

Abstract
Recently, it was discovered that exposure to mainstream antibiotics activate numerous bacterial riboregulators that control antibiotic resistance genes including metabolite-binding riboswitches and other transcription attenuators. However, the effects of commonly used antibiotics, many of which exhibit RNA-binding properties, on the widespread T-box riboswitches, remain unknown. In Staphylococcus aureus, a species-specific glyS T-box controls the supply of glycine for both ribosomal translation and cell wall synthesis, making it a promising target for next-generation antimicrobials. Here, we report that specific protein synthesis inhibitors could either significantly increase T-box-mediated transcription antitermination, while other compounds could suppress it, both in vitro and in vivo. In-line probing of the full-length T-box combined with molecular modelling and docking analyses suggest that the antibiotics that promote transcription antitermination stabilize the T-box:tRNA complex through binding specific positions on stem I and the Staphylococcal-specific stem Sa. By contrast, the antibiotics that attenuate T-box transcription bind to other positions on stem I and do not interact with stem Sa. Taken together, our results reveal that the transcription of essential genes controlled by T-box riboswitches can be directly modulated by commonly used protein synthesis inhibitors. These findings accentuate the regulatory complexities of bacterial response to antimicrobials that involve multiple riboregulators.
INTRODUCTION
Riboswitches represent a widespread class of polymorphic gene-regulatory RNA structures found in the 5′UTR of numerous mRNAs. Riboswitches control the transcription or translation of downstream genes through a RNA conformation switch induced upon binding or release of small molecule metabolites such as amino acids, nucleosides, vitamins, cations or anions, etc. (1,2). The presence or absence of the metabolite in question directly influence the folding of the malleable RNA into a dichotomy of two mutually exclusive conformations, which ultimately determine the fate of downstream gene expression. Such genetic switching can occur at the level of transcription antitermination/termination, translation initiation, splicing, etc. (3–5). Almost all prominent human pathogens use riboswitches to swiftly respond to various environmental or intracellular signals and to adjust their metabolic programs. Since their early discovery, they have attracted considerable attention as novel antibacterial targets that hold great promise as potential new arsenal against the global rise of antibiotic resistance (6). Recently, new molecules that structurally mimic the natural ligands of riboswitches have been identified and proved effective both in vitro and in vivo (3,7–10). However, very recent studies raised concerns on the role of riboswitch-mediated regulation in the presence of currently used antibiotics (11,12). It has been suggested that protein synthesis inhibitors like aminoglycosides or lincomycin, besides blocking bacterial ribosomes as their prominent molecular target, can additionally bind to riboswitches, thus affecting the expression of antibiotic resistance genes. The same studies raised concerns not only regarding the complexity of bacterial response to various environmental stimuli (including antibiotics) but also regarding the possible direct or indirect synergistic action of antibiotics and riboswitches as response mechanisms, beyond the currently known. Finally, they revealed a broader contribution of RNA-mediated regulation regarding several genes related to antibiotic resistance and implied the existence of additional riboregulators which might play roles in drug-resistance mechanisms. These studies come in support of previous observations underlining the involvement of a number of small regulatory non-coding RNAs that modulate antibiotic resistance (13,14).
T-box riboswitches are among the first discovered transcription attenuators (15). They are the only known riboswitches that switch conformations upon binding of a tRNA molecule, instead of a small ligand (16). T-boxes control mainly transcription of genes related to amino acid transport, metabolism and aminoacylation and they are found in almost all prominent Gram-positive human pathogens (17,18). T-boxes are efficient sensors of amino acid availability by sensing the aminoacylation status of their cognate tRNAs that directly associate with them. T-boxes comprise of three major domains: (i) a stem I, which initially binds and selects a cognate tRNA by base-pairing to its anticodon and stacking against its elbow, (ii) a linker of variable length, sequence and structure and (iii) an antiterminator/terminator domain, which includes the conserved T-box bulge responsible for the recognition of tRNA’s conserved 3′CCA end and sensing its aminoacylation status (19). In stem I, a codon-like trinucleotide presented from the so-called specifier loop (SL) recognizes the tRNA’s anticodon via Watson–Crick pairs similarly to the decoding process during translation. To increase binding avidity and/or specificity, the apical region of stem I makes additional stacking interactions with the flat surface of the tRNA elbow, where T- and D-loops pair and intercalate. After a cognate uncharged tRNA is accommodated on the T-box stem I via interactions to its anticodon and elbow, the tRNA’s 3′ end is positioned to interact with the nascent antiterminator through base pairing and coaxial stacking, thereby stabilizing it against the formation of the thermodynamically more stable terminator hairpin (ΔΔG ∼16 kcal/mol). Stabilization of the antiterminator conformation allows the transcribing RNA polymerase to traverse the T-box region and to transcribe downstream open reading frames. Binding of an aminoacylated tRNA, in contrast, creates steric hindrance (from the esterified amino acid itself) to the intimate tRNA–antiterminator coaxial stacking, resulting in destabilization of the antiterminator conformation and attendant formation of an intrinsic terminator. Intrinsic transcription terminators dismantle the otherwise highly stable and processive elongation complexes by pulling on the nascent transcript and laterally shearing the DNA–RNA hybrid on weak, homooligomeric dA-rU tracks (19,20).
Until recently, the knowledge of T-boxes was coming from studies on a handful of species belonging mainly to the Bacilli class. Although there is a certain degree of overall conservation shared among all T-boxes, individual T-boxes exhibit strong clade- and species-specific variations (17,18,21). Recent studies focused on the characterization of T-boxes from pathogenic bacteria revealed unexpected complexities of T-box systems both at the structural and biochemical level. T-boxes are no longer considered of single tRNA specificity and can contain species-specific structural features, like SL with tandem or overlapping codons (22,23). Certain T-boxes, exemplified by those in Actinobacteria, control translation initiation instead of transcription, in comparable genomic contexts (24). The 3D structure of a complete T-box:tRNA complex remains unavailable and therefore it remains elusive how an uncharged tRNA exactly directs the conformational switch of the antiterminator/terminator domain. Nonetheless, the recent crystal structures of stem I from the glyS T-box of O. iheyensis and G. kaustophilus in complex with the cognate tRNAGlyGCC, together with NMR data, provide key insights into this phylogenetically conserved class of riboregulators (25–28). T-boxes seem to utilize a universal mechanism, where stem I serves as a flexible molecular caliper to measure the length between the tRNA anticodon and the elbow. Specific interactions with nucleotides of the T/D-loops appear important to secure binding and correct orientation of the tRNA’s 3′ CCA end to interact with the T-box antiterminator domain (29–32).
Recently, we characterized an unusual glyS T-box (GT-box), which in S. aureus controls the transcription of a single gene encoding a glycyl-tRNA synthetase (23). This GT-box senses the availability of glycine not only as substrate for ribosomal protein synthesis, but also as substrate for the exo-ribosomal synthesis of pentaglycines that stabilize the S. aureus cell wall (33). Its secondary structure encompasses species-specific structural features, including a very short interdomain linker and a characteristic antiterminator-terminator domain with an insertion of 42 nucleotides, termed stem Sa. Interestingly, transcription antitermination of glyS is induced by all five existing tRNAGly isoacceptors, but with different binding affinities, which depend on specific interactions with the stem I’s apical loop and stem Sa. The dependency of two metabolic pathways on the GT-box and its regulatory nuances make for an ideal target for screening against current and new antibiotics, since interference of T-box regulation could result in impairment of not one, but two essential pathways (23).
So far, the possible effects of protein synthesis inhibitors on the T-box-mediated transcription antitermination are unknown. Previous reports, using a T-box antiterminator model RNA (consisting of 29 nts) identified binding sites for neomycin B and oxazolidinone derivatives, but analyses in the context of a complete riboswitch have yet to be performed (34–42). Therefore, we decided to examine the effects of various protein synthesis inhibitors on the S. aureus GT-box-controlled transcription antitermination, using in vitro read-through assays with the full-length T-box. Remarkably, dose-response assays revealed that several antibiotics strongly activate T-box directed gene expression, in an unprecedented observation for a T-box riboswitch. These effects are antibiotic-specific, as other antibiotics exhibited strong inhibitory effects on T-box transcription, or exerted no discernible effect. Subsequent structure-probing analyses and molecular modeling revealed that these antibiotics influence the critical interactions between stem I, stem Sa and tRNA to achieve this modulation. The in vitro effects of antibiotics targeting protein synthesis on T-box transcription were verified in vivo, where we used an appropriate Escherichia coli strain complemented with plasmids bearing the GT-box and the cognate tRNAGly. Our results demonstrate that protein synthesis inhibitors can directly modulate T-box riboswitch-controlled gene expression, both in vitro and in vivo. Finally, our observations are consistent with recent reports that protein synthesis inhibitors unexpectedly interfere with a panel of riboregulators besides their primary targets on the ribosomal RNA, some of which may be involved in novel antibiotic-resistance mechanisms.
