

Abstract
Eusociality represents a major transition in evolution and is typified by cooperative brood care and reproductive division of labor between generations. In bees, this division of labor allows queens and workers to phenotypically specialize. Worker traits associated with helping are thought to be crucial to the fitness of a eusocial lineage, and recent studies of honey bees (genus Apis) have found that adaptively evolving genes often have worker-biased expression patterns. It is unclear however if worker-biased genes are disproportionately acted on by strong positive selection in all eusocial insects. We undertook a comparative population genomics study of bumble bees (Bombus) and honey bees to quantify natural selection on queen- and worker-biased genes across two levels of social complexity. Despite sharing a common eusocial ancestor, genes, and gene groups with the highest levels of positive selection were often unique within each genus, indicating that life history and the environment, but not sociality per se, drives patterns of adaptive molecular evolution. We uncovered differences in the contribution of queen- and worker-biased genes to adaptive evolution in bumble bees versus honey bees. Unlike honey bees, where worker-biased genes are enriched for signs of adaptive evolution, genes experiencing positive selection in bumble bees were predominately expressed by reproductive foundresses during the initial solitary-founding stage of colonies. Our study suggests that solitary founding is a major selective pressure and that the loss of queen totipotency may cause a change in the architecture of selective pressures upon the social insect genome.
Keywords: sociality, natural selection, kin selection, fitness
Introduction
Within a hymenopteran eusocial colony, labor is divided between the queens—responsible for most of the reproduction—and their workers—responsible for all aspects of colony upkeep including brood care, nest defense, and foraging (Wheeler 1910; Wilson 1985; Winston 1987; Hölldobler and Wilson 1990; Sagili etal. 2011; Wray etal. 2011). The separation and subsequent specialization of these roles is the result of Darwinian selection that has acted directly on mutations contributing to queen phenotypes and indirectly on mutations that influence worker traits (Hamilton 1964a, 1964b; Wilson 1985; Sagili etal. 2011; Wray etal. 2011). We do not yet have an understanding of the relative role of queen or worker phenotypes to the fitness of eusocial lineages, a knowledge gap that has hindered our ability to understand the evolutionary processes responsible for caste divergence across different stages of social evolution and the resulting changes in social complexity.
Until recently, it was impossible to objectively compare the fitness effects of mutations influencing queen and worker traits. However, advances in population and functional genomics of social insects have allowed researchers to identify genes that are associated with worker and queen traits and quantify adaptive evolution in social lineages (Hasselmann etal. 2015; Kent and Zayed 2015).
The first population genomic study of a social insect demonstrated genes with worker biased expression being more commonly under positive selection in the eusocial honey bee, Apis mellifera (Harpur etal. 2014). However, the relative levels of adaptive evolution of worker genes in other eusocial species is not well understood and may be substantially different as a result of variation in social lifestyles and life histories among taxa. In the honey bees (Apis spp.), for example, colonies are perennial and contain thousands of individual workers that are morphologically distinct from their single queen (Michener 1974; Rehan and Toth 2015). In contrast, most bumble bees (Bombus spp.) have small, annual colonies, and have a solitary worker-less phase that precedes early colony development (Michener 1974; Winston 1987; Rehan and Toth 2015). During the worker-less phase, foundresses (future queens) are solely responsible for the success of a future colony’s output and perform all or a subset of the behavioral repertoire of workers to provision their first brood (Alford 1969; Crespi and Yanega 1995; Gadagkar 1997; Bourke 2011; Rehan and Toth 2015). In these annual eusocial societies, the success of a colony may be more influenced by traits expressed by foundresses and queens than those expressed by workers (Michener 1974).
The corbiculate bees are an ideal group to study the relative contribution of queen-acting and worker-acting mutations to fitness because of their considerable variation in social organization (Rehan and Toth 2015). Moreover, honey bees and bumble bees share a common social ancestor, and consequently, have been subject to the potential genomic impacts of social evolution for the same length of time (Romiguier etal. 2016). We carried out a comparative population genomics study of bumble bees and honey bees to identify and characterize genes with signatures of adaptive evolution in the two lineages, and compared the fitness effects of nonsynonymous mutations influencing queen and worker phenotypes in the bumble bees relative to the perennially eusocial honey bees.
