


Abstract
Antimicrobial resistance within a wide range of pathogenic bacteria is an increasingly serious threat to global public health. Among these pathogenic bacteria are the highly resistant, versatile and possibly aggressive bacteria, Staphylococcus aureus. Lincosamide antibiotics were proved to be effective against this pathogen. This small, albeit important group of antibiotics is mostly active against Gram-positive bacteria, but also used against selected Gram-negative anaerobes and protozoa. S. aureus resistance to lincosamides can be acquired by modifications and/or mutations in the rRNA and rProteins. Here, we present the crystal structures of the large ribosomal subunit of S. aureus in complex with the lincosamides lincomycin and RB02, a novel semisynthetic derivative and discuss the biochemical aspects of the in vitro potency of various lincosamides. These results allow better understanding of the drugs selectivity as well as the importance of the various chemical moieties of the drug for binding and inhibition.
INTRODUCTION
Antimicrobial resistance of a wide range of pathogenic bacteria is increasingly on the rise, thus poses a severe threat to global public health. A post-antibiotic era, in which common infections and minor injuries can be fatal, is far from being an apocalyptic fantasy, as it seems to become a reality (1). Staphylococcus aureus (2,3), especially methicillin-resistant S. aureus (MRSA) is among the highly resistant, versatile Gram-positive pathogens, which are a major cause of nosocomial infections and impose serious economic burden on health organizations worldwide.
The lincosamides are a small, albeit important class of antibiotics that proved to be effective against this pathogen (4–6). These compounds are produced by several Streptomyces species. Their chemical structure consists of an amino acid moiety (propyl hygric acid) and a sugar moiety (α-methylthiolincosamine, α-MTL) (Figure 1) (7). Lincosamides are mostly active against Gram-positive pathogens and against selected Gram-negative anaerobes and protozoa (8). All of the semi-synthetic derivatives of this class originate from the natural product lincomycin (Figure 1). Of a large number of lincosamide derivatives reported to date, clindamycin (Figure 1), the chlorinated analog of lincomycin, is the only semisynthetic lincosamide that is in clinical use, thus further alleviating the need for structural information that may lead for the development of novel lincosamides with superior clinical performance.
Figure 1.

Chemical structures of lincosamides. Lincomycin (natural antibiotic) and its semi-synthetic derivatives clindamycin and RB02. The chemical structure of Lincomycin’s sugar moiety, α-methylothiolincosamine is also shown.
S. aureus resistance to lincosamides is usually acquired by 23S rRNA modification. Remarkably, S. aureus strains that were resistant to lincosamides were also found to be cross-resistant to macrolides and streptogramins B antibiotics (9). Resistance to MLSB (macrolide—lincosamide—streptogramin B) is commonly caused by rRNA mutations of nucleotide A2058 and by the activity of a methyltransferase enzyme, ermC. This enzyme methylates 23S rRNA at the N6 position of adenosine A2058 (E. coli numbering throughout) (9–11), a pivotal nucleotide for the binding of MLSB antibiotics (12). An additional methyltransferase enzyme, cfr, was found to cause multidrug resistance in S. aureus to chloramphenicol, lincosamides, oxazolidinones, pleuromutilins and streptogramin A (PhLOPSA) (13–15) by methylation of the C8 position of the 23S rRNA nucleotide A2503 (16). This gene is mainly found in Gram-positive bacteria and sporadically in Gram-negative bacteria such as E. coli (17). In addition to the mechanisms of resistance in bacteria with multiple rrna operons, such as Staphylococcus species, several mutations in rRNA and rProteins that cause cross-resistance in S. aureus, were identified; A2058G and A2058U mutations (mentioned above) in the 23S rRNA were associated with MLSB resistance (18), similar to those already reported for other organisms (19). A2059G mutation is associated with macrolide-lincosamide resistance (18); however the insusceptibility to lincosamides seemed to be moderate, as previously reported for Helicobacter pylori and Streptococcus pneumoniae (19,20).
Structural studies have greatly advanced our understanding of the inhibitory mechanisms of antibiotics. Among them are some that bind to the peptidyl transferase center (PTC), including clindamycin. In this study, we aimed to shed light on the dominant chemical determinants for lincosamides inhibition mechanism on ribosomes from Gram-positive pathogenic bacteria. The structures of the large ribosomal subunit of S. aureus in complex with the lincosamides lincomycin and RB02 (Figure 1), a novel semi synthetic lincomycin derivative, were determined alongside biochemical assays to assess the in vitro potency of lincomycin, clindamycin, RB02 and their common sugar scaffold lincosamine were performed. We discuss the significance of the various chemical moieties of the drugs for binding and inhibition of prokaryotic translation.