MATERIALS AND METHODS
Chemicals, enzymes, plasmid vectors and bacterial strains
All chemicals and antibiotics were purchased from Sigma. The primers used for primer extension analysis have been previously described (23). Enzymes were purchased from New England Biolabs or Takara, except for RNase T1 used for enzymatic probing (Life Technologies). DMS reagent used for chemical modification experiments was from Sigma. Plasmid DNA was prepared using the NucleoSpin Plasmid Mini Kit and PCR products were purified by NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel). [γ-32P] ATP (6000 Ci/mmol) and [α-32P] UTP (800Ci/mmol) were from Izotop (Hungary). Radiolabeled RNA elution and desalting was performed using mini Quick Spin RNA Columns (Roche). Purification of RNA molecules was performed using a gel filtration chromatography column Superdex 200 10/300 GL from GE Healthcare Life Sciences attached to an ÄKTA FPLC system (Amersham Biosciences).
Phylogenetic analysis of T-boxes
The GT-box sequences that are found upstream the regulated coding region were identified by using the RegPrecise database (http://regprecise.lbl.gov) (43; Supplementary Table S1). The broader full-length riboswitch region (including the Specifier Loop's sequence) was further verified using the KEGG database and ‘The T-box search pattern’ tool (44). All multiple sequence alignments were created using the T-Coffee server algorithm, based on the full-length T-box riboswitch sequences, including the Specifier Loop (SL) and the terminator-antiterminator stem. Sequences of tRNAGly isoacceptors were obtained from the genomic tRNA database (45,46; Supplementary Table S1). The phylogenetic tree of GT-boxes was constructed by using the ClustalW2 algorithm and was visualized by using the Phylogeny.fr online tool (47,48).
In vitro transcription antitermination assays
In vitro transcription antitermination assays were performed as previously described (23). Detailed conditions for in vitro transcription, purification procedures and quality assessment of the transcripts that are used in the present study are described in the Supplementary Materials and Methods section and elsewhere (23). The transcription of the GT-box was initiated by omitting the G nucleotide, incubated at 37°C for 15min, using [α-32P] UTP (800Ci/mmol; 0.25 μM) to visualize the size of GT-box conformation products during transcription by 1 U of recombinant E. coli RNAP holoenzyme (New England Biolabs). The initiation step is paused with heparin (20 μg/ml) and elongation of transcription is allowed by the addition of MgCl2 (28 mM), KCl (86 mM), and all nucleotides in a final concentration of 5 μM each, in the presence of the cognate tRNAGlyGCC (P1). Antibiotics concentration range in dose-response experiments was between 50 and 500 μM. Elongation reactions were carried out at 37°C for 15 min. All reactions were performed twice in duplicates and error bars represent ± SD values from the corresponding experiments. Bands corresponding to transcripts in termination (T) and readthrough (RT) conformations were visualized by scanning on phosphoimager (Fujifilm FLA 3000 platform) and were quantified using AIDA image analyzer software (version 5.0).
Enzymatic and chemical probing
In-line probing was performed as previously described (23). For the enzymatic probing, the full-length S. aureus glyS T-box transcript (20 pmol of unlabeled transcript mixed with 0.2 pmol of [γ-32P] ATP 5′-end labeled transcript) was digested with RNase T1 (0.1 U, ThermoFischer Scientific) according to the manufacturer's protocol, in denaturing (20 mM sodium citrate, pH 5.0, 1 mM EDTA, 7 M urea; AMBION) or native conditions (10 mM Tris–HCl, pH 7.1, 100 mM KCl and 10 mM MgCl2). The same digestion protocol was used for analysis of P1 tRNAGly isoacceptor in complex with the GT-box. Both transcripts were mixed simultaneously (3:1 stoichiometry of radiolabeled to non-labeled transcript), denatured at 65°C for 3 min, and incubated for 1 h at 25°C under native conditions. Alkaline hydrolysis conditions of either tRNA or GT-box transcript (50 mM sodium carbonate, pH 9.2, and 1 mM EDTA) was used for ladder construction. Digestion reactions under both native and denaturing conditions were incubated at 25°C for 15 min. For the denaturing conditions, we performed pre-incubation of the transcript at 50°C for 5 min. Alkaline hydrolysis reactions were performed at 95°C for 10 min. All reactions were stopped by placing reaction mixtures on ice and the samples were ethanol precipitated, denatured at 80°C for 5 min and analyzed on a 10%/8M urea PAGE. Modifications were introduced in the presence of DMS (dimethyl sulfate) as previously described, followed by analysis by primer extension using previously used [γ-32P] labeled primers, which anneal at the antitermination stem (stem III, including the T-box bulge and stem Sa, Figure 3C). Extension reactions were performed in the presence of 20 mM Tris-acetate, pH 8.3, 10 mM Mg(OAc)2, 5 mM DTT, 1 mM of each dNTP, and 5U AMV reverse transcriptase at 47°C for 1 h. Reaction products were ethanol precipitated and analyzed on 6%/8 M urea PAGE after denaturation at 80°C for 2 min. Both primer extension and enzymatic probing analysis were visualized and analyzed as described above.
Figure 3.

Probing analysis of the whole S. aureus glyS T-box in the presence of neomycin B, tigecycline, linezolid. (A) Chemical probing analysis of the full-length S. aureus glyS T-box (20 pmol) in the presence or the absence of tRNAGlyGCC (100 pmol) and/or increasing concentrations of neomycin B, tigecycline or linezolid. TbGl_6 primer was used for the terminator/antiterminator primer extension analysis. Red, black or magenta arrows indicate DMS base modification in the presence neomycin B, tigecycline and linezolid, respectively. (B) Enzymatic probing analysis; (D) indicates denaturing conditions, (N) native conditions and (L) alkaline hydrolysis ladder. Red, black or magenta arrows correspond to base protection from RNase T1 cleavage in the presence of neomycin B, tigecycline and linezolid, respectively. (C) Illustration of the S. aureus glyS T-box secondary structure. The binding sites of neomycin B, tigecycline and linezolid on the glyS T-box are shown; Red, black and magenta arrows indicate interacting points of neomycin B, tigecycline and linezolid, respectively. Yellow labelled nucleotides indicate the conserved T-box bulge region.
Molecular modeling of neomycin binding
Docking calculations were performed using the Oceanobacillus iheyensis glyQ stem I in complex with its cognate tRNA as target structure (PDB ID: 4LCK) (25). Chains B and C were extracted from the PDB file, excluding the bacterial K-turn binding protein YbxF. Missing and polar hydrogen atoms were added and Gasteiger charges were applied using AutoDockTools v1.5.4 (49). For neomycin (NMY) we employed five different conformations extracted from crystallographic complexes with PDB IDs: 3C7R, 1I9V and 4LFB, and prepared using VIDA v4.2 (OpenEye Scientific, http://eyesopen.com). The search space was defined by two overlapping cubes of 50 Å edges, to include the entire T-box interface with tRNA. Docking calculations were performed using AutoDock Vina, by setting the exhaustiveness level and number of output modes to 20 (50). The bound conformations were examined using VMD v1.9 (51). The four top-ranked bound conformations of neomycin (NMY1–4) displayed binding affinities that were estimated in the range of –9.6 to –10.5 kcal/mol. Molecular illustrations were rendered using MacPymol.
