species in the pathogenesis of early diabetic nephropathy. J Diabetes Investig. 2015;6:137–139. doi: 10.1111/jdi.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu S, Sun G, Zhang Y, Li X, Wang R. Involvement of the NF-kappaB signaling pathway in the renoprotective effects of isorhamnetin in a type 2 diabetic rat model. Biomed Rep. 2016;4:628–634. doi: 10.3892/br.2016.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu W, Yang Y, Liu Y, Lu X, Guo S, Wu M, Wang M, Yan L, Wang Q, Zhao X, Tong X, Hu J, Li Y, Hu R, Stanton RC, Zhang Z. Exogenous kallikrein protects against diabetic nephropathy. Kidney Int. 2016;90:1023–1036. doi: 10.1016/j.kint.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 51.Ono T, Shikata K, Obika M, Miyatake N, Kodera R, Hirota D, Wada J, Kataoka H, Ogawa D, Makino H. Factors associated with remission and/or regression of microalbuminuria in type 2 diabetes mellitus. Acta Med Okayama. 2014;68:235–241. doi: 10.18926/AMO/52789. [DOI] [PubMed] [Google Scholar]
- 52.Gnudi L, Coward RJ, Long DA. Diabetic nephropathy: perspective on novel molecular mechanisms. Trends Endocrinol Metab. 2016;27:820–830. doi: 10.1016/j.tem.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 53.Gluhovschi C, Gluhovschi G, Petrica L, Timar R, Velciov S, Ionita I, Kaycsa A, Timar B. Urinary biomarkers in the assessment of early diabetic nephropathy. J Diabetes Res. 2016;2016:4626125. doi: 10.1155/2016/4626125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ognibene A, Grandi G, Lorubbio M, Rapi S, Salvadori B, Terreni A, Veroni F. KDIGO 2012 clinical practice guideline CKD classification rules out creatinine clearance 24 hour urine collection? Clin Biochem. 2016;49:85–89. doi: 10.1016/j.clinbiochem.2015.07.030. [DOI] [PubMed] [Google Scholar]
- 55.de Zeeuw D. Albuminuria: a target for treatment of type 2 diabetic nephropathy. Semin Nephrol. 2007;27:172–181. doi: 10.1016/j.semnephrol.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 56.Hillege HL, Fidler V, Diercks GF, van Gilst WH, de ZD, van Veldhuisen DJ, Gans RO, Janssen WM, Grobbee DE, de Jong PE. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation. 2002;106:1777–1782. doi: 10.1161/01.cir.0000031732.78052.81. [DOI] [PubMed] [Google Scholar]
- 57.Miranda-Diaz AG, Pazarin-Villasenor L, Yanowsky-Escatell FG, Andrade-Sierra J. Oxidative stress in diabetic nephropathy with early chronic kidney disease. J Diabetes Res. 2016;2016:7047238. doi: 10.1155/2016/7047238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caramori ML, Fioretto P, Mauer M. Low glomerular filtration rate in normoalbuminuric type 1 diabetic patients: an indicator of more advanced glomerular lesions. Diabetes. 2003;52:1036–1040. doi: 10.2337/diabetes.52.4.1036. [DOI] [PubMed] [Google Scholar]
- 59.Thajudeen B, Budhiraja P, Meister E, Popovtzer M. Improvement in glomerular filtration rate may decrease mortality among type-2 diabetics with chronic kidney disease lacking proteinuria: a retrospective study. Saudi J Kidney Dis Transpl. 2015;26:702–707. doi: 10.4103/1319-2442.160148. [DOI] [PubMed] [Google Scholar]
- 60.Tsalamandris C, Allen TJ, Gilbert RE, Sinha A, Panagiotopoulos S, Cooper ME, Jerums G. Progressive decline in renal function in diabetic patients with and without albuminuria. Diabetes. 1994;43:649–655. doi: 10.2337/diab.43.5.649. [DOI] [PubMed] [Google Scholar]
- 61.Chavers BM, Bilous RW, Ellis EN, Steffes MW, Mauer SM. Glomerular lesions and urinary albumin excretion in type I diabetes without overt proteinuria. N Engl J Med. 1989;320:966–970. doi: 10.1056/NEJM198904133201503. [DOI] [PubMed] [Google Scholar]