MATERIALS AND METHODS
RB02 synthesis
p-Nitrobenzoic acid (233 mg, 1.4 mmol) was dissolved in dry dimethylformamide (DMF) (13 ml) under argon and treated with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (663 mg, 1.75 mmol) and N,N-Diisopropylethylamine (DIPEA) (0.73 ml, 4.2 mmol) and stirred for 10 min at 0°C. To the reaction mixture, α-Methylthiolincosamine (200 mg, 0.7 mmol) was then added and the solution was stirred at room temperature for 24 h (α-Methylthiolincosamine was synthesized according to a previously reported procedure by Schroeder et al. (21). The reaction was monitored by thin-layer chromatography (TLC) (dichloromethane/methanol, 2:8). The product was extracted with ethyl acetate and brine. The organic layers were washed with H2O (3 × 10 ml), dried over MgSO4 and concentrated to give the crude product. The product was isolated by column chromatography on SiO2 using dichloromethane/methanol 15:85 as eluent to afford the product compound RB02 (168 mg, 60%) as white powder. HRESI-MS m/z calculated for C16H22N2O8SNa, 425.0995; found for [M+Na]+,425.0988. 1H NMR (400 MHz, CD3OD) δ 8.32 (d, J = 8.8 Hz, 2H, H-11), 8.01 (d, J = 8.8 Hz, 2H, H-10), 5.28 (d, J = 5.6 Hz, 1H, H-1), 4.44 (m, 2H, H-5, H-6), 4.10 (m, 3H, H-2, H-4, H-7), 3.60 (dd, J = 10.1, 3.3 Hz, 1H, H-3), 2.10 (s, 3H, H-9), 1.26 (d, J = 6.4 Hz, 1H, H-8). 13C NMR (100 MHz, CD3OD) δ 167.21 (C-14), 149.71 (C-13), 140.14 (C-12), 128.31 (C-10), 123.26 (C-11), 88.41 (C-1), 70.60 (C-5), 69.50 (C-3), 68.66 (C-4), 68.10 (C-2), 67.09 (C-7), 56.03 (C-6), 17.92 (C-8), 11.99 (C-9). See also Supplementary Material and Figures S7–9.
NMR analysis
1H-Nuclear magnetic resonance (NMR) spectra (including 1D-TOCSY) were recorded on Bruker Avance™ 400 spectrometers and chemical shifts (reported in ppm) were calibrated to CD3OD (d = 3.31) when CD3OD was the solvent. 13C-NMR spectra were recorded on Bruker Avance™ 400 spectrometers at 100 MHz. Multiplicities are reported using the following abbreviations: b = broad, s = singlet, d = doublet, dd = doublet of doublets, m = multiplet. Coupling constants (J) are given in Hertz. High-resolution electron spray ionization (HR-ESI) mass spectra were measured on a Waters Synapt instrument. Chemical reactions were monitored by TLC (Merck, Silica gel 60 F254). Visualization was achieved using a Cerium-Molybdate stain (NH4)2Ce(NO3)6 (5 g)), (NH4)6Mo7O24•4H2O (120 g), H2SO4 (80 ml) and H2O (720 ml)). All chemicals, unless otherwise stated, were obtained from commercial sources. Compounds were purified by flash chromatography (SiO2, Merck, Kieselgel 60).
S. aureus growth and cell wall disruption
S. aureus strain RN4220 (American Type Culture Collection 35556) (22) was grown and disrupted as described previously (22).
Ribosome purification, crystallization and compound soaking experiments
Ribosomes were purified as described previously (22). SA50S was crystallized at 20°C by the hanging-drop vapor diffusion technique. The crystallization drop contained 0.166% 2-Methyl-2,4-pentanediol (MPD), 0.333% ethanol, Buffer A (10 mM MgCl2, 60 mM NH4Cl, 15 mM KCl) + 20 mM Hepes (pH range, 6.8–7.8), 5 mM spermidine, 0.5 mM MnCl2 and 1–1.6 mg/ml SA50S subunits. The reservoir solution contained 15% of 1:2 ethanol-MPD and buffer A + 110 mM Hepes (pH range, 6.8–7.8) as previously described (22). For obtaining SA50S antibiotic complexes, SA50S crystals were soaked in stabilization solutions containing 22 μg/ml RB02 or lincomycin for 6 h before flash-freezing and data collection.
Data collection and processing
Before data collection, the crystals were immersed in cryo-protectant solution of 20% MPD, 15% ethanol, buffer A + 110 mM Hepes and 0.5 mM MnCl2. Crystallographic data were collected at the ID23–1 and ID29 beamlines at the European Synchrotron Radiation Facility in Grenoble, France. X-ray diffraction data were collected from hexagonal crystals at 100 K. Up to 30 crystals were needed to yield complete datasets of SA50S complexes using 0.1° oscillations. Data were processed with HKL2000 (23) and the CCP4 package suite (24).
Map calculation, model building and refinement
The native SA50S structure (PDB ID: 4WCE) was used as a starting model for calculating Fo-Fc and 2Fo-Fc difference electron density maps using PHENIX. Once initial phases were obtained, rigid body and positional refinement were performed using Phenix.refine (25). For R-free calculations during refinement cycles, a random 5% of the data were omitted during refinement cycles. Compounds were positioned based on the unbiased difference electron density obtained in each experiment using Coot (26,27). Figures were generated using Chimera (28).
Inhibition assays
The inhibition effect RB02 and lincomycin on E. coli and S. aureus ribosomes was tested in a bacterial coupled transcription/translation assay system, where the expression of the luciferase gene was measured (29). The luciferase gene was inserted into a plasmid with T7 RNA polymerase promoter. The reaction mixture contained: 160 mM Hepes-KOH (pH 7.5), 6.5% polyethylene glycol (PEG) 8 K, 0.074 mg/ml tyrosine, 1.3 mM adenosine triphosphate (ATP), 0.86 mM cytidine triphosphate (CTP), guanosine triphosphate (GTP) and uridine triphosphate (UTP), 208 mM potassium glutamate, 83 mM creatine phosphate, 28 mM NH4OAc, 0.663 mM cyclic adenosine monophosphate (cAMP), 1.8 mM dithiothreitol (DTT), 0.036 mg/ml folinic acid, 0.174 mg/ml E. coli tRNA mix, 1 mM amino acid, 0.25 mg/ml creatine kinase, 0.027 mg/ml T7 RNA polymerase, E. coli cell-free extract, 0.003 μg/μl luciferase plasmid and serial dilutions of lincosamides ranging from 2.3 mg/ml to 0.05 ng/ml. When the effect of these compounds was tested against S. aureus ribosomes, the E. coli cell-free extract was replaced with ribosome-free E. coli cell-free extract and S. aureus ribosomes were added at a final concentration of 300 nM. The reaction mixture was incubated at 37°C for 1 h and terminated by the addition of erythromycin at a final concentration of 8 μM. To quantify the reaction products, luciferin assay reagent (LAR, Promega) at 5:3 (luciferase: reaction mix) volume ratio was added to the mixture and luminescence was measured. The results were plotted and IC50 values were calculated with the program GraFit 7 (30).