Construction of recombinant plasmids and β-galactosidase measurements
The glyS T-box-lacZ-pRB382-amp (the T-box sequence was placed before the lacZ gene under the control of a constitutive veglI promoter) and the P1 tRNAGlyGCC-pBAD18-Kan (bearing an inducible l-arabinose promoter) were produced after cloning the respective sequences via PCR using the appropriate restriction sites. The recombinant plasmids were verified through sequencing (Macrogen, South Korea). The expression of P1 tRNAGlyGCC is induced in the presence of 0.1% l-arabinose. Under conditions where glycine is depleted from the medium, the tRNA binds on the glyS T-box, which in our case was placed upstream of the lacZ gene, and induces its transcription in vivo (Figure 6A). The produced β-galactosidase activity was measured using ONPG (2-nitrophenyl β-d-galactopyranoside) as substrate. In the present study, a previously used and appropriate E. coli M5154 ΔlacZ strain (generous gift of Prof. H.D. Becker, University of Strasbourg, France; F− ΔlacZ39, λ−, trpA49(Am), recA11, relA1, rpsL150(strR), spoT1) was transformed with both recombinant plasmids (22). For the experiments in the presence of tigecycline and linezolid, the IC50 values were measured prior to further experimentation (52). For tigecycline the IC50 was found 0.103 μg/ml and for linezolid was 85.43 μg/ml (Supplementary Figure S2). The strain was then used for subsequent experimentation to measure the activity of β-galactosidase which is produced. In the presence of minimal growth medium, where glycine is depleted, the non-aminoacylated P1 tRNAGlyGCC binds the T-box and favors the transcription antitermination conformation (Figure 6A). The same strain transformed only with the T-box construct was used as additional negative control to measure basal β-galactosidase activity. Both baseline activities were comparable under all the conditions tested. All the strains were handled as described in similar previous studies (22,53,54). Overnight cultures in LB medium (5 ml) were used to inoculate minimal medium (50 ml M9 broth supplemented with 25 μg/ml l-tryptophan) and they were incubated at 37°C until the early log growth phase was reached (A600 = 0.4–0.6). Cells were collected by centrifugation at 6000 × g for 10 min, and re-suspended in 60 ml of minimal medium at 4°C. Cultures were divided into two different batches (30 ml each). 50 μg/ml of glycine was added in the first one to enrich the medium and in contrast to the very low concentration (5 μg/ml), which was added in the second one to induce starvation conditions. Both cultures under starvation and non-starvation condition were further incubated after the addition of 0.1% (w/v) l-arabinose for induction of tRNAGlyGCC expression. The same conditions were repeated in presence of specific concentrations of antibiotics (half-IC50, IC50 and IC90). Cultures were incubated at 37°C for 4 h, followed by harvest of cells after measured at A595 and pelleted by centrifugation at 14 000 × g for 5 min at 4°C prior storage at –80°C. The measurement of β-galactosidase activity was performed essentially as described previously (53,55). Cell pellets were suspended in 1 ml buffer Z (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM Mg2SO4) and lysed under vigorous shaking in vortex apparatus for 1 min in the presence of toluene (1%). Subsequently, 0.4 ml of the lysate were mixed with 0.6 ml Z buffer supplemented with 38 mM β-mercaptoethanol. The reaction was initiated by addition of 0.2 ml ONPG (4 mg/ml) and mixtures were incubated at 37°C for 5 min until the development of light yellow color and stopped by addition of 0.5 ml of 1 M Na2CO3. Cell debris were pelleted by centrifugation at 14 000 × g for 1 min and supernatants were measured (A420), using as blank a lysed cell sample without addition of ONPG substrate (negative control). The β-galactosidase activity was calculated in Miller units using the following equation: (A420 × 1000)/(A595 × 0.4 ml × min), where A595 indicates the cell growth of each time point and min represents the recorded time of β-galactosidase reaction after substrate addition. All measurements were performed in duplicates and each experiment was performed at least three times. Error bars represent ± SD values from the corresponding experiments.
Figure 6.

In vivo glyS T-box-mediated transcription antitermination assay. (A) Schematic representation of the glyS T-box-dependent lacZ expression in E. coli. The full length (FL) glyS T-box and the tRNAGlyGCC were cloned into pRB382 and pBAD18 plasmid, respectively. Both constructs or pRB382-T-box alone were transformed into a ΔlacZ E. coli strain. T corresponds to T-box terminator conformation and anti-T to T-box antiterminator conformation. Diagram of the β-galactosidase activity in the presence of tigecycline (B) and linezolid (C). The bars represent β-gal activity relative to cell density, in Miller Units. Error bars represent ± SD values from three independent experiments. The β-galactosidase activity was measured after 4 h of culture in either rich (+Gly) or minimal media (–Gly) in the presence or in the absence of tigecycline or linezolid (concentrations tested half-IC50, IC50 and IC90) and after expression induction of S. aureus tRNAGlyGCC.
RESULTS
Idiosyncratic features of GT-boxes among pathogens
To get a broader view of GT-box distribution among pathogens, we performed phylogenetic analysis of pathogenic and non-pathogenic Gram-positive bacteria and a handful of Gram-negative species. We sought to detect species-specific sequence and structural features, such as specifier loops that contain tandem or overlapping codons, recurring deletions or insertions in the T-boxes, in correlation with the number of tRNAGly isoacceptors and the type of the corresponding anticodon triplets that exist in each organism (Figure 1A and B and Supplementary Table S1). Besides the typical GGC SL codon triplet that exists in all GT-boxes, this analysis identified overlapping glycine GGA and GGG SL triplets in GT-boxes from Clostridia, Deinococci and Chloroflexi (Figure 1A and B). Distinct GT-boxes were found in L. innocua and L. monocytogenes that harbor a 7nt SL and in S. saprophyticus that harbors a 9nt SL sequence. The variation in the specifier loop of GT-boxes is reminiscent of the codon ambiguity proposed for the C. acetobutylicum NT-box, where more than one tRNAs can compete for binding to the same T-box specifier loop (22). The phylogenetic analysis showed diversity even among closely related species within the same phylum. For example, relative species within Firmicutes can be divided into three separated subgroups; Clostridia, Streptococci-Staphylococci and Bacilli-Enterococci. The latest are clustered with Deinococci/Chloroflexi, which include also species like H. aurantiacus (Figure 1A). H. aurantiacus, despite its classification among Gram-negative bacteria, does not synthesize lipopolysaccharides and thus does not possess outer membranes, resembling the Gram-positive cell wall. In addition, species that belong to the Deinococci/Chloroflexi (such as H. aurantiacus and D. geothermalis) exhibit divergent SL sequence (possible tandem SL and overlapping glycine codons; Figure 1B). On the other hand, organization of tRNAGly isoacceptors bearing different anticodons is restricted among species within the same clade, but without any obvious correlation regarding the downstream genes under T-box control (Figure 1B). At least two tRNAGly isoacceptors were identified in each organism with different anticodon triplets that could interact with the corresponding SL codon triplet (Figure 1B). Overall, the analysis suggests that distinct species-specific T-box patterns do exist, and is concomitant with specific contexts of tRNA isoacceptors. This observation presumably reflects genome-specific metabolic adaptations of each organism. Interestingly, although C. difficile contains a glyQ/glyS operon, the 5′UTR region of the corresponding mRNA does not seem to fold in to a typical T-box conformation, when analyzed using current bioinformatics tools like RegPrecise and Pacific prediction algorithms. The particularity of each GT-box/tRNA set for each phylogenetic group calls for further experimentation, in order to develop narrow-spectrum antibacterial compounds.
Figure 1.

(A) Phylogenetic tree constructed after analysis of the predicted GT-boxes based on the mRNA leader region from the SL until the T-box conserved sequence, along with the SL specificity (indicated with C for the GGC triplet, A for the GGA and G for the GGG) and the tRNAGly isoacceptor specificity. The number of proteinogenic (P) and non-proteinogenic (NP) isoacceptors and their corresponding anticodon triplet is shown (G for GCC, U for UCC, and C for CCC). (B) GT-boxes distribution among different pathogens, the potential SL regions and the tRNA anticodon triplets and the number of tRNAGly isoacceptors. In the specifier loop column, the red underlined sequences indicate the potential SL sequence and the bold letters correspond to nucleotides of the SL codon-like triplets for glycine. H.aur: Herpetosiphon aurantiacus, D.geo: Deinococcus geothermalis, B.sub: Bacillus subtilis, B.cer: Bacillus cereus, C.tet: Clostridium tetani, C.bot: Clostridium botulinum, C.ace: Clostridium acetobutylicum, C.dif: Clostridium difficile, E.fae: Enterococcus faecalis, S.aur: Staphylococcus aureus, S.epi: Staphylococcus epidermidis, S.sap: Staphylococcus saprophyticus, S.san: Streptococcus sanguinis, S.mut: Streptococcus mutans, S.pne: Streptococcus pneumoniae, S.aga: Streptococcus agalactiae, L.inn: Listeria innocua, L.mon: Listeria monocytogenes.
Effect of protein synthesis inhibitors on transcription antitermination in vitro
The effects of commonly used antibiotics on T-box structure and transcription antitermination are essentially unknown. This information is crucial because Gram-positive bacteria make up a major fraction of human gut and oral microbiome and most of them employ several T-boxes to control essential genes in amino acid metabolism. Previous reports tested a series of oxazolidinone derivatives exhibiting affinity for a minimal RNA construct, which only partially resembled the antiterminator region (a T-box bulge construct) of the B. subtilis glyS T-box (34,37–42). Furthermore, it was shown that the aminoglycoside neomycin B exhibits affinity for specific nucleotides on the same T-box bulge construct (35). Although those studies used a relatively small portion of the antiterminator region and not the full-length riboswitch, they nonetheless suggested that specific T-box sites could support binding of the compounds. Moreover, the antiterminator bulge was suggested to contain a metal ion-binding pocket, which possibly explains the affinity for neomycin B. Interestingly, in vitro antitermination assays identified one compound that led to reduced tRNA-dependent antitermination and another that lead to increased tRNA-independent antitermination (36).