Materials and Methods
Sampling, Sequencing, Alignment, and SNP Calling
We sampled haploid males from populations of the bumble bees B. impatiens (Toronto, Canada; N = 10 and B. melanopygus (Oregon, United States; N = 3; supplementary table S1, Supplementary Material online), both in subgenus Pyrobombus, and B. terrestris (subgenus Bombus s.s.) (Norwich, United Kingdom; N = 8). Each bee sample was paired-end sequenced (150 bp) with Illumina Hi-Seq Sequencing at either Génome Québec’s Innovation Centre or the Penn State Huck Institutes of the Life Sciences’ Genome Core Facility to an average read-depth at each SNP of 16.5 × (supplementary table S1, Supplementary Material online). All reads were aligned to the B. impatiens genome assembly v 2.0 and annotated with Official Gene Set v 2.0 (Sadd etal. 2015) using the default parameters of BWA v 7.5 and SAMtools v 1.19 (Li etal. 2009; Li and Durbin 2010). Because sequences were diverse and divergent relative to the reference genome, we remapped each bee’s sequence using STAMPY v 1.0 (Lunter and Goodson 2011) at a substitution rate of 0.02. We subsequently realigned with GATK v 3.1 RealignerTargetCreator followed by IndelRealigner to reduce any potential erroneous alignments close to indels (DePristo etal. 2011). VCF files were created using GATK UnifiedGenotyper for both Single Nucleotide Polymorphisms (SNPs) and indels using –ploidy 1. We used three filters to reduce the chance of making erroneous genotype or variant calls. First, we removed all SNPs within 10 bp of an indel using GATK’s –maskExtension command. Second, we removed all SNPs in areas of outlier depth using a 1.5× Inter Quartile Range cutoff. Third, we broadly removed all SNPs within repetitive or potentially paralogous areas of the genome. To perform this filter, we performed a BLAST of 150 bp sequences across the B. impatiens genome and excluded any SNP within an area with multiple BLAST best-matches (E-value cut-off of 1−6). Finally, we removed any SNP that could potentially be misgenotyped due to its local sequence complexity. We performed this filter by allowing GATK to call SNP genotypes for three randomly selected B. impatiens samples using the –ploidy 2 option. Because all of our samples were haploid, any site called as heterozygotic is erroneous. We compiled a list of such sites and removed all SNP calls within 5 bp from all samples we sequenced.
We identified all SNPs within protein coding genes and identified if those SNPs were nonsynonymous or synonymous using SNPEFF v 3.6 (Cingolani etal. 2012) and excluded all genes lacking start codons, lacking stop codons, or containing premature stop codons (N = 1,018 genes excluded). Because OGS v.2 contains isoforms of each gene, we included either the longest isoform or, in the case of isoforms being the same size, we randomly selected a single isoform for our analyses.
Relatedness and Population Structure
Because we sampled individuals of the species B. impatiens and B. terrestris within the same municipality, we tested whether samples within each species were siblings or close relatives. We used the program RELATEDNESS 4.2 (Queller and Goodnight 1989) to determine the average relatedness of individuals within each species using the genotypes at 127 randomly selected SNPs with MAF > 0.1 over 10 runs, each selecting a new set of 127 random SNP genotypes. No two individuals within any of these two Bombus species had significant evidence of being closely related (relatedness not significantly different from 0; P > 0.25 for all comparisons). To ensure each sample was indeed a member of its designated species and to ensure there was no evidence of population structure within species, we used the program ADMIXTURE v 1.22 (Alexander etal. 2009). Within each species we estimated K, the number of groups within a data set, by randomly selecting 10% of SNPs with MAF > 0.1 and estimating K = 1 to N - 2 where N is the number of samples for a given species. We tested each value of K 5 times with different sets of randomly selected SNPs. We used the cross-validation (CV) method to determine the optimal value for K. We used the same method above, but for SNPs shared across all species to ensure our sampling represented three distinct bumble bee lineages. There was no evidence of population structure within any species using ADMIXTURE (K = 1 with all species individually).