RESULTS
The crystal structures of the large ribosomal subunit of S. aureus (SA50S) in complex with lincomycin (SA50S–linc) and with RB02 (SA50S–RB02) were determined (Table 1). The calculated electron density maps allowed for the unambiguous assignment of these compounds within the SA50S structure (Supplementary Figure S1).
Table 1. Crystallographic data and refinement statistics of SA50S in complex with lincomycin (SA50S–linc) and SA50S in complex with RB02 (SA50S–RB02). Values for the highest-resolution shells are in parentheses.
| SA50S–linc | SA50S–RB02 | |
|---|---|---|
| Crystal information | ||
| Space group | P6522 | P6522 |
| a = b [Å] | 280.8 | 279.8 |
| c [Å] | 873.5 | 873.3 |
| α,β,γ [°] | 90, 90, 120 | 90, 90, 120 |
| Diffraction data statistics | ||
| X-ray source (ESRF) | ID29, ID23–1 | ID29, ID23–1 |
| Wavelength [Å] | 0.971 | 0.971 |
| Number of crystals | 30 | 17 |
| Resolution [Å] | 50–3.64 (3.70–3.64) | 50–3.45 (3.51–3.45) |
| Observed reflections | 2230088 | 2863682 |
| Unique reflections | 223602 | 254888 |
| Redundancy | 10.0 (4.4) | 11.2 (3.6) |
| Completeness [%] | 98.0 (78.1) | 96.3 (72.8) |
| <I>/<σ(I)> | 9.08 (1.37) | 10.28 (1.36) |
| R-merge [%] | 23.7 (97.2) | 18.8 (77.2) |
| Refinement statistics | ||
| R-factor [%] | 18.74 | 19.00 |
| R-free [%] | 23.4 | 23.30 |
| rmsd bonds [Å] | 0.009 | 0.0086 |
| Rmsd angles [°] | 1.438 | 1.388 |
Structure of the SA50S–lincomycin complex
The SA50S–lincomycin (SA50S–linc) structure shows that the propyl hygric acid group of the drug is positioned at the PTC and interferes with the accommodation of the A-site tRNA 3′ end, whereas its α-MTL moiety points toward the exit tunnel. The α-MTL possesses three hydroxyl groups that form hydrogen bonds with the 23S rRNA (Figure 2 A and B). Specifically, the O2 group forms hydrogen bonds with N4 of C2611 and N1 of A2058, the O3 group interacts with N6 of A2058 and with the phosphate’s oxygens of G2505 and the O4 group forms a hydrogen bond with N6 of A2059 and the O2 of G2503’s sugar. In addition to the α-MTL-mediated interactions, the bridging amide NH forms a hydrogen bond with O4′ of G2505. All other lincomycin interactions with the rRNA nucleotides G2061, A2451, C2452, U2504 and U2506 are van der Waals contacts (Supplementary Figure S3A).
Figure 2.

Lincosamides interaction with their binding site within the SA50S complex. (A and B) Lincomycin (orange) and the network of hydrogen bonds (black dashes): it forms with the 23S rRNA (green) and spermidine (yellow). (C and D) RB02 (magenta) and the network of hydrogen bonds (black dashes) and electrostatic interactions (black dots): it forms with the 23S rRNA (blue). The two conformations of A2062 are designated as A’ and B’ with an arrow suggestion the movement between conformations.
Interestingly, in SA50S–linc electron density map an additional density was observed between lincomycin and nucleotide A2062 that can accommodate a molecule of spermidine (an additive to the crystallization solution) (Figure 2 B, Supplementary Figure S1 A). This spermidine interacts with the 7-hydroxy group of lincomycin and with A2062 thus stabilizing A2062 such that it is slightly different from the apo SA50S structure (Supplementary Figure S2). Based on chemical footprinting and spectroscopy experiments it has been suggested that clindamycin interacts with ribosomes in a biphasic fashion; in the first phase, clindamycin binds to the ribosome and blocks the A-site. Following this step clindamycin slowly shifts toward the P-site (31–33). Substantial protection of nucleotides A2058 and A2059 in both phases suggests that the orientation of the α-MTL moiety remains the same and the propyl hygric group rotates from the A- to P-site .The same study also suggested that polyamines such as spermine and spermidine may bind at the of vicinity clindamycin binding pocket, which negatively affects the drug’s binding to the P-site, thus favoring the first phase orientation. In the SA50S–linc structure presented here, lincomycin’s binding configuration coincides with the first step of the lincosamides biphasic interaction hypothesis (31) and the presence of spermidine in the binding pocket is rationalized. Our structural study offers the first evidence for the possible presence of the polyamine, spermidine, in the lincosamides binding pocket.