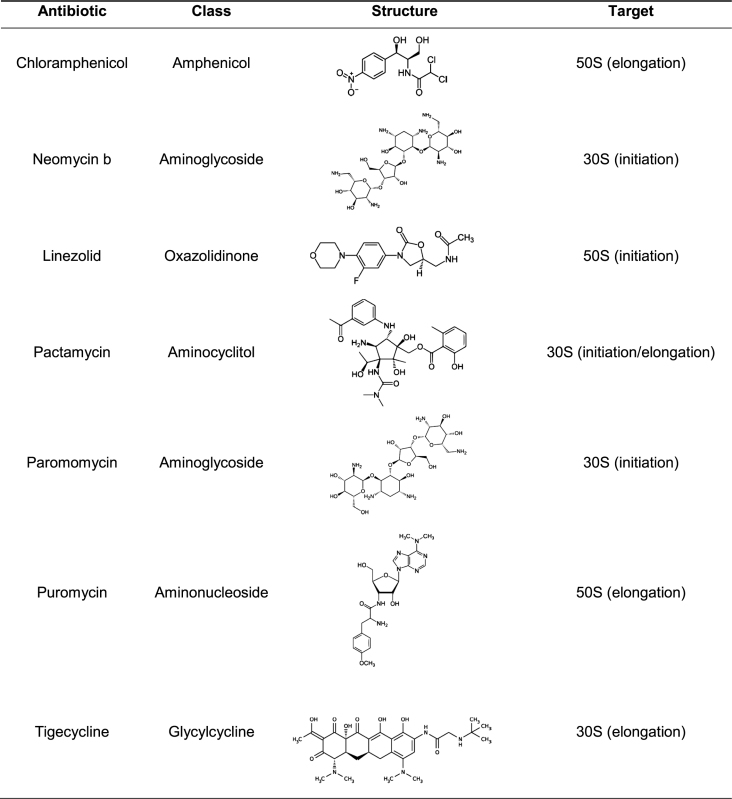
To evaluate the effect of protein synthesis inhibitors on S. aureus GT-box mediated transcription antitermination, we performed in vitro read-through assays using the full-length T-box sequence and the cognate P1 tRNAGlyGCC. The antibiotics were selected based on their known mechanisms of action on the ribosome, affecting either interactions with the tRNA’s anticodon loop or the 3′CCA end (Table 1) (56). Both tRNA features are essential for transcription antitermination on a T-box (25,27). We tested the ability of neomycin B, paromomycin, tigecycline, pactamycin (targeting the 30S ribosomal subunit) puromycin, chloramphenicol and linezolid (targeting the 50S ribosomal subunit) to modulate transcription antitermination of glyS in an in vitro assay, at concentrations ranging between 50 and 500 μM. We observed that neomycin B, tigecycline, pactamycin and paromomycin significantly increased tRNA-dependent transcription antitermination to levels substantially higher than the control reactions, in a dosage-dependent manner (Figure 2, upper panel). The effect was evident even in the presence of the lowest concentration tested. Only in the presence of the highest concentration of tigecycline (500 μM) we could not detect the formation of the transcription product, an observation that indicates possible off-target inhibition of the RNA polymerase. Notably, a weak, nonspecific inhibition of the RNA polymerase alone does not usually lead to substantial changes in the levels of readthrough. This is consistent with the notion that the T-box mechanism is more dependent on the structural partition of the competing RNA conformers, than on the kinetic properties of the RNA polymerases. For instance, the E. coli RNA polymerase effectively operates B. subtilis T-box riboswitches, despite drastic differences in how these enzymes respond to hairpin-dependent pauses (57). In contrast, chloramphenicol and linezolid that perturb the interactions of tRNA’s 3′CCA end in the peptidyl transferase center, exhibited inhibitory effect, again in a dosage-dependent manner (Figure 2, lower panel). The dosage-response curves for all antibiotics were characteristic of specific binding, exhibiting saturation above 200 μM. Finally, puromycin which mimics tRNA's aminoacylated 3′CCA end had no discernible effect on the level of readthrough (Figure 2, lower panel). Overall, the response of the S. aureus glyS T-box-controlled transcription to various protein synthesis inhibitors clearly showed that T-box riboswitches can be targeted and differentially modulated by these compounds.
Table 1. Protein synthesis inhibitors tested.
|
Figure 2.

Dose-response curves showing the effect of increasing concentration of antibiotics on the S. aureus glyS T-box transcription antitermination, in vitro. The values for the curves were extracted after analysis of representative autoradiograms (insets) as described in Materials and Methods. All reactions were performed twice in duplicates and error bars represent ± SD from the corresponding experiments. T and RT correspond to transcription termination and transcription readthrough, respectively.
Commonality and variability in binding sites on the T-box:tRNA complex
The fact that neomycin B (an aminoclycoside widely used in similar studies), tigecycline (a recently introduced glycylcycline and a tetracycline derivative) and linezolid (an oxazolidinone) can differentially modulate GT-box-controlled transcription, prompted us to identify the underlying binding sites on the RNAs. The full-length T-box RNA (275nt) was either treated enzymatically by RNase T1 (cleaving 3′ of single-stranded guanosines) or chemically modified by DMS (modifying strongly adenosines and moderately cytosines) in the presence or absence of the cognate tRNAGlyGCC and increasing concentrations of antibiotics (see Materials and Methods). The analysis of the T-box:tRNA complex in the presence of neomycin B revealed specific binding sites on stem I and stem Sa. Specifically, we observed strong protection at positions G60 and G68 of the AG bulge and at positions G82 (a conserved guanosine in the apical loop), G109 and G110 (the first two bases of the SL’s codon triplet) (Figure 3B). The primer extension analysis of the antiterminator domain showed protection at positions A182, A196, A209 and A210 (Figure 3A). All these positions are part of the staphylococcal-specific stem Sa, which is inserted in the canonical antiterminator-terminator domain (Figure 3C). The protected positions were identical in the case of antibiotic alone and the presence of tRNA was intensifying the protection effect in almost all cases (Figure 3A and B). Interestingly, tigecycline protected the same positions as neomycin B, an observation that suggests that the antibiotics that stabilize the T-box:tRNA complex share a common set of binding sites. As a result, the enhanced stability of the stem I:tRNA interaction binding induces a stable antitermination conformation, which seems to be further favored via interactions with stem Sa, ultimately resulting in higher levels of transcription readthrough. The only difference between the two antibiotics was an additional protection site by tigecycline at position A162. This position is part of the T-box bulge and is thought to base pair with U73, the discriminator base for tRNAGlyGCC (Figure 3A and B).
On the other hand, structural probing of the T-box in the presence of linezolid revealed that although some protection sites overlap with those for neomycin B and tigecycline, several striking differences also emerged. The common protection sites include the conserved G82 (of the apical loop) and G109 and G110 (of the SL’s anticodon). The most important difference, is the strong protection at position G93, a semi-conserved base of the AG bulge. Although this position is base-paired in the crystal structure (corresponds to U75, which base pairs with A33), in the case of S. aureus glyS T-box G93 is found in a single-stranded region opposite the AG bulge, and could thus affect the presentation of one of the two interdigitated T-loop from this bulge. Another important difference was observed with the strong protection of G105 at the 5′ end of the SL. G105 (corresponding to position G84 in the crystal structure) occupies an important location at the top layer of the loop E (or S turn) motif. The phylogenetically conserved loop E motif, located just distal to the specifier codon (some T-boxes contain a short spacer in between the two), participates in the structural presentation of the specifier codon to properly engage the tRNA anticodon (Figure 3, magenta arrows and Figure 7) (20). Further, in the G. kaustophilus glyQ T-box structure, A85, located just under the loop E in the spacer, is seen making a hydrogen bond with the backbone of the G39 of the tRNA anticodon stem loop (20,27). The putative linezolid interaction with G105 could distort the loop E structure and /or alter tRNA contacts in this region, thus interfering with the T-box activation. Finally, we could not observe any protected nucleotides on stem Sa in the presence of linezolid, an observation which suggests that linezolid does not bind to this domain (Figure 3C and Figure 7). Taken together, our data suggest that a set of common binding sites for all the antibiotics tested exist, which presumably reflect interactions with surface-exposed regions that exhibit certain structural features or electrostatic patterns. In addition, we identified different binding sites for neomycin B and tigecycline, which increase tRNA-dependent transcription antitermination compared to linezolid, which inhibits it. Interestingly, the differential binding patterns are correlated with the contrasting effects on the genetic outcome observed in the antitermination assay. In particular, antibiotics that promote transcription readthrough (like neomycin B and tigecycline) and presumably provide extra stabilization of T-box:tRNA complex, tend to bind on stem Sa. Linezolid on the other hand, is just the opposite and it does not bind to stem Sa.