Analysis of Positive Selection
We estimated the strength of selection within the Bombus genus for 10,048 genes using a Bayesian implementation of the McDonald–Kreitman test (Eilertson etal. 2012). After identifying synonymous and nonsynonymous mutations (above), we classified mutations as being fixed or polymorphic within species pairs within the genus (e.g. within and between B. impatiens and B. terrestris). We ran the Bayesian implementation of SNIPRE for 15,000 iterations after 100,000 burnin steps. After a Bayesian-equivalent False Discovery Rate correction, SNIPRE outputs estimates of the scaled selection coefficient, γ (2Nes) and its 95% confidence interval. We also performed the same analyses above using B. impatiens and B. melanopygus to derive counts of fixed and polymorphic SNPs within exons. Our estimates of selection were highly correlated between the two potential outgroups (t10,006 = 85; r = 0.65, P < 2.2 × 10−6). Finally, to validate our estimates of γ, we calculated α, a measure of the proportion of nonsynonymous mutations fixed by selection (Eyre-Walker 2006) to ensure our results were consistent across methodologies. We found that γ and α correlated significantly (r = 0.80, P < 2.2 × 10−16) and that high γ genes tended to also have α ≫ 0.
Caste-Biased Genes and Genes Expressed during Diapause
We followed the same procedure used by our group previously to identify differential expression among castes of honey bees (Harpur etal. 2014) using the Honey Bee Protein Atlas (Chan etal. 2013). The Atlas provides a list of proteins which were found to be differentially expressed consistently across 26 tissues of queens and workers. We identified Bombus genes with caste-biased expression patterns by using results kindly provided to us from previous micro-array analyses that examined brain gene expression of B. terrestris within queens, workers, foundresses, and gynes (Woodard etal. 2014). By comparing gene expression among castes, Woodard etal. (2014) were able to identify differentially expressed genes between reproductive and nonreproductive castes and brood-caring versus nonbrood-caring castes. We classified genes as functioning in reproduction or brood care by making use of an ANOVA model performed previously that classified genes as being over or underexpressed in castes performing either function (Woodard etal. 2014). We were able to calculate γ on 5,643 genes within this data set and found that 24.5% of these genes have significant evidence of having caste-bias expression patterns. To explore which reproductive-expressed genes were acted on by selection within Bombus, we analyzed another transcriptomic data set from the fat bodies of virgin or mated female reproductives, diapausing female reproductives, and egg-laying foundresses (Amsalem etal. 2015 see supplementary table S3a, Supplementary Material online therein). This data set compared expression using a pairwise approach between all life-history stages and treatments. We tallied the total number of differentially expressed genes within each pairwise comparison (N = 17 comparisons) acted on by positive selection. On average, 13% of differentially expressed genes had evidence of positive selection (γ > 1). We then compared the total proportion of genes acted on by selection to those comparisons exclusively differentially expressed in all foundress (“FC” or “FD”) comparisons.
GO Analysis
To identify functional relevance of genes with evidence of positive selection, we used GO analysis as executed by DAVID v 6.7. We followed the same procedure used by our group previously (Harpur etal. 2014) in order to compare GO terms between the current study and work examining selection within the genus Apis. For both the Apis and Bombus data sets, we identified putative fly orthologues using a BlastP best match (E-value 1−6) and used this list as our background gene set. We used default parameters but report only MF_FAT, CC_FAT, and BP_FAT and significant GO results and KEGG pathways, following correction for False Discovery Rate (Bonferroni < 0.05).