Structure of the SA50S–RB02 complex
As the crystallographic data suggest, the propyl hygric acid of clindamycin and of lincomycin only interacts with the rRNA of their binding site via van der Waals contacts, we designed RB02, a semi-synthetic derivative of lincomycin, in which the propyl hygric acid of the parent lincomycin was replaced by the hydrophobic and less sterically hindered para nitrobenzamide group. The SA50S–RB02 crystal structure, similar to the SA50S–linc structure, clearly shows that RB02 is bound at the PTC, blocking the binding site of the 3′ end of the A-site tRNA. The network of hydrogen bonds between the RB02 and the S. aureus ribosome is also similar to the SA50S–linc structure, where the three hydroxyl groups of RB02 α-MTL form hydrogen bonds with the 23S rRNA (Figure 2 C and D); the O2 group forms hydrogen bonds with N4 of C2611 and N1 of A2058. The O3 group interacts with N6 of A2058 and with the phosphate oxygens of G2505 and the O4 group forms a hydrogen bond with N6 of A2059 and the O2 of G2503’s ribofuranose. All other interactions of RB02 with the 23S rRNA nucleotides G2061, A2451, C2452, U2506 and C2611 are van der Waals-based contacts (Supplementary Figure S3 B).
In addition to the sugar-mediated interactions, O7 of RB02 interacts with N6 of A2062 and the bridging amide NH forms a hydrogen bond with O4′ of G2505. Notably, the purine base of A2062 displays an additional conformation that does not lead to interactions of this ribonucleotide with RB02. Even though the geometry of the RB02 OH7 group and the bound A2062 (A2062A’) favors hydrogen bond formation, the co-existence of an alternative conformation (A2062B’) may suggest that RB02 interact with A2062A’ via electrostatic interactions. The para nitrobenzamide of RB02 is engaged only in weak van der Waals contacts with the 23S rRNA; in the SA50S–RB02 structure, this segment can rotate freely around its axis as supported by the low occupancy of this group.
Structural comparison of ribosomal complexes with different lincosamides
Comparison between the SA50S–linc and SA50S–RB02 structures and other available crystal structures of ribosomal particles in complex with clindamycin: G2058A Haloarcula marismortui (H50S-CLY) (34), Deinococcus radiodurans 50S (D50S-CLY) (35) and E. coli 70S (E70S-CLY) (36) indicates that all lincosamides bind to the large ribosomal subunit at the same pocket in a rather similar conformation. However, in D50S-CLY the lincosamide is positioned somewhat different, so that its propyl hygric acid moiety is pointing 102° away from its position in the other available structures (Figure 3). Moreover, the para nitrobenzamide of RB02 overlaps with the position that is occupied by the propyl hygric acid of lincomycin in the SA50S–linc as well as clindamycin in the H50S-CLY and E70S-CLY structures.
Figure 3.

Overlay of the structures of various ribosome–lincosamides complexes. SA50S–linc (orange; this study), SA50S–RB02 (magenta; this study), H50S-CLY (PDB ID: 1YJN) (green), D50S-CLY (gray) (PDB ID: 1JZX) and E70SCLY (sky blue) (PDB ID: 3OFZ). The color coding of the rRNA components of the various lincosamide-binding pockets is the same as that of the corresponding licosamides molecules.
A2062 is a known flexible nucleotide (37) that adopts different orientation in different ribosome structures. This nucleotide was previously identified as nascent-peptide sensor in the ribosome exit tunnel to relay the stalling signal to the PTC (38). In the complex of H50S with virginiamycin M (SA), A2062 appears to have undergone a conformational change, namely rotation of ∼90° compared with the prevalent orientation found in the native structure of H50S, which seems to contribute to the inhibitory effect of the drug (39). Furthermore, in the structure of E. coli 70S in complex with streptogramin A alone and in complex with synercid® (streptogramin A and B) A2062 undergoes a conformational change upon drug binding (40). A similar movement was observed in the structure of D. radiodurans complex with synercid® (41). The enhanced synergistic inhibition, compared to the individual contributions of each of the components, appears to be a direct consequence of interactions between the two streptogramin components, which includes the fixation of A2062 in an orientation permitting simultaneous binding of both compounds that contributes significantly to the synergistic activity. Similarly, in the current SA50S structures, the position of this nucleotide is restricted by its bond with the mentioned above spermidine (in the SA50S–linc structure) or with RB02. Moreover, in the SA50S–RB02 structure, the orientation of A2062A’ (that interacts with RB02) is similar that observed in the synercid® bound structures of E. coli 70S and D. radoidurans 50S. This type of interaction was not observed in the ribosome complexes with clindamycin since this lincosamide comprised of a chlorine atom instead of the hydroxyl group, which points to opposite direction due to the inverted C7 stereochemistry in clindamycin versus lincomycin.
Lincosamides show stronger inhibition of in-vitro translation in S. aureus compared to E. coli
Lincosamides are effective mainly against Gram-positive bacteria (9). The results of the cell-free in vitro inhibition assay of various lincosamides using both E. coli and S. aureus ribosomes (Figure 4), presented here, support previously described minimum inhibitory concentration (MIC) results (42,43); lincomycin inhibit protein synthesis with an IC50 of 0.03 ± 0.002 μg/ml in an S. aureus in vitro translation system (SATS), whereas in the E. coli system (ECTS) lincomycin has an IC50 of 1.7 ± 0.2 μg/ml i.e. lincomycin is ∼50× more potent against S. aureus than against E. coli (Figure 4A). A similar trend was observed for clindamycin as the IC50 value in SATS was 0.07 ± 0.002 μg/ml and 0.6 ± 0.06 μg/ml for ECTS (Figure 4B). The cell-free in vitro inhibitory effect of lincomycin and clindamycin is rather similar, however, clinically clindamycin exhibits higher potency against Gram-positive bacteria (8). This suggests that the substitution of the OH group at C7 with chlorine, with inverted stereochemistry of C7, increases clindamycin potency due to non-ribosomal factors such as cell permeability. Interestingly, when examining the activity of the compound RB02 on both systems, the SATS IC50 value of RB02 was 19.2 ± 1.05 μg/ml; however in the ECTS the IC50 value could not have been accurately calculated as it exhibited very low inhibitory properties (Figure 4C). Furthermore, at the highest RB02 concentration, this compound was ∼100× less effective on ECTS than on the SATS (Supplementary Figure S4 C).