Figure 7.

Illustration of the proposed model of neomycin B, tigecycline and linezolid binding sites on the S. aureus glyS T-box:tRNAGlyGCC complex (A) and the secondary structure of the S. aureus tRNAGlyGCC. The protected bases on the S. aureus glyS T-box:tRNAGlyGCC in the presence of neomycin B (B), tigecycline (C) and linezolid (D) are indicated with different colored arrows; red arrows correspond to neomycin B interaction, black arrows to tigecycline and magenta arrows to linezolid. Green or red stars show the enhancement or blocking of the glyS T-box read-through transcription by the antibiotics tested, respectively. Orange lines show the GCC codon-anticodon like interaction and the orange dashed line the wobble pairing. Numbers indicate the position of each nucleotide on the S. aureus glyS T-box or the tRNAGlyGCC (in green). Red nucleotides at the apical loop show the bases that interact with the tRNA elbow based on the crystal structure (25) (Right panel); based on the available crystal structure, red nucleotides are important for interaction with the Stem I. Red circled nucleotides and red arrows correspond to the binding sites of neomycin B, and black circled nucleotides and black arrows show and tigecycline on the tRNAGlyGCC.
Neomycin B and tigecycline exhibit differential binding to tRNA
In addition to identifying putative antibiotics binding sites on the T-box RNA that underlie their modulations on the T-box, we also searched for possible binding sites on the tRNA. Enzymatic analysis of P1 tRNAGlyGCC using RNase T1 (cleaves single-stranded RNA after guanosines) either free or in complex with the T-box showed that in the presence of neomycin B, the position G57 is protected (Figure 4A, lanes 5–7). This specific position lies underneath G19 and C56 and is not accessible for cleavage when the tRNA is incubated alone (Figure 4A, lane 5). When the tRNA is bound to the T-box, G57 is presumably exposed and can be cleaved (Figure 4A, lane 5, Figure 7 and Supplementary Figure S1). In the presence of neomycin B and the tRNA but in the absence of the T-box, G57 appears again susceptible to RNase T1, an observation, which suggests that neomycin B could bind tRNA in a different position that could lead to a conformational change (Figure 4A, lane 8). Indeed, previous studies reported that aminoglycosides, like neomycin B, can bind tRNAs and displace divalent metal ions, especially in the D-loop and the variable loop (58). In the present study, a weak protection was observed at position G46 (variable loop) that could account for such a behavior. When neomycin B was present during the formation of the T-box:tRNA complex, G57 was found protected, indicating that the protection is a result of the antibiotic binding on the apical loop, at position G82 (see above) and not to the tRNA elbow (Figure 4A, lanes 5–7 and Supplementary Figure S1). When the same analysis was repeated for tigecycline, the position that was found protected was G19 (D-loop), which is directly involved in the platform-stacking interaction between the tRNA elbow and the stem I apical loop (Figure 4B, lanes 6 and 7) (25). In free tRNA, G19 appears exposed and susceptible to cleavage by RNase T1 (Figure 4B, lane 4). The same behavior is also observed when either the T-box or tigecycline is present separately in the reaction mixture (Figure 4B, lanes 5 and 8, Figure 7 and Supplementary Figure S1). However, when the T-box:tRNA complex is formed in the presence of tigecycline, position G19 is again protected (Figure 4B, lane 7). A common protection by both antibiotics was observed at position G34 of the anticodon triplet (wobble position), with neomycin B exerting a larger effect than tigecycline. This observation suggests that the strength of SL codon:tRNA anticodon interaction could be modulated in the presence of either neomycin or tigecyline. The fact that neomycin B mimics the anticodon-protective effect of T-box is an intriguing one. The activating effect of neomycin B for the T-box indicates that it does not compete with T-box for tRNA binding. Then it stands to reason that neomycin B may induce a conformation of the anticodon stem loop that is conducive to T-box binding. Thus, neomycin B may also stabilize the tRNA:T-box interaction at the anticodon by restricting the anticodon stem loop in a binding-productive conformation. Finally, the results from the footprinting analysis are consistent with the previously observed binding mode of both antibiotics on the apical loop of stem I (Figure 5 and Figure 7B and C).
Figure 4.

Probing analysis of tRNAGlyGCC in the presence of neomycin B (A) or tigecycline (B). Enzymatic probing analysis of the tRNAGlyGCC (20 pmol) in the presence of the full-length S. aureus glyS T-box (60 pmol) and/or increasing concentrations of neomycin B or tigecycline. Red and black arrows indicate base protection from RNase T1 cleavage in the presence of neomycin B and tigecycline, respectively. Red bar shows a weak base protection in the presence of neomycin B. (D) indicates denaturing conditions, (N) native conditions and (L) alkaline hydrolysis ladder. (Bottom panel) The secondary structure of the S. aureus tRNAGlyGCC is shown (C); based on the available crystal structure, red nucleotides are important for interaction with the Stem I (25). Red circled nucleotides and red arrows show the binding sites of neomycin B and black circled nucleotides and black arrows the binding sites of tigecycline on the tRNAGlyGCC.
Figure 5.

Molecular modelling of antibiotics’ binding sites onto the glyS T-box:tRNA complex. (A) Schematic representation of the T-box stem I complex with tRNA, illustrating potential interaction sites of neomycin (NMY). The structure of the complex is based on the X-ray structure of T-box riboswitch of Oceanobacillus iheyensis glyQ (blue) in complex with its cognate tRNA (green) from PDB ID 4LCK (25) NMY is shown as CPK model with yellow C, blue N and red O atoms. Close-up views of the four binding sites NMY1 (B), NMY2 (C), NMY3 (D) and NMY4 (E).
Molecular modeling of neomycin B binding to the T-box:tRNA complex
To computationally simulate the binding of protein synthesis inhibitors to the T-box riboswitch at the structural level, we performed in silico docking analyses. Known conformations of neomycin B (NMY) extracted from the Protein Data Bank (PDB) were docked to the co-crystal structure of O. iheyensis glyQ T-box riboswitch stem I in complex with its cognate tRNA (PDB ID: 4LCK). The docking analysis identified a number of potential binding sites for neomycin with estimated binding energies between –9.6 and –10.5 kcal/mol (Figure 5, NMY1–4). The top-ranked docked conformations of neomycin B (NMY) revealed that the antibiotic could potentially bind at the specifier-anticodon region (Figure 5, NMY1) and close to the T1 and T2 loops (Figure 5, NMY2) of the glyQ stem I, without requiring any conformational change of the complex with its cognate tRNA. This is also the case for the two putative binding sites of NMY on the tRNA (NMY3 and NMY4), which might stabilize further the stem I:tRNA complex by acting on the tRNA. One of the most prominent simulated binding sites (NMY1) is located in the ‘neck’ region that connects the interdigitated T-loops module located at the apex of stem I to the body of stem I, consisting of mostly regular A-form RNA duplexes (Figure 5A and B). Neomycin B binding to this otherwise unoccupied space is expected to stabilize the local structure of stem I apical region and potentially improves the presentation of the conserved base triple that directly stacks against the tRNA elbow. This sequence-nonspecific, structure-selective stacking interaction accounts for about a third of total area buried by T-box stem I-tRNA binding (333 out of 802 Å2). Disruption of this interface either from the tRNA or the T-box drastically reduces the binding affinity. Consistent with this, neomycin site NMY2 is located close to this interface on the tRNA side (Figure 5C). This projected binding site is congruent with the strong protection of tRNA G57 from DMS modification by neomycin (Figures 4, 7 and Supplementary Figure S1). NMY3 binds in a crevice at the junction of coaxially stacked anticodon stem loop (ASL) and D stem loop (DSL), in immediate proximity of the non-canonical, Watson–Crick-like G26•A44 base pair. G26•A44 is a prominent hinge about which the two halves of tRNA can swivel, up to 70° while the tRNA transits through the ribosome (Figure 5D). Binding of T-box stem I bends the tRNA about the G26•A44 hinge by ∼20° compared to the free tRNA. Neomycin binding at this strategic location could stabilize this necessary bend and reduce the entropic cost of pre-organizing the tRNA conformation for improved stem I binding. Finally, NMY4 binds along the major groove of the T-box stem I, placing its D-neosamine moiety directly behind the specifier codon triplet. Thus, neomycin B binding here could help position the stacked single-stranded conformation of the specifier triplet to stably base pair with the incoming tRNA anticodon (Figure 5E). Owing to the short duplex length, effective Watson–Crick base pairing between the T-box codon and tRNA anticodon requires stabilization by the flanking conserved purines (tRNA 37 and T-box G112). Similarly, neomycin B binding from behind the T-box codon could provide lateral stabilization of the short duplex (Figure 7).