Statistical Analyses and Data Accession
All analyses and pipelines can be found on the first author’s GitHub (https://github.com/harpur/Bombus; last accessed September 12, 2017), including all supplementary Data, Supplementary Material online used in this study. We performed analyses using all values of γ, as well as for γ > 1, which we termed “high gamma”. Where appropriate, we used parametric models for all statistical tests, unless otherwise stated. All sequence data have been deposited with NCBI’s Short-Read Archive (BioProject PRNJA347806; supplementary table S1, Supplementary Material online).
Results and Discussion
Adaptively Evolving Genes in Bombus and Apis Are Largely Different
We used population genomic approaches (Hasselmann etal. 2015; Kent and Zayed 2015) to estimate the strength of selection acting on genes in Bombus and genes in Apis over the ∼5–25 Ma of divergence between the studied species within each genus (Arias and Sheppard 1996; Cameron etal. 2007; Hines 2008; Kotthoff etal. 2013). The bumble bee data set comprised 21 newly resequenced genomes representing Bombus impatiens, B. terrestris, and B. melanopygus sequenced at high depth (16.5×, supplementary table S1, Supplementary Material online; Materials and Methods). We compared this data set to a recently published Apis population genomic study (39 A. mellifera and 1 A. cerana) that was sequenced and analyzed using similar methods (Harpur etal. 2014). Within the new Bombus data set, we were able to identify 60,887 polymorphic sites and 353,250 fixed sites in genes (supplementary Data S1, Supplementary Material online) including 136,985 nonsynonymous mutations and 277,152 synonymous mutations. We used a Bayesian implementation of the McDonald–Kreitman test (Eilertson etal. 2012) to estimate γ, the average selection coefficient of nonsynonymous mutations scaled by the effective population size, for 10,008 protein-coding genes in Bombus and 12,303 protein-coding genes in Apis (Materials and Methods; fig. 1). This method is able to identify instances of positive selection, but may miss the most extreme cases of positive selection due to elevated divergence in homologous sequences between sister taxa. Therefore, our approach likely underestimates the total number of adaptively evolving genes.
Fig. 1.

—Distribution of the selection coefficient of replacement mutations scaled by the effective population size (γ = 2Nes) for protein-coding genes within Bombus (top) and Apis (bottom).
We found evidence of strong positive selection acting on sets of genes within both lineages (fig. 1): 17.8% of Bombus genes and 9.3% of genes within Apis genes (Harpur etal. 2014) had evidence of strong positive selection (γ > 1; fig. 1) and both groups had ∼2% of genes with γ > 2. The positively selected genes in honey bees and bumble bees are largely unique to each lineage. There was a weak positive correlation between the selection coefficient (γ) between all genes in Bombus and their putative orthologous in Apis (Pearson Correlation, r = 0.17, P < 2.2 × 10−16). However, this likely reflects shared patterns of evolutionary constraint on protein-coding sequences in both groups. As the selection coefficient increases in either lineage, the correlation coefficient between γ in Apis and Bombus rapidly becomes significantly negative then nonsignificant (fig. 2). Further, among the 7,005 orthologs with estimates of γ, we found only 3% with γ > 1 in both lineages and 0.2% with γ > 2 in both lineages. While this overlap is statistically higher than expected (1.7% of γ > 1 genes and 0.05% of γ > 2 genes expected to overlap by chance; P < 0.001 for both comparisons), it is small in absolute terms: 83.5% of Bombus genes with positive selection are not adaptively evolving in Apis. This is consistent with two recent studies that found little evidence for common patterns of accelerated amino acid evolution across independently derived social lineages (Woodard etal. 2011; Kapheim etal. 2015). Given that honey bees and bumble bees share a common social ancestor, our finding that genes with high levels of positive selection were largely unique to each genus indicates that adaptive divergence involves very different genes in social lineages.
Fig. 2.

—Genes acted on by strong positive selection within Apis are not generally the same as those acted on by strong positive selection within Bombus: as the selection co-efficient increases in both species, genes with strong signs of positive selection in one species tend to be neutrally evolving in the other. Red line is x = y line. Insert shows correlation coefficient and its significance for different γ cutoffs.