- 62.Ekinci EI, Jerums G, Skene A, Crammer P, Power D, Cheong KY, Panagiotopoulos S, McNeil K, Baker ST, Fioretto P, Macisaac RJ. Renal structure in normoalbuminuric and albuminuric patients with type 2 diabetes and impaired renal function. Diabetes Care. 2013;36:3620–3626. doi: 10.2337/dc12-2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas MC, Burns WC, Cooper ME. Tubular changes in early diabetic nephropathy. Adv Chronic Kidney Dis. 2005;12:177–186. doi: 10.1053/j.ackd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Vaidya VS, Niewczas MA, Ficociello LH, Johnson AC, Collings FB, Warram JH, Krolewski AS, Bonventre JV. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-beta-D-glucosaminidase. Kidney Int. 2011;79:464–470. doi: 10.1038/ki.2010.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol. 2012;32:452–462. doi: 10.1016/j.semnephrol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nauta FL, Boertien WE, Bakker SJ, van GH, van OW, de Jong PE, Bilo H, Gansevoort RT. Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care. 2011;34:975–981. doi: 10.2337/dc10-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abd El Dayem S, El Bohy Ael M, El Shehaby A. Value of the intrarenal arterial resistivity indices and different renal biomarkers for early identification of diabetic nephropathy in type 1 diabetic patients. J Pediatr Endocrinol Metab. 2016;29:273–279. doi: 10.1515/jpem-2014-0397. [DOI] [PubMed] [Google Scholar]
- 68.Fiseha T. Urinary biomarkers for early diabetic nephropathy in type 2 diabetic patients. Biomark Res. 2015;3:16. doi: 10.1186/s40364-015-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim SS, Song SH, Kim IJ, Yang JY, Lee JG, Kwak IS, Kim YK. Clinical implication of urinary tubular markers in the early stage of nephropathy with type 2 diabetic patients. Diabetes Res Clin Pract. 2012;97:251–257. doi: 10.1016/j.diabres.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 70.Marketou NP, Chrousos GP, Kanaka-Gantenbein C. Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis and diagnosis. Diabetes Metab Res Rev. 2017:33. doi: 10.1002/dmrr.2841. [DOI] [PubMed] [Google Scholar]
- 71.Petrica L, Petrica M, Vlad A, Jianu DC, Gluhovschi G, Ianculescu C, Firescu C, Dumitrascu V, Giju S, Gluhovschi C, Bob F, Gadalean F, Ursoniu S, Velciov S, Bozdog G, Milas O. Proximal tubule dysfunction is dissociated from endothelial dysfunction in normoalbuminuric patients with type 2 diabetes mellitus: a crosssectional study. Nephron Clin Pract. 2011;118:c155–c164. doi: 10.1159/000320038. [DOI] [PubMed] [Google Scholar]
- 72.Waring WS, Moonie A. Earlier recognition of nephrotoxicity using novel biomarkers of acute kidney injury. Clin Toxicol (Phila) 2011;49:720–728. doi: 10.3109/15563650.2011.615319. [DOI] [PubMed] [Google Scholar]
- 73.Rosenberg ME, Silkensen J. Clusterin: physiologic and pathophysiologic considerations. Int J Biochem Cell Biol. 1995;27:633–645. doi: 10.1016/1357-2725(95)00027-m. [DOI] [PubMed] [Google Scholar]
- 74.Nguan CY, Guan Q, Gleave ME, Du C. Promotion of cell proliferation by clusterin in the renal tissue repair phase after ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2014;306:F724–F733. doi: 10.1152/ajprenal.00410.2013. [DOI] [PubMed] [Google Scholar]
- 75.Brott DA, Furlong ST, Adler SH, Hainer JW, Arani RB, Pinches M, Rossing P, Chaturvedi N. Characterization of renal biomarkers for use in clinical trials: effect of preanalytical processing and qualification using samples from subjects with diabetes. Drug Des Devel Ther. 2015;9:3191–3198. doi: 10.2147/DDDT.S78792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conti M, Moutereau S, Zater M, Lallali K, Durrbach A, Manivet P, Eschwege P, Loric S. Urinary cystatin C as a specific marker of tubular dysfunction. Clin Chem Lab Med. 2006;44:288–291. doi: 10.1515/CCLM.2006.050. [DOI] [PubMed] [Google Scholar]