Modulation of T-box controlled transcription by antibiotics in vivo
To verify the in vitro observation that specific antibiotics can strongly modulate tRNA-dependent transcription antitermination, we tested these effects in vivo. We measured the effect of tigecycline, and linezolid using the previously described lacZ deficient E. coli M5154 strain complemented with two plasmids (Figure 6A, see Materials and Methods). The first plasmid (pRB382) uses the vegII constitutive promoter, which is appropriate for recognition by the E. coli RNA polymerase and was engineered to place the transcription of the lacZ gene under the control of the S. aureus glyS T-box. The second plasmid (pBAD18) bears an inducible promoter and was used for cloning of the P1 tRNAGlyGCC gene. The tRNA expression was induced in the presence of l-arabinose. The specific binding of uncharged P1 tRNA to the glyS T-box results in increased transcription of its downstream lacZ gene and elevated β-galactosidase activity (Figure 6A). The cultures were grown in minimal media, in the presence of limited and excess concentration of glycine, under three different antibiotic concentrations corresponding to half-IC50, IC50 and IC90 (Materials and Methods and Supplementary Figure S2). A significant reduction of cell growth was observed after 4 h in the presence of both antibiotics, indicating that were effective. The in vivo effects of of tigecycline and linezolid on the expression of lacZ as reported by the β-galactosidase activity correlate with the in vitro effects reported by the fraction of transcriptional readthrough. When tRNA expression was induced, the β-galactosidase activity was significantly higher in the presence of tigecycline (Figure 6B). Similarly, β-galactosidase activity was reduced in the presence of linezolid. Although the reduced β-galactosidase activity could be in part attributable to the overall inhibitory effect of antibiotics on cell growth, the observation that tigecycline stimulates T-box-mediated transcription in vivo despite its cytotoxicity, points to a specific effect in addition to ribosomal inhibition. Consistent with a functional T-box system in vivo, the presence of glycine in the growth medium suppressed the T-box controlled gene expression, reducing the β-galactosidase activity.
DISCUSSION
An increasing number of recent studies have shown that the expression of a number of bacterial regulatory ncRNAs are modulated by exposure to antibiotic compounds (59). For example, the expression of four sRNAs was enhanced in Salmonella enterica upon exposure to tigecycline or tetracycline. One of these sRNAs is known to be involved in tigecycline resistance (60). Another study showed that the exposure of S. aureus to antibiotics like linezolid and tigecycline alters the expression of specific sRNAs (61). Our knowledge on the effects of antibiotics on riboswitches was enriched recently by studies showing that mainstream antibiotics can rapidly activate the expression of resistance genes, many of which are now known to be under control of riboregulators (11,12). Most antibiotics acting as protein synthesis inhibitors possess intrinsic RNA binding properties and it has been suggested that bacterial RNA-based regulation could be modulated in the presence of external signals like antibiotics (62). These recent discoveries underscore the prevalence of riboregulators as probable secondary targets of commonly used antibiotics. Alternatively, these riboregulators have evolved to integrate metabolic input (to which they are canonically known to respond) with the presence or absence of RNA-binding antibiotics, to make improved regulatory decisions that confer enhanced fitness in complex nutritional and chemical environments. In either case, these studies provide essential insights on the versatility of bacterial RNA-based regulation and the possible crosstalk and co-evolution of riboregulation and drug-resistance mechanisms.
In the present study, we investigated the effect of several protein synthesis inhibitors on transcription antitermination controlled by the full-length glyS T-box riboswitch from S. aureus. S. aureus is a prominent pathogen with a variety of RNA-mediated gene expression mechanisms, many of which respond to specific intracellular or extracellular signals to link virulence to stress adaptation and metabolism (63). The diversity of GT-boxes among pathogens as revealed by the phylogenetic analysis, as well as the variations in the number and sequences of tRNA ligands, are likely results of the evolution of species-specific T-box riboswitches, as driven by selective environmental pressures, particularly in the nosocomial environments where antibiotic-resistant strains rapidly evolve and thrive. The recent studies on the effect of mainstream protein synthesis inhibitors on the levels of riboswitch-controlled transcription in pathogens and microbiome strains, raise questions on the role of T-box riboswitches both as regulatory systems and molecular targets (12). Interestingly, many GT-boxes possess SL loops that contain tandem or overlapping glycine codons rather than single codons found in canonical T-boxes. This SL ambiguity alone can permit differential, nuanced anticodon recognition in distinct tRNAGly isoacceptors, which can vary under changing metabolic needs. The T-box RNA does not just decipher the tRNA anticodon trinucleotide. It employs multiple points and types of contacts to recognize the cognate tRNA as a three-dimensional object, which has characteristic structural variations and idiosyncrasies that are specific to the bacterial genome, type of the amino acid, and subtype of tRNA, etc. Indeed, a comparative analysis of the two co-crystal structures of T-box stem I-tRNA complexes from two closely related species within the same Bacillaceae family, namely Oceanobacillus and Geobacillus, revealed substantial structural differences (20,25,27). Along this same vein of T-box sequence and structural diversity, S. aureus GT-box provides a more dramatic example where an entire new domain, Stem Sa, is inserted into the antiterminator domain.
Many more examples of such species-specific T-box structural features may have evolved to either fine-tune T-box regulation or confer novel functions. For instance, one can hypothesize the existence of chimeric T-box riboswitches that may use extra domains (such as Stem Sa) to directly bind small molecule metabolites or antibiotics, thus integrating the signals of nutritional state (tRNA charging) with the chemical and metabolic state (e.g. antibiotics).
During this study, we observed that mainstream protein synthesis inhibitors directly modulate T-box controlled transcription antitermination, outside of their known roles in ribosomal inhibition. In addition to aminoglycosides, which can engage non-specific electrostatic interactions with RNA, tigecycline, a newly developed tetracycline derivative intended to fight antibiotic resistance, strongly enhanced T-box transcription antitermination in vitro and in vivo. The binding positions of either neomycin B or tigecycline are almost identical and contribute to the stabilization of the T-box:tRNA complex (Figure 7). The stabilization appears to be achieved through modulations near known binding interfaces between the two RNAs, including the SL loop, the apical loop of stem I and stem Sa, which together directs transcription of the downstream gene under T-box control. Our results expand the recent studies reporting that antibiotics can modulate riboswitch-controlled transcription, which now includes the widespread T-box riboswitches class. Previous studies have tested libraries of 4,5-disubstituted oxazolidinones for their effectiveness, but a key limitation was the use of a T-bulge construct (a minimal RNA which only partially resembled the antiterminator region) instead of a full-length T-box. Similar to our results, those studies identified compounds that could either induce or disrupt antitermination conformation suggesting that transcription antitermination controlled by T-boxes can indeed be modulated by antibiotics (34,37–42). Specifically, neomycin B exhibited affinity for nucleotides located in the bulged region of antiterminator domain via electrostatic attraction (35). In the present study, we observed that neomycin B mimics the anticodon-protective effect of T-box binding to tRNA. Neomycin B presumably induces a conformation that stabilizes the tRNA:T-box interaction at the anticodon-specifier interface by restricting the anticodon stem loop in a binding-productive conformation (Figure 7A and B).
The study of a full-length T-box allowed for a more complete and biologically relevant view of the myriad of T-box-tRNA interactions and conformational changes that are important for sensing esterified amino acid and conditional switching. Only three common positions were found between neomycin B and tigecycline, which increase transcription antitermination and linezolid, which decreases it. The first one is the semi-conserved G82 (G63 of the crystal structure) a conserved guanosine of the apical loop that interacts with C44 and A56 and faces G19 of the P1 tRNAGlyGCC elbow (Figure 7 and Supplementary S1). This position is considered very important for the overall local conformation upon tRNA binding, although it is replaced by U19 in the remaining four tRNAGly isoacceptors in S. aureus. The second (G109) and third positions (G110) are nucleotides of the SL, known to be important for the interaction and the recognition of tRNA’s anticodon. Interestingly, all three common positions are directly or indirectly involved with structural rearrangements that facilitate induced fit of the tRNA anticodon or elbow, both of which are important for recognition by stem I to ensure binding and recognition of the cognate tRNA ligand (Figure 7; 20,25). In addition, the protection of G34 in the tRNA by both neomycin B and tigecycline indicates that both antibiotics could favor a stronger interaction at the wobble position (Supplementary Figure S1).