Adaptively Evolving Gene Functions in Bombus and Apis
Whereas the genes acted on by the strongest positive selection within Bombus are largely not the same as those acted on by strong positive selection within Apis, there may be overlap in the biological, molecular, or cellular functions of these genera-specific positively-selected genes if eusocial societies face similar selective pressures. We explored this hypothesis by identifying the Gene Ontology (GO) terms (Huang etal. 2008) associated with genes underlying adaptive evolution in both genera. Similar to our gene-specific analysis, we found little overlap between the functions of genes underlying adaptive evolution in bumble bees and honey bees. Within Bombus, we found 53 significantly enriched GO terms that were mostly involved in metabolic processes: The most significant terms included mitochondrion (GO: 0005739), oxidative reduction (GO: 0055114), mitochondrial organization (GO: 0007005), NADH dehydrogenase activity (GO: 0003954), and mitochondrial ATP synthesis coupled electron transport (GO: 0042775; supplementary table S2, Supplementary Material online). As we removed all mitochondrial genome scaffolds from our analyses (Materials and Methods), these genes represent adaptively evolving nuclear genes that are involved in mitochondrial function. In contrast, positively selected genes within Apis were enriched for 41 significant GO terms often related to behavior, including sensory perception (GO: 0007600), sensory perception of smell (GO: 0007608), cofactor binding (GO: 0048037), channel-activity (GO: 0015267), and cognition (GO: 0050890). We found very little overlap between GO terms acted on by selection within Apis and Bombus: Only two terms were enriched within each of the Biological and Molecular Functions between genera (supplementary table S2, Supplementary Material online). If we examine only genes with very strong evidence of positive selection (γ > 2), there is no overlap in the GO terms acted on by selection within Apis and Bombus. Overall, this indicates that, for at least the strongest instances of selection within these two social genera, much of the adaptive protein evolution is lineage-specific.
Adaptive Evolution of Queen-Biased versus Worker-Biased Genes in Apis and Bombus
We found little overlap in the genes experiencing the strongest positive selection in both genera, suggesting that the traits underlying adaptive evolution differ between the two. We directly tested this hypothesis and found a shift in the relative levels of adaptive evolution of genes with an expression bias towards adult workers between these two eusocial genera. We made use of several data sets that quantified gene expression across caste (Materials and Methods) and extracted those with caste-biased expression patterns. We used these caste-biased genes as a surrogate for genes involved in generating caste-biased traits (Jasper etal. 2015). We had previously reported, and here replicated, that worker-biased genes in honey bees have higher selection coefficients relative to queen-biased and nondifferentially expressed genes in Apis and have a higher proportion of genes acted on by positive selection (Harpur etal. 2014; fig. 3). Using a similar data set for Bombus that examined differential brain gene expression across each life-history stage and caste (Woodard etal. 2014), we found that the genes associated with Bombus female reproductives had significantly higher selection coefficients and a higher proportion of genes acted on by positive selection than genes biased in nonreproductives and those that are not differentially expressed (fig. 3).
Fig. 3.
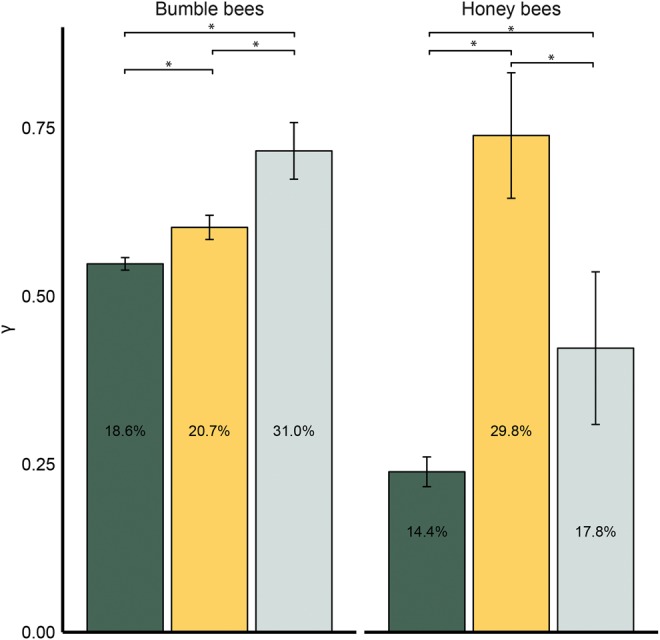
—In Apis (right) genes associated with nonreproductive phenotypes (gold bars) show signs of adaptive evolution relative to genes expressed in reproductives (silver bars; F2,1688 = 11.97; P = 0.0000007; Tukey P < 0.01 for all comparisons); however, in Bombus (left), this pattern in reversed and genes expressed in female reproductive castes show signs of positive selection greater than those expressed in workers (F2,5640 = 10.7; P = 0.00002; Tukey P < 0.03 for all comparison). Error bars denote SEM; Percentages within bars are percent genes with high gamma (γ > 1). Green = nondifferentially expressed genes.