Figure 4.
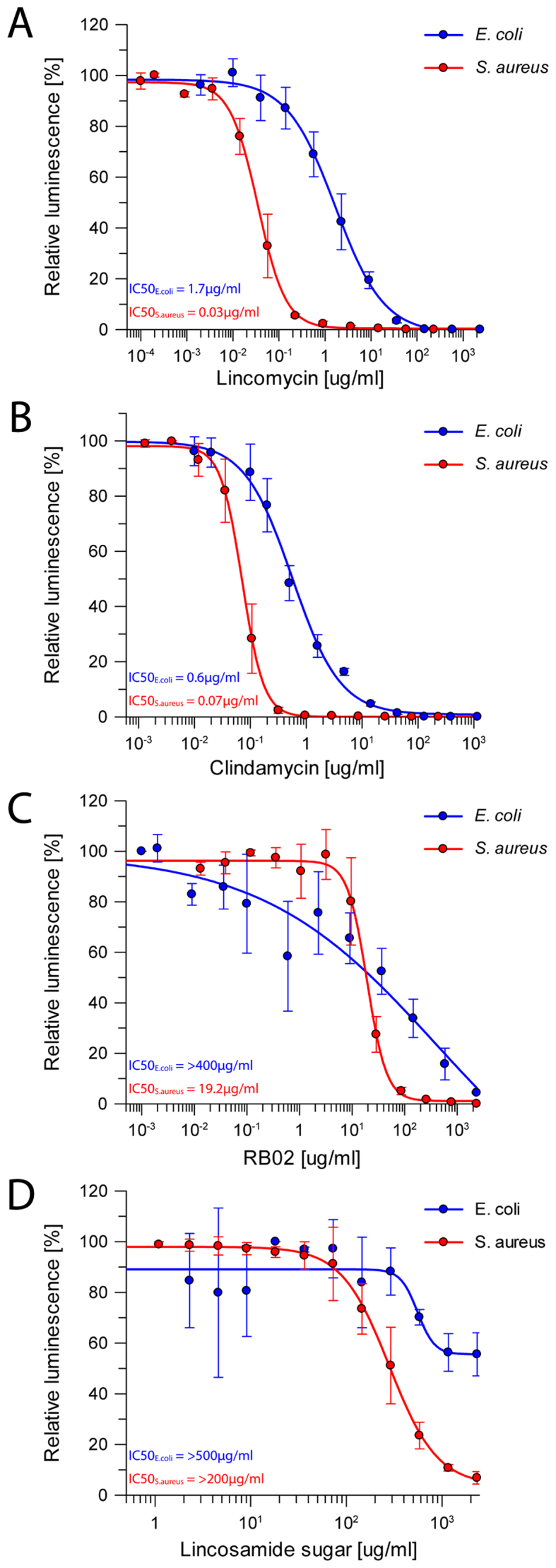
Protein synthesis inhibition by lincosamides. Inhibition results of increasing concentrations of (A) lincomycin, (B) clindamycin, (C) RB02 and (D) α-methylothiolincosamine measured by the luminescence resulting from the translation of firefly luciferase on both S. aureus in vitro translation system (red) and E. coli in vitro system (blue). The error bars represent the standard deviations from the mean for triplicate experiments and the luminescence is normalized relative to that measured in the absence of any inhibitor, which was assigned as 100%.
As seen in the ribosome structures in complexes with various lincosamides, the majority of the interactions are formed through the sugar moiety of the compound. In order to better decipher the factors dominating lincosamides inhibition, the impact of α-MTL on the drugs activity was tested by preforming cell-free inhibition assay of this moiety (obtained by the cleavage of the propyl hygric acid of lincomycin), on both SATS and ECTS. In both systems, the sugar possesses a limited inhibitory capability (Figure 4D), hence it is more likely to serve as the anchor for the rest of the antibiotic. Once the sugar moiety of the lincosamides binds to the tunnel entrance, it may not preclude polypeptide chain elongation mainly owing to its limited dimensions, as can be observed by superposition of the erythromycin bound D50S crystal structure on that of SA50S–linc (Figure 5B). These results suggest that the propyl hygric acid of clindamycin and lincomycin that merely form relatively week van der Waals contacts at its binding site plays a key role in sterically blocking the tunnel entrance. This hypothesis is further supported by the fact that RB02 with the less hindered para benzamide group is a significantly less potent inhibitor of in vitro translation as compared to the parent natural lincosamide lincomycin.
Figure 5.

Binding sites for antibiotics in the PTC and peptide exit tunnel. (A) Erythromycin (PDB ID: 1JZY) (blue), lincomycin (orange) and RB02 (pink) are shown as stick models. Ribbons denote the sugar phosphate backbone of 23S rRNA (gray), the acceptor ends of A-site tRNA (blue) and P-site tRNA (green). Lincomycin and RB02 are bound at the PTC and interferes with A-site tRNA positioning and erythromycin is bound at the entrance of the exit tunnel. Superimposing these structures shows that the sugar moiety of the lincosamides overlaps the desosamine sugar of erythromycin elucidating the MLSB cross-resistance. (B) Surface representation of erythromycin (PDB ID: 1JZY) (blue) superimposed on α-methylothiolincosamine (pink) in their binding pockets. The sugar phosphate backbone of 23S rRNA is denoted as ribbons (colored in gray).