We observed a striking correlation between the ability of a given antibiotic to enhance transcription antitermination and its binding to stem Sa. It was shown previously that probably stem Sa provides kinetic discrimination between the cognate P1 tRNAGlyGCC and the four remaining tRNAGly isoacceptors in S. aureus (23). The involvement of stem Sa in antibiotic binding could also provide a means for further stabilization of the antitermination conformation. Common binding positions for neomycin B and tigecycline, such as A182 and A196, may engage direct interactions with the tRNA acceptor stem through A-minor interactions (23). These and other adenosines that are protected by antibiotic binding are single-stranded, making them available to engage the minor groove of the tRNA acceptor stem, which may play key roles in stabilizing the T-box:tRNA complex in the antitermination conformation (Figure 7A). Both neomycin B and tigecycline bind to the T-box through specific interactions, in the presence of P1 tRNAGlyGCC. The binding sites that were identified both on the T-box and the tRNA indicate that transcription antitermination is increased through the stabilization of RNA-RNA interactions at one or more of the three interfaces: the anticodon-specifier helix stabilized by cross-strand stacking, the platform-platform stacking interaction between tRNA elbow and the stem I apical base triple, and the hypothesized A-minor interactions between tRNA acceptor stem and stem Sa (Figure 7B and C). Finally, our observation that medically relevant tigecycline concentrations present in cultures modulate T-box activity in vivo, is in agreement with previous observations showing that antibiotics that target translation can also affect RNA-mediated regulation of gene expression (11,12,61). The same studies suggested that besides affecting their primary targets, such as the ribosome, antibiotics present in the growth environment of pathogens can also induce broad and pleiotropic effects through resistance mechanisms. The stimulation of S. aureus glyS transcription by tigecycline in vivo could represent a similar response mechanism. Although tigecycline is ultimately bacteriocidal, it might help pathogens to induce early-response mechanisms, which could be effective during a very narrow time frame to allow emergence of random resistance mutations during replication.
The expanding knowledge and deepening mechanistic understanding of bacterial regulatory ncRNAs, including riboswitches, support the urgent need to discover new alternative molecular targets (63). For instance, PC1 (2,5,6-triaminopyrimidin-4-one), a novel pyrimidine derivative, was designed to target a purine-sensing riboswitch in S. aureus. This riboswitch controls the expression of GuaA GMP synthase, involved in pathogen's survival during infection (64). Moreover, the recent discovery of ribocil, a synthetic mimic of the flavin mononucleotide, represents an innovative strategy to target specific bacterial riboswitches (10). As our knowledge of the structure and function of T-box riboswitches is enriched, development of novel antimicrobial agents against the T-box system becomes more feasible. T-box riboswitches represent ideal targets because they regulate expression of many essential genes in amino acid metabolism, exert strong impact on protein translation, and can regulate multiple metabolic pathways. Phylogenetic evidence suggests that strong selective pressure exists to preserve the T-boxes from random mutagenesis, which is the common cause of antibiotic resistance. Genetic, biochemical, and structural analyses reveal a high degree of sequence and structural conservation that are concentrated on a small number of nucleosides in key locations. As the T-boxes are ‘ON’ switches, their activation, and concomitant downstream gene expression, require the proper alignment and engagement of multiple points of contact between the uncharged tRNA and the T-box riboswitch. Therefore, T-box function is easily disrupted by, and less tolerant of even single substitutions near any of these interfaces. Random mutagenesis of many T-box positions has a high probability (and occurrence) of disrupting the productive interaction between the uncharged tRNA and the T-box riboswitch, which leads to an inability to express essential genes in amino acid biosynthesis, import, aminoacylation etc., ultimately resulting in lethality, or at least much reduced competitive fitness. The observation that mainstream antibiotics can modulate T-box riboswitches among several other riboregulators calls for further biochemical and structural characterization of additional T-box riboswitches from representative pathogens. In particular, future work using native, host RNA polymerases from pathogenic bacteria that have co-evolved with their cognate T-box riboswitches are expected to uncover additional regulatory nuances that fine-tune the interplay between the sequence, structure, and energetics of the T-boxes and thermodynamic and kinetic properties of the host RNA polymerases (65,66). The anticipated results will provide much-needed knowledge and insights that inform the development of novel efficient and specific antibacterial compounds.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Konstantina Papakonstantinou for help with the constructions of vectors, Lena Kaliatsi and Nikoleta Giarimoglou for help with the in vivo experiments, Dr George Dinos (University of Patras) for discussions on antibiotics and Dr Hubert D. Becker (University of Strasbourg) for sharing materials, protocols and constructive discussions.
Footnotes
Present address: Maria Apostolidi, Department of Cellular and Molecular Physiology, School of Medicine, Yale University, Systems Biology Institute, 300 Heffernan drive, West Haven, CT 06516, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Fondatión Santé Grants 2016 [E515 to C.S.]; Intramural Research Program of National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the U.S. National Institutes of Health (NIH) (to J.Z.). Katerina Lamprinou is a fellowship recipient from Lilian Voudouri Foundation. Funding for open access charge: Fondatión Sante.
Conflict of interest statement. None declared.
REFERENCES
- 1. Breaker R.R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012; 4:a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sherwood A.V., Henkin T.M.. Riboswitch-mediated gene regulation: novel RNA architectures dictate gene expression responses. Annu. Rev. Microbiol. 2016; 70:361–374. [DOI] [PubMed] [Google Scholar]
- 3. Mellin J.R., Cossart P.. Unexpected versatility in bacterial riboswitches. Trends Genet. 2015; 31:150–156. [DOI] [PubMed] [Google Scholar]
- 4. Breaker R.R. Prospects for riboswitch discovery and analysis. Mol. Cell. 2011; 43:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henkin T.M. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008; 22:3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blount K.F., Breaker R.R.. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006; 24:1558–1564. [DOI] [PubMed] [Google Scholar]
- 7. Deigan K.E., Ferre-D’Amare A.R.. Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Acc. Chem. Res. 2011; 44:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colameco S., Elliot M.A.. Non-coding RNAs as antibiotic targets. Biochem. Pharmacol. 2017; 133:29–42. [DOI] [PubMed] [Google Scholar]
- 9. Mulhbacher J., Brouillette E., Allard M., Fortier L.C., Malouin F., Lafontaine D.A.. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 2010; 6:e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howe J.A., Wang H., Fischmann T.O., Balibar C.J., Xiao L., Galgoci A.M., Malinverni J.C., Mayhood T., Villafania A., Nahvi A. et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015; 526:672–677. [DOI] [PubMed] [Google Scholar]
- 11. Jia X., Zhang J., Sun W., He W., Jiang H., Chen D., Murchie A.I.. Riboswitch control of aminoglycoside antibiotic resistance. Cell. 2013; 152:68–81. [DOI] [PubMed] [Google Scholar]
- 12. Dar D., Shamir M., Mellin J.R., Koutero M., Stern-Ginossar N., Cossart P., Sorek R.. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science. 2016; 352:aad9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lalaouna D., Eyraud A., Chabelskaya S., Felden B., Massé E.. Regulatory RNAs involved in bacterial antibiotic resistance. PLoS Pathog. 2014; 10:e1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Howden B.P., Beaume M., Harrison P.F., Hernandez D., Schrenzel J., Seemann T., Francois P., Stinear T.P.. Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob Agents Chemother. 2013; 57:3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grundy F.J., Henkin T.M.. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell. 1993; 74:475–482. [DOI] [PubMed] [Google Scholar]