This shift in the strength of selection on workers versus reproductives may reflect fundamental differences in the life histories of these two lineages. For example, the solitary founding phase in Bombus imposes a strong selective filter on reproductive individuals (Free and Butler 1959; Goulson 2010). At this life history stage, the foundress is solely responsible for the success of a future colony’s output and there are strong metabolic demands to produce eggs, forage, and maintain the colony (Free and Butler 1959; Goulson 2010; Woodard etal. 2013). We predicted that genes expressed at this life history stage may be those contributing to the patterns of positive selection we have detected on the Bombus genome. To test this prediction, we analyzed a recent transcriptomic data set from the fat bodies of virgin or mated female reproductives, diapausing female reproductives, and egg-laying foundresses for signatures of selection (Amsalem etal. 2015). We found that genes differentially-expressed by foundresses had a significantly higher proportion of genes acted on by strong positive selection (21.8%) relative to genes highly expressed during any other life history stage in this study (13.2% of genes across all other stages; Fisher Exact tests; P < 0.0001). This analysis suggests that genes expressed by foundresses early in the colony cycle are the major source of adaptive evolution in bumble bees.
Our study explored the evolution of genes within Apis and Bombus in greater detail than any to date. Two broader studies comparing rates of protein evolution among bees echoed our findings that sociality does not lead to correlated patterns of protein sequence divergence (Woodard etal. 2011; Kapheim etal. 2015). In both studies, there was little overall overlap in genes rapidly evolving in social bee species. However, examining lineage-specific patterns of selection within species of Bombini and Allodapini—both with annual life histories and a founding phase—Woodard etal. (2011) found that although the positively-selected genes of these two groups were largely different as a set, they tended to be associated with reproductive function, similar to our findings of positive selection on genes associated with queens and reproductives based on expression data.
Our study used the best available transcriptomic data to identify caste-biased expression patterns, but, clearly, the honey bee data sets are much larger in scope relative to Bombus. For example, we used the Honey Bee Protein Atlas (Chan etal. 2013), a resource that identifies proteins that are differentially expressed consistently across 26 tissues of queens and workers. In contrast, the bumble bee data sets we used were only derived from two tissues (Woodard etal. 2014; Amsalem etal. 2015). Studies of adaptive evolution in bumble bees will surely improve as richer transcriptomic data sets become available for the genus.
Conclusions
Our analyses provide unique insights into the factors that influence adaptive caste divergence in social organisms. The stark differences in the adaptive evolution of queen and worker genes between bumble bees and honey bees is particularly intriguing because it suggests that the evolution of eusociality per se does not necessary lead to conditions that render worker phenotypes to be of primary importance for the adaptive evolution of eusocial lineages. Honey bee workers are present during the entire life cycle and thus can continuously influence the fitness of a colony. However, bumble bee workers are present only after colony founding and, according to our results; their overall contribution to adaptive evolution may be smaller relative to queens, perhaps as a result of the strong selective pressure on queens during the solitary founding stage.