DISCUSSION
All lincosamides studied so far bind to the same binding pocket in the bacterial large ribosomal subunit. The main determinant for lincosamides targeting to the ribosome is their α-MTL segment as they interact with 23S nucleotides mainly via this moiety.
In all studied ribosomal crystal structures, the sugar part of the drug is positioned at the exit tunnel entrance overlapping with the site occupied by the desosamine sugar of macrolides (Figure 5A). Thus, the cross-resistance between them and the macrolides can be rationalized. Furthermore, the comparison with the macrolide bound ribosome structure offers an explanation to the lack of inhibition by the lincosamine sugar moiety alone (Figure 5B).
In both SA50S–linc and SA50S–RB02, the nucleotide A2062 is interacting with the lincosamides, either directly or indirectly. Such interaction could not be established with clindamycin as its chlorine substitution on C7 and its stereochemistry could not facilitate hydrogen bond formation. However, in the cell-free in vitro inhibition assay presented here, lincomycin and clindamycin exhibit similar inhibitory effect thus the addition interaction of lincomycin to the ribosome via nucleotide A2062 may not be a pivotal determinant for the drugs potency. Furthermore, clindamycin is clinically more potent against Gram-positive infection than lincomycin which leads us to conclude that the chlorine substitution on C7 improves clindamycin’s pharmacokinetics properties rather than add beneficial interaction with the ribosome binding site.
This study offers the first evidence for the presence of polyamines in the lincosamides binding pocket as was suggested by Kouvela et al. (32). Their studies were performed mainly on clindamycin; however, spermidine was not found in none of the available ribosome structure in complex with clindamycin, even though it was also an additive to all crystallization drops. Since clindamycin’s C7 stereochemistry is inverse compared to that of lincomycin C7, the interactions of the C7 moiety with its environment is diverse among the two lincosamides. It is plausible that spermidine interact with clindamycin by transient electrostatic interaction through its C7 methyl group in a manner that supports the binding of clindamycin to the ribosome but may be hard to observe in a crystal structure that represents a stable bound state. As for RB02, although spermidine was present in the crystallization drop, electron density for it could not be identified in the SA50S–RB02 electron density map. Since lincomycin and RB02 are involved in a somewhat different network of van der Waals contacts with the surrounding rRNA nucleotides (Supplementary Figure S3), resulting in a slightly different orientation of the C7 which may favor interaction with A2062 and consequently interfere with spermidine binding.
The SA50S–linc complexes reported in this study and previous ribosome structures in complex with clindamycin demonstrate that the interactions of the propyl hygric acid segment of lincosamides with the 23S nucleotides are solely based on van der Waals forces and that this appendage may be responsible for the proper steric blockage of the tunnel entrance. Indeed, substitution of this segment which occupies a volume of 290 Å3 with the less bulk para nitrobenzamide group (with a smaller volume of 201 Å3calculated by 3V: volume assessor (44)) drastically decreased the in vitro potency of the compound.
The binding pocket of lincosamides and its surroundings shells in S. aureus and E. coli are highly conserved in terms of sequence (Supplementary Figure S5) and structure, with the exception of A2062. This nucleotide exhibits different orientations in lincosamide-bound ribosome structures; however, these differences seems to be induced by the compounds C7 substituent. Yet, significant differences in IC50 values were observed between the SATS and ECTS. Since their inhibition was examined in vitro, effects of cell permeability and efflux mechanisms could not offer an explanation to this. Both S. aureus and E. coli can acquire resistance to lincosamides by nucleotidyltransferases, which deactivates the drug although each of these bacteria acquires its own unique genes of these nucleotidyltransferases (9,45–48). However, the difference in activity of lincosamides among S. aureus and E. coli cannot be attributed to these drug modification mechanisms as both the in vitro translation systems include E. coli extract in the reaction mixture (the E. coli ribosomes were removed from the extract when inhibition was tested against S. aureus).
Offering a structural explanation to these observations is not straightforward due to the high structural similarities of the lincosamides binding pocket among S. aureus and E. coli. Nevertheless, the rRNA modification pattern is not conserved among different bacterial species. In E. coli, these rRNA modifications are well studied; however only limited data on the modification pattern of S. aureus ribosome are available. Both E. coli and S. aureus nucleotide A2503 is post-transcriptionally modified to m2A by an indigenous methytransferase (49,50) which by itself cause minimal antibiotic resistance (16). However, additional methylations on A2503 C8 by cfr confer resistance to PhLOPSA antibiotics. U2504 that is also part of the lincosamides binding pocket is pseudouridinilated in E. coli but whether or not this nucleotide is modified in S. aureus is unknown. The current complex structures are reaching 3.45–3.6 Å resolution therefore modifications could not be observed in the electron density maps thus it remains to be investigated if the difference in in vitro activity against ribosome from both species is due to differences in the modification pattern.
An additional explanation to the different in vitro inhibition levels between E. coli and S. aureus may lay further away from the binding pocket. Several rRNA nucleotide that are located 17–35 Å away from the lincosamides binding pocket, differ in both sequence identity and conformation between the two ribosomes, can be observed (Supplementary Figure S6). Thus, a cascade of interactions may account for the diverse inhibition of lincosamides against different species. It is unclear if these differences are meaningful in regard to lincosamides potency however, previous studies suggested that resistance to ribosomal antibiotics can be conferred by mutations at nucleotide that are distal to the binding pocket through remote interactions (22,51,52).
Whether or not these distal structural differences affect the drugs ability to inhibit the ribosome, it seems that there might be more to species-specificity than the binding pocket itself and its immediate vicinity.