- 16. Henkin T.M. The T box riboswitch: A novel regulatory RNA that utilizes tRNA as its ligand. Biochim. Biophys. Acta. 2014; 1839:959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gutiérrez-Preciado A., Henkin T.M., Grundy F.J., Yanofsky C., Merino E.. Biochemical features and functional implications of the RNA-based T-box regulatory mechanism. Microbiol. Mol. Biol. Rev. 2009; 73:36–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vitreschak A.G., Mironov A.A., Lyubetsky V.A., Gelfand M.S.. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA. 2008; 14:717–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Ferre-D’Amare A.R.. Direct evaluation of tRNA aminoacylation status by the T-box riboswitch using tRNA-mRNA stacking and steric readout. Mol. Cell. 2014; 55:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J., Ferré-D’Amaré A.R.. Structure and mechanism of the T-box riboswitches. Wiley Interdiscip. Rev. RNA. 2015; 6:419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wels M., Groot Kormelink T., Kleerebezem M., Siezen R.J., Francke C.. An in silico analysis of T-box regulated genes and T-box evolution in prokaryotes, with emphasis on prediction of substrate specificity of transporters. BMC Genomics. 2008; 9:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saad N.Y., Stamatopoulou V., Braye M., Drainas D., Stathopoulos C., Becker H.D.. Two-codon T-box riboswitch binding two tRNAs. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:12756–12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Apostolidi M., Saad N.Y., Drainas D., Pournaras S., Becker H.D., Stathopoulos C.. A glyS T-box riboswitch with species-specific structural features responding to both proteinogenic and nonproteinogenic tRNAGly isoacceptors. RNA. 2015; 21:1790–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherwood A.V., Grundy F.J., Henkin T.M.. T box riboswitches in Actinobacteria: translational regulation via novel tRNA interactions. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J., Ferré-D’Amaré A.R.. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013; 500:363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grigg J.C., Chen Y., Grundy F.J., Henkin T.M., Pollack L., Ke A.. T box RNA decodes both the information content and geometry of tRNA to affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7240–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grigg J.C., Ke A.. Structural determinants for geometry and information decoding of tRNA by T box leader RNA. Structure. 2013; 21:2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang A.T., Nikonowicz E.P.. Solution NMR determination of hydrogen bonding and base pairing between the glyQS T box riboswitch Specifier domain and the anticodon loop of tRNA(Gly). FEBS Lett. 2013; 587:3495–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J., Ferré-D’Amaré A.R.. The tRNA elbow in structure, recognition and evolution. Life (Basel). 2016; 12:E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J., Ferré-DAmaré A.R.. Trying on tRNA for size: RNase P and the T-box riboswitch as molecular rulers. Biomolecules. 2016; 6:E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lehmann J., Jossinet F., Gautheret D. A universal RNA structural motif docking the elbow of tRNA in the ribosome, RNAse P and T-box leaders. Nucleic Acids Res. 2013; 41:5494–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grigg J.C., Ke A.. Sequence, structure, and stacking: specifics of tRNA anchoring to the T box riboswitch. RNA Biol. 2013; 10:1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giannouli S., Kyritsis A., Malissovas N., Becker H.D., Stathopoulos C.. On the role of an unusual tRNAGly isoacceptor in Staphylococcus aureus. Biochimie. 2009; 91:344–351. [DOI] [PubMed] [Google Scholar]
- 34. Means J., Katz S., Nayek A., Anupam R., Hines J.V., Bergmeier S.C.. Structure-activity studies of oxazolidinone analogs as RNA-binding agents. Bioorg. Med. Chem. Lett. 2006; 16:3600–3604. [DOI] [PubMed] [Google Scholar]
- 35. Anupam R., Denapoli L., Muchenditsi A., Hines J.V.. Identification of neomycin B-binding site in T box antiterminator model RNA. Bioorg. Med. Chem. 2008; 16:4466–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anupam R., Nayek A., Green N.J., Grundy F.J., Henkin T.M., Means J.A., Bergmeier S.C., Hines J.V.. 4,5-Disubstituted oxazolidinones: high affinity molecular effectors of RNA function. Bioorg. Med. Chem. Lett. 2008; 18:3541–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Acquaah-Harrison G., Zhou S., Hines J.V., Bergmeier S.C.. Library of 1, 4-disubstituted 1, 2, 3-triazole analogs of oxazolidinone RNA-binding agents. J. Comb. Chem. 2010; 12:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maciagiewicz I., Zhou S., Bergmeier S.C., Hines J.V.. Structure-activity studies of RNA-binding oxazolidinone derivatives. Bioorg. Med. Chem. Lett. 2011; 21:4524–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Orac C.M., Zhou S., Means J.A., Boehm D., Bergmeier S.C., Hines J.V.. Synthesis and stereospecificity of 4, 5-disubstituted oxazolidinone ligands binding to T-box riboswitch. RNA. 2011; 54:6786–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou S., Acquaah-Harrison G., Bergmeier S.C., Hines J.V.. Anisotropy studies of tRNA-T box antiterminator RNA complex in the presence of 1, 4-disubstituted 1, 2, 3-triazoles. Bioorg. Med. Chem. Lett. 2011; 21:7059–7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maciagiewicz I., Zhou S., Bergmeier S.C., Hines J.V.. Structure-activity studies of RNA-binding oxazolidinone derivatives. Bioorg. Med. Chem. Lett. 2011; 21:4524–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou S., Means J.A., Acquaah-Harrison G., Bergmeier S.C., Hines J.V.. Characterization of a 1, 4-disubstituted 1, 2, 3-triazole binding to T box antiterminator RNA. Bioorg. Med. Chem. 2012; 20:1298–1302. [DOI] [PubMed] [Google Scholar]
- 43. Novichkov P.S., Kazakov A.E., Ravcheev D.A., Leyn S.A., Kovaleva G.Y., Sutormin R.A., Kazanov M.D., Riehl W., Arkin A.P., Dubchak I. et al. RegPrecise 3.0—a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics. 2013; 14:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanabe M., Kanehisa M.. Using the KEGG database resource. Curr. Protoc. Bioinformatics. 2012; doi:10.1002/0471250953.bi0112s11. [DOI] [PubMed] [Google Scholar]
- 45. Notredame C., Higgins D.G., Heringa J.. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000; 302:205–217. [DOI] [PubMed] [Google Scholar]
- 46. Schattner P., Brooks A.N., Lowe T.M.. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005; 33:W686–W689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. et al. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England). 2007; 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 48. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M. et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008; 36:W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J.. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Comput. Chem. 2009; 16:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trott O., Olson A.J.. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010; 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 52. Wiegand I., Hilpert K., Hancock R.E.. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008; 3:163–175. [DOI] [PubMed] [Google Scholar]
- 53. Henkin T.M. Analysis of tRNA-directed transcription antitermination in the T box system in vivo. Methods Mol. Biol. 2009; 540:281–290. [DOI] [PubMed] [Google Scholar]
- 54. Saad N.Y., Schiel B., Braye M., Heap J.T., Minton N.P., Durre P., Becker H.D.. Riboswitch (T-box)-mediated control of tRNA-dependent amidation in Clostridium acetobutylicum rationalizes gene and pathway redundancy for asparagine and asparaginyl-trnaasn synthesis. J. Biol. Chem. 2012; 287:20382–20394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Griffith K.L., Wolf R.E. Jr. Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 2002; 290:397–402. [DOI] [PubMed] [Google Scholar]
- 56. Arenz S., Wilson D.N.. Blast from the past: reassessing forgotten translation inhibitors, antibiotic selectivity, and resistance mechanisms to aid drug development. Mol. Cell. 2016; 61:3–14. [DOI] [PubMed] [Google Scholar]
- 57. Artsimovitch I., Svetlov V., Anthony L., Burgess R.R., Landick R.. RNA polymerases from Bacillus subtilis and Escherichia coli differ in recognition of regulatory signals in vitro. J. Bacteriol. 2000; 182:6027–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mikkelsen N.E., Johansson K., Virtanen A., Kirsebom L.A.. Aminoglycoside binding displaces a divalent metal ion in a tRNA-neomycin B complex. Nat. Struct. Biol. 2001; 8:510–514. [DOI] [PubMed] [Google Scholar]
- 59. Lalaouna D., Eyraud A., Chabelskaya S., Felden B., Massé E.. Regulatory RNAs involved in bacterial antibiotic resistance. PLoS Pathog. 2014; 10:e1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu J., Schneiders T.. Tigecycline challenge triggers sRNA production in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2012; 12:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Howden B.P., Beaume M., Harrison P.F., Hernandez D., Schrenzel J., Seemann T., Francois P., Stinear T.P.. Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob. Agents Chemother. 2013; 57:3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sommer M.O., Suess B.. RIBOSWITCHES. (Meta-)genome mining for new ribo-regulators. Science. 2016; 352:144–145. [DOI] [PubMed] [Google Scholar]
- 63. Fechter P., Caldelari I., Lioliou E., Romby P.. Novel aspects of RNA regulation in Staphylococcus aureus. FEBS Lett. 2014; 588:2523–2529. [DOI] [PubMed] [Google Scholar]
- 64. Mulhbacher J., Brouillette E., Allard M., Fortier L.C., Malouin F., Lafontaine D.A.. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 2010; 6:e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang J., Landick R.A. Two-way street: Regulatory interplay between RNA polymerase and nascent RNA structure. Trends Biochem. Sci. 2016; 41:293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang J., Lau M.W., Ferré-D’Amaré A.R.. Ribozymes and riboswitches: modulation of RNA function by small molecules. Biochemistry. 2010; 49:9123–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.