- 77.Lock EA. Sensitive and early markers of renal injury: where are we and what is the way forward? Toxicol Sci. 2010;116:1–4. doi: 10.1093/toxsci/kfq128. [DOI] [PubMed] [Google Scholar]
- 78.Javanmardi M, Azadi NA, Amini S, Abdi M. Diagnostic value of cystatin C for diagnosis of early renal damages in type 2 diabetic mellitus patients: the first experience in Iran. J Res Med Sci. 2015;20:571–576. doi: 10.4103/1735-1995.165960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lassus J, Harjola VP. Cystatin C: a step forward in assessing kidney function and cardiovascular risk. Heart Fail Rev. 2012;17:251–261. doi: 10.1007/s10741-011-9242-6. [DOI] [PubMed] [Google Scholar]
- 80.Goa KL, Haria M, Wilde MI. Lisinopril. A review of its pharmacology and use in the management of the complications of diabetes mellitus. Drugs. 1997;53:1081–1105. doi: 10.2165/00003495-199753060-00010. [DOI] [PubMed] [Google Scholar]
- 81.Newman DJ, Mattock MB, Dawnay AB, Kerry S, McGuire A, Yaqoob M, Hitman GA, Hawke C. Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol Assess. 2005;9:iii–vi. xiii–163. doi: 10.3310/hta9300. [DOI] [PubMed] [Google Scholar]
- 82.Muslih AI. Reduction of mean arterial pressure and proteinuria by the effect of ACEIs (Lisinopril) in Kurdish hypertensive patients in Hawler City. Glob J Health Sci. 2012;4:14–19. doi: 10.5539/gjhs.v4n5p14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol. 2012;74:351–375. doi: 10.1146/annurev-physiol-020911-153333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noonan WT, Shapiro VM, Banks RO. Renal glucose reabsorption during hypertonic glucose infusion in female streptozotocin-induced diabetic rats. Life Sci. 2001;68:2967–2977. doi: 10.1016/s0024-3205(01)01090-6. [DOI] [PubMed] [Google Scholar]
- 85.Fioretto P, Zambon A, Rossato M, Busetto L, Vettor R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care. 2016;39(Suppl 2):S165–S171. doi: 10.2337/dcS15-3006. [DOI] [PubMed] [Google Scholar]
- 86.Kojima N, Williams JM, Slaughter TN, Kato S, Takahashi T, Miyata N, Roman RJ. Renoprotective effects of combined SGLT2 and ACE inhibitor therapy in diabetic Dahl S rats. Physiol Rep. 2015:3. doi: 10.14814/phy2.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schoolwerth AC, Sica DA, Ballermann BJ, Wilcox CS. Renal considerations in angiotensin converting enzyme inhibitor therapy: a statement for healthcare professionals from the council on the kidney in cardiovascular disease and the council for high blood pressure research of the American Heart Association. Circulation. 2001;104:1985–1991. doi: 10.1161/hc4101.096153. [DOI] [PubMed] [Google Scholar]
- 88.Thorp ML, Ditmer DG, Nash MK, Wise R, Jaderholm PL, Smith JD, Chan W. A study of the prevalence of significant increases in serum creatinine following angiotension-converting enzyme inhibitor administration. J Hum Hypertens. 2005;19:389–392. doi: 10.1038/sj.jhh.1001832. [DOI] [PubMed] [Google Scholar]
- 89.Van Buren PN, Adams-Huet B, Toto RD. Effective antihypertensive strategies for high-risk patients with diabetic nephropathy. J Investig Med. 2010;58:950–956. doi: 10.231/JIM.0b013e3181ff46a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Galle J. Reduction of proteinuria with angiotensin receptor blockers. Nat Clin Pract Cardiovasc Med. 2008;5(Suppl 1):S36–S43. doi: 10.1038/ncpcardio0806. [DOI] [PubMed] [Google Scholar]