ACCESSION NUMBERS
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number 5HKV and 5NRG.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff members at Beamlines ID23–1 of the European Synchrotron Radiation Facility/European Molecular Biology Laboratory for their assistance during data collection. We thank Dr Moshe Peretz, Shoshana Tel-Or, Miriam Lachever and Maggie Kessler for theis interest and experimental support; Astrid Gruss, Dr Susanne Paukner and Dr Rosemarie Riedl from Nabriva Therapeutics AG,Vienna, Austria for S. aureus growth.
Footnotes
Present address: Prof. Ada Yonath, Department of Structural Biology, The Weizmann Institute of Science, Rehovot 7610001, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council Grants [322581 (NOVRIB)]; Kimmelman Center for Macromolecular Assemblies; Adams Fellowship (to M.K.); Martin S. and Helen Kimmel Professorial Chair at the Weizmann Institute of Science (to A.Y.). United States–Israel Binational Science Foundation Grant [2012007 to M.F.]. Funding for open access charge: European Research Council Grants [322581 (NOVRIB)].
Conflict of interest statement. None declared.
REFERENCES
- 1. World Health Organization Antimicrobial Resistance: Global Report on Surveillance. 2014; Geneva: World Health Organization. [Google Scholar]
- 2. Kurosu M., Siricilla S., Mitachi K.. Advances in MRSA drug discovery: where are we and where do we need to be. Expert Opin. Drug Discov. 2013; 8:1095–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lowy F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998; 339:520–532. [DOI] [PubMed] [Google Scholar]
- 4. Marcinak J.F., Frank A.L.. Treatment of community-acquired methicillin-resistant Staphylococcus aureus in children. Curr. Opin. Infect. Dis. 2003; 16:265–269. [DOI] [PubMed] [Google Scholar]
- 5. Johnson M.D., Decker C.F.. Antimicrobial agents in treatment of MRSA infections. Dis. Mon. 2008; 54:793–800. [DOI] [PubMed] [Google Scholar]
- 6. Champney S.W., Tober L.C.. Specific inhibition of 50S ribosomal subunit formation in Staphylococcus aureus cells by 16-membered macrolide, lincosamide, and Streptogramin B antibiotics. Curr. Microbiol. 2000; 41:126–135. [DOI] [PubMed] [Google Scholar]
- 7. Hoeksema H., Bannister B., Birkenmeyer R.D., Kagan F., Magerlein B.J., MacKellar F.A., Schroeder W., Slomp G., Herr R.R.. Chemical studies on lincomycin. I. The structure of lincomycin. J. Am. Chem. Soc. 1964; 86:4223–4224. [Google Scholar]
- 8. Rezanka T., Spizek J., Sigler K.. Medicinal use of lincosamides and microbial resistance to them. Anti Infect. Agents Med. Chem. 2007; 6:133–144. [Google Scholar]
- 9. Spížek J., Řezanka T.. Lincomycin, clindamycin and their applications. Appl. Microbiol. Biotechnol. 2004; 64:455–464. [DOI] [PubMed] [Google Scholar]
- 10. Skinner R., Cundliffe E., Schmidt F.J.. Site of action of a ribosomal RNA methylase responsible for resistance to erythromycin and other antibiotics. J. Biol. Chem. 1983; 258:12702–12706. [PubMed] [Google Scholar]
- 11. Lüthje P., Schwarz S.. Molecular basis of resistance to macrolides and lincosamides among staphylococci and streptococci from various animal sources collected in the resistance monitoring program BfT-GermVet. Int. J. Antimicrob. Agents. 2007; 29:528–535. [DOI] [PubMed] [Google Scholar]
- 12. Douthwaite S. Interaction of the antibiotics clindamycin and lincomycin with Escherichia coli 23S ribosomal RNA. Nucleic Acids Res. 1992; 20:4717–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kehrenberg C., Schwarz S., Jacobsen L., Hansen L.H., Vester B.. A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol. Microbiol. 2005; 57:1064–1073. [DOI] [PubMed] [Google Scholar]
- 14. Long K.S., Poehlsgaard J., Kehrenberg C., Schwarz S., Vester B.. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 2006; 50:2500–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zurenko G.E., Yagi B.H., Schaadt R.D., Allison J.W., Kilburn J.O., Glickman S.E., Hutchinson D.K., Barbachyn M.R., Brickner S.J.. In vitro activities of U-100592 and U-100766, novel oxazolidinone antibacterial agents. Antimicrob. Agents Chemother. 1996; 40:839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giessing A.M.B., Jensen S.S., Rasmussen A., Hansen L.H., Gondela A., Long K., Vester B., Kirpekar F.. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA. 2009; 15:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng H., Sun J., Ma J., Li L., Fang L.-X., Zhang Q., Liu Y.-H., Liao X.-P.. Identification of the multi-resistance gene cfr in Escherichia coli isolates of animal origin. PLoS One. 2014; 9:e102378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prunier A.L., Malbruny B., Tande D., Picard B., Leclercq R.. Clinical isolates of Staphylococcus aureus with ribosomal mutations conferring resistance to macrolides. Antimicrob. Agents Chemother. 2002; 46:3054–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vester B., Douthwaite S.. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 2001; 45:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tait-Kamradt A., Davies T., Appelbaum P.C., Depardieu F., Courvalin P., Petitpas J., Wondrack L., Walker A., Jacobs M.R., Sutcliffe J.. Two new mechanisms of macrolide resistance in clinical strains of Streptococcus pneumoniae from Eastern Europe and North America. Antimicrob. Agents Chemother. 2000; 44:3395–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schroeder W., Bannister B., Hoeksema H.. Lincomycin. III. Structure and stereochemistry of the carbohydrate moiety. J. Am. Chem. Soc. 1967; 89:2448–2453. [DOI] [PubMed] [Google Scholar]