Solitary nest founding is a common feature of most primitively eusocial insects and among many highly eusocial ant species. Our results suggest this life history trait leads to faster rates of adaptive evolution in genes expressed by reproductives relative to workers. The finding that strong selection acts on different genes within the genomes of Apis and Bombus and that those genes with the strongest signs of selection in each group likely act on traits relevant to different castes is of considerable sociobiological importance. It suggests that the loss of queen totipotency causes a dramatic change in the architecture of selection pressures upon the social insect genome. Switches from eusociality to solitary behavior have occurred many times but there seem to have been few switches from swarm to independent colony founding among social insects (Packer 1997; Noll 2002; Cronin etal. 2013). Our results suggest that divergent selection regimes may have made the latter transition much less likely.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This study was supported by a Garfield Weston Foundation grant (L.P. and A.Z.), a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (A.Z.), a NSERC Alexander Graham Bell PGS (B.A.H.), and a National Science Foundation CAREER grant DEB – 1453473 (H.M.H.).
Literature Cited
- Alexander DH, Novembre J, Lange K.. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19(9):1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford D. 1969. A study of the hibernation of bumblebees (Hymenoptera: Bombidae) in southern England. J Anim Ecol. 38(1):149–170. [Google Scholar]
- Amsalem E, Galbraith DA, Cnaani J, Teal PE, Grozinger CM.. 2015. Conservation and modification of genetic and physiological toolkits underpinning diapause in bumble bee queens. Molec Ecol. 24(22):5596–5615. [DOI] [PubMed] [Google Scholar]
- Arias MC, Sheppard WS.. 1996. Molecular phylogenetics of honey bee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequence. Molec Phylogenet Evol. 5(3):557–566. [DOI] [PubMed] [Google Scholar]
- Bourke AFG. 2011. Principles of social evolution. Oxford; New York: Oxford University Press. [Google Scholar]
- Cameron SA, Hines HM, Williams PH.. 2007. A comprehensive phylogeny of the bumble bees (Bombus). Biol J Linnean Soc. 91(1):161–188. [Google Scholar]
- Chan QW, Chan MY, Logan M, Fang Y, Higo H, Foster LJ.. 2013. Honey bee protein atlas at organ-level resolution. Genome Res. 23:1951–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, et al. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi BJ, Yanega D.. 1995. The definition of eusociality. Behav Ecol. 6(1):109–115. [Google Scholar]
- Cronin AL, Molet M, Doums C, Monnin T, Peeters C.. 2013. Recurrent evolution of dependent colony foundation across eusocial insects. Ann Rev Entomol. 58(1):37–55. [DOI] [PubMed] [Google Scholar]
- DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilertson KE, Booth JG, Bustamante CD, Kosakovsky Pond SL.. 2012. SnIPRE: selection inference using a Poisson random effects model. PLoS Comput Biol. 8(12):e1002806.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. 2006. The genomic rate of adaptive evolution. Trends Ecol Evol. 21(10):569–575. [DOI] [PubMed] [Google Scholar]
- Free JB, Butler CG.. 1959. Bumblebees. London: Collins. [Google Scholar]
- Gadagkar R. 1997. The evolution of caste polymorphism in social insects: genetic release followed by diversifying evolution. J Genet. 76(3):167–179. [Google Scholar]
- Goulson D. 2010. Bumblebees: behaviour, ecology, and conservation. Oxford; New York: Oxford University Press. [Google Scholar]
- Hamilton WD. 1964a. Genetical evolution of social behaviour 2. J Theor Biol. 7:17–52. [DOI] [PubMed] [Google Scholar]
- Hamilton WD. 1964b. Genetical evolution of social behaviour I. J Theor Biol. 7:1–16. [DOI] [PubMed] [Google Scholar]
- Harpur BA, et al. 2014. Population genomics of the honey bee reveals strong signatures of positive selection on worker traits. Proc Natl Acad Sci U S A. 111(7):2614–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmann M, Ferretti L, Zayed A.. 2015. Beyond fruit-flies: population genomic advances in non-Drosophila arthropods. Brief Funct Genomics. 14(6):424–431. [DOI] [PubMed] [Google Scholar]
- Hines HM. 2008. Historical biogeography, divergence times, and diversification patterns of bumble bees (Hymenoptera: Apidae: Bombus). Syst Biol. 57(1):58–75. [DOI] [PubMed] [Google Scholar]
- Hölldobler B, Wilson EO.. 1990. The ants. Cambridge: Belknap Press of Harvard University Press. [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA.. 2008. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4(1):44–57. [DOI] [PubMed] [Google Scholar]
- Jasper WC, et al. 2015. Large-scale coding sequence change underlies the evolution of postdevelopmental novelty in honey bees. Molec Biol Evol. 32(2):334–346. [DOI] [PubMed] [Google Scholar]
- Kapheim KM, et al. 2015. Genomic signatures of evolutionary transitions from solitary to group living. Science 348(6239):1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent CF, Zayed A.. 2015. Chapter nine-population genomic and phylogenomic insights into the evolution of physiology and behaviour in social insects. Adv Insect Physiol. 48:293–324. [Google Scholar]
- Kotthoff U, Wappler T, Engel MS.. 2013. Greater past disparity and diversity hints at ancient migrations of European honey bee lineages into Africa and Asia. J Biogeogr. 40:1832–1838. [Google Scholar]
- Li H, Durbin R.. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Goodson M.. 2011. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21(6):936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michener CD. 1974. The social behavior of the bees; a comparative study. Cambridge: Belknap Press of Harvard University Press. [Google Scholar]
- Noll FB. 2002. Behavioral phylogeny of corbiculate Apidae (Hymenoptera; Apinae), with special reference to social behavior. Cladistics 18(2):137–153. [DOI] [PubMed] [Google Scholar]
- Packer L. 1997. The relevance of phylogenetic systematics to biology: examples from medicine and behavioural ecology. In: Grandcolas P, editor. The origin of biodiversity in insects: phylogenetic tests of Evolutionary Scenarios Museum National d'Histoire Naturelle. p. 11–29.
- Queller DC, Goodnight KF.. 1989. Estimating relatedness using genetic-markers. Evolution 43(2):258–275. [DOI] [PubMed] [Google Scholar]
- Rehan SM, Toth AL.. 2015. Climbing the social ladder: the molecular evolution of sociality. Trends Ecol Evol. 30(7):426–433. [DOI] [PubMed] [Google Scholar]
- Romiguier J, et al. 2016. Phylogenomics controlling for base compositional bias reveals a single origin of eusociality in corbiculate bees. Molec Biol Evol. 33(3):670–678. [DOI] [PubMed] [Google Scholar]
- Sadd BM, et al. 2015. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 16:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagili RR, Pankiw T, Metz BN.. 2011. Division of labor associated with brood rearing in the honey bee: how does it translate to colony fitness? PLoS ONE. 6(2):e16785.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler WM. 1910. Ants; their structure, development and behavior. New York: Columbia University Press. [Google Scholar]
- Wilson EO. 1985. The sociogenesis of insect colonies. Science 228(4707):1489–1495. [DOI] [PubMed] [Google Scholar]
- Winston ML. 1987. The biology of the honey bee. Cambridge: Harvard University Press. [Google Scholar]
- Woodard SH, Bloch G, Band MR, Robinson GE.. 2013. Social regulation of maternal traits in nest-founding bumble bee (Bombus terrestris) queens. J Exp Biol. 216(Pt 18):3474–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard SH, Bloch GM, Band MR, Robinson GE.. 2014. Molecular heterochrony and the evolution of sociality in bumblebees (Bombus terrestris). Proc R Soc B-Biol Sci. 281(1780):20132419.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard SH, et al. 2011. Genes involved in convergent evolution of eusociality in bees. Proc Natl Acad Sci U S A. 108(18):7472–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray MK, Mattila HR, Seeley TD.. 2011. Collective personalities in honeybee colonies are linked to colony fitness. Anim Behav. 81(3):559–568. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.