- 22. Eyal Z., Matzov D., Krupkin M., Wekselman I., Paukner S., Zimmerman E., Rozenberg H., Bashan A., Yonath A.. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E5805–E5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 24. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 29. Murray R.W., Melchior E.P., Hagadorn J.C., Marotti K.R.. Staphylococcus aureus cell extract transcription-translation assay: firefly luciferase reporter system for evaluating protein translation inhibitors. Antimicrob. Agents Chemother. 2001; 45:1900–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leatherbarrow R.J. Erithacus Software Limited. 2009; 7.0 edn, Horley. [Google Scholar]
- 31. Kostopoulou O.N., Papadopoulos G., Kouvela E.C., Kalpaxis D.L.. Clindamycin binding to ribosomes revisited: foot printing and computational detection of two binding sites within the peptidyl transferase center. Pharmazie. 2013; 68:616–621. [PubMed] [Google Scholar]
- 32. Kouvela E.C., Petropoulos A.D., Kalpaxis D.L.. Unraveling new features of clindamycin interaction with functional ribosomes and dependence of the drug potency on polyamines. J. Biol. Chem. 2006; 281:23103–23110. [DOI] [PubMed] [Google Scholar]
- 33. Verdier L., Bertho G., Gharbi-Benarous J., Girault J.P.. Lincomycin and clindamycin conformations. A fragment shared by macrolides, ketolides and lincosamides determined from TRNOE ribosome-bound conformations. Bioorg. Med. Chem. 2000; 8:1225–1243. [DOI] [PubMed] [Google Scholar]
- 34. Tu D., Blaha G., Moore P.B., Steitz T.A.. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 2005; 121:257–270. [DOI] [PubMed] [Google Scholar]
- 35. Schlunzen F., Zarivach R., Harms J., Bashan A., Tocilj A., Albrecht R., Yonath A., Franceschi F.. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001; 413:814–821. [DOI] [PubMed] [Google Scholar]
- 36. Dunkle J.A., Xiong L., Mankin A.S., Cate J.H.. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:17152–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fulle S., Gohlke H.. Statics of the ribosomal exit tunnel: implications for cotranslational peptide folding, elongation regulation and antibiotics binding. J. Mol. Biol. 2009; 387:502–517. [DOI] [PubMed] [Google Scholar]
- 38. Vazquez-Laslop N., Thum C., Mankin A.S.. Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell. 2008; 30:190–202. [DOI] [PubMed] [Google Scholar]
- 39. Hansen J.L., Moore P.B., Steitz T.A.. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003; 330:1061–1075. [DOI] [PubMed] [Google Scholar]
- 40. Noeske J., Huang J., Olivier N.B., Giacobbe R.A., Zambrowski M., Cate J.H.. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrob. Agents Chemother. 2014; 58:5269–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harms J.M., Schlunzen F., Fucini P., Bartels H., Yonath A.. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004; 2:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmalreck A.F., Kottmann I., Reiser A., Ruffer U., Schlenk R., Vanca E., Wildfeuer A.. Susceptibility testing of macrolide and lincosamide antibiotics according to DIN guidelines. Deutsches Institut fur Normung. J. Antimicrob. Chemother. 1997; 40:179–187. [DOI] [PubMed] [Google Scholar]
- 43. Le Goffic F. Structure activity relationships in lincosamide and streptogramin antibiotics. J. Antimicrob. Chemother. 1985; 16(Suppl. A):13–21. [DOI] [PubMed] [Google Scholar]
- 44. Voss N.R., Gerstein M.. 3V: cavity, channel and cleftvolume calculator andextractor. Nucleic Acids Res. 2010; 38:W555–W562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wallace A.C., Laskowski R.A., Thornton J.M.. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995; 8:127–134. [DOI] [PubMed] [Google Scholar]
- 46. Brisson-Noel A., Delrieu P., Samain D., Courvalin P.. Inactivation of lincosaminide antibiotics in Staphylococcus. Identification of lincosaminide O-nucleotidyltransferases and comparison of the corresponding resistance genes. J. Biol. Chem. 1988; 263:15880–15887. [PubMed] [Google Scholar]
- 47. Heir E., Lindstedt B.-A., Leegaard T.M., Gjernes E., Kapperud G.. Prevalence and characterization of integrons in blood culture enterobacteriaceae and gastrointestinal Escherichia coli in Norway and reporting of a novel class 1 integron-located lincosamide resistance gene. Ann. Clin. Microbiol. Antimicrob. 2004; 3:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Levings R.S., Hall R.M., Lightfoot D., Djordjevic S.P.. linG, a new integron-associated gene cassette encoding a lincosamide nucleotidyltransferase. Antimicrob. Agents Chemother. 2006; 50:3514–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Toh S.-M., Xiong L., Bae T., Mankin A.S.. The methyltransferase YfgB/RlmN is responsible for modification of adenosine 2503 in 23S rRNA. RNA. 2008; 14:98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kowalak J.A., Bruenger E., McCloskey J.A.. Posttranscriptional modification of the central loop of domain V in Escherichia coli 23 S ribosomal RNA. J. Biol. Chem. 1995; 270:17758–17764. [DOI] [PubMed] [Google Scholar]
- 51. Davidovich C., Bashan A., Auerbach-Nevo T., Yaggie R.D., Gontarek R.R., Yonath A.. Induced-fit tightens pleuromutilins binding to ribosomes and remote interactions enable their selectivity. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:4291–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Long K.S., Vester B.. Resistance to linezolid caused by modifications at its binding site on the ribosome. Antimicrob. Agents Chemother. 2012; 56:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.