

Abstract
Mycobacterium avium subsp. hominissuis (MAH) is one of the most common nontuberculous mycobacterial species responsible for chronic lung disease in humans. Despite increasing worldwide incidence, little is known about the genetic mechanisms behind the population evolution of MAH. To elucidate the local adaptation mechanisms of MAH, we assessed genetic population structure, the mutual homologous recombination, and gene content for 36 global MAH isolates, including 12 Japanese isolates sequenced in the present study. We identified five major MAH lineages and found that extensive mutual homologous recombination occurs among them. Two lineages (MahEastAsia1 and MahEastAsia2) were predominant in the Japanese isolates. We identified alleles unique to these two East Asian lineages in the loci responsible for trehalose biosynthesis (treS and mak) and in one mammalian cell entry operon, which presumably originated from as yet undiscovered mycobacterial lineages. Several genes and alleles unique to East Asian strains were located in the fragments introduced via recombination between East Asian lineages, suggesting implication of recombination in local adaptation. These patterns of MAH genomes are consistent with the signature of distribution conjugative transfer, a mode of sexual reproduction reported for other mycobacterial species.
Keywords: NTM pulmonary disease, homologous recombination, pan genome, Mycobacterium intracellulare, genetic population structure, DCT
Introduction
In many industrialized countries, the societal impact of mycobacterial infections recently underwent a change due to a decrease in tuberculosis (TB) and a simultaneous increase in diseases caused by nontuberculous mycobacteria (NTM) (Thomson 2010; Prevots and Marras 2015). The incidence of NTM lung diseases has been increasing worldwide over the past few decades (Prevots and Marras 2015), causing an increase in the public awareness of the pathogen as a global health threat. In fact, it was reported that NTM diseases would lead to a greater disease burden than TB in the US, Canada, Japan, Korea, Australia, and United Kingdom (Thomson 2010; Adjemian etal. 2012; Koh etal. 2013; Namkoong etal. 2016; Shah etal. 2016). The annual incidence rates of NTM diseases in England, Wales, and Northern Ireland increased from 5.6/100,000 in 2007 to 7.6/100,000 in 2012 (Shah etal. 2016), whereas in Japan it increased from 5.6/100,000 in 2007 to 14.7/100,000 in 2015 (Namkoong etal. 2016). Furthermore, NTM diseases are likely underdiagnosed and underreported in developing countries, where TB and AIDS are necessarily the major focus of the healthcare system (Gopinath and Singh 2010).
NTM are a diverse group of microorganisms with over 160 different species belonging to the Mycobacterium genus (http://www.bacterio.net). The Mycobacterium avium complex (MAC), consisting of M. avium, M. intracellulare, and several other rarer species, is the most common NTM species worldwide (Simons etal. 2011; Hoefsloot etal. 2013; Prevots and Marras 2015). The MAC lung disease is a treatment-resistant, chronic disease (Maekura etal. 2005; Griffith 2007; Xu etal. 2014), and was initially thought to be an opportunistic infectious disease, particularly in patients with AIDS. However, recent studies have reported MAC infection in patients with no apparent risk factors (Turenne etal. 2007; Inagaki etal. 2009). In addition, it can cause progressive lung disease, leading to respiratory failure and even death, in elderly patients with no history of lung disease or immunodeficiency (Field etal. 2004; Maekura etal. 2005; Griffith 2007).
The main pathogenic agent of the MAC lung disease is M. avium, a slow-growing mycobacterium found in various environments such as water, soil, and even as part of biofilms on showerheads and bathtub inlets (Nishiuchi etal. 2007; Lahiri etal. 2014a; Falkinham 2015). Mycobacterium avium currently comprises four subspecies, M. avium subspecies avium (MAA), M. avium subspecies silvaticum (MAS), M. avium subspecies paratuberculosis (MAP), and M. avium subspecies hominissuis (MAH), each named based on the main host species (Turenne etal. 2006; Rindi and Garzelli 2014). Among these, MAH induces MAC lung disease in humans. It also causes lymphadenitis in pigs, and can thus severely affect the meat industry (Álvarez etal. 2011). Thus, understanding the source of infection, transmission route, survival strategy, and genetic diversity of MAH is essential to maintain public health and agricultural productivity.
So far, a number of MAC isolates have been classified into MAH based on identified DNA sequences of several conserved genes (Turenne etal. 2006, 2008; Radomski etal. 2010). In addition, multiple-locus variable-number tandem repeat analysis (MLVA) has been widely used to infer genetic relatedness within MAH populations in order to identify the source of infection (Inagaki etal. 2009; Iwamoto etal. 2012). Nevertheless, it is unclear whether these conserved gene sequences accurately reflect the whole genome-scale genetic relatedness in the MAH population. Furthermore, due to the lack of population-wide genomic data, it is difficult to elucidate the unique adaptive strategy, or to identify potential clinical and epidemiological markers. To overcome this limitation, it was necessary to substantially add to the whole genome information available.
To elucidate the adaptation mechanisms of MAH, we conducted comprehensive population genomics analyses on the genomic sequences of 36 isolates, including 12 Japanese isolates sequenced in this study. In addition, MLVA data from 692 isolates were used to infer the genetic population structure of MAH and the worldwide distribution pattern of the population groups. We detected genome-wide occurrence of recombination between the different MAH lineages. We observed allelic variations in the loci associated with cell surface structure and the allele types unique to East Asian populations, and detected their transmission among lineages. Together, these findings suggest that recombination facilitates local adaptation of MAH.
Materials and Methods
Mycobacterial Strains
For genome sequencing, we collected 12 MAH strains isolated from nine immunocompetent patients with pulmonary MAC infection, two pigs with visible tuberculous lymph node lesions, and the bathroom of a healthy volunteer’s home (supplementary data S1, Supplementary Material online). These sources were selected to cover a wide variety of genotypes and isolation sources, and to include previously reported microsatellite polymorphisms (Iwamoto etal. 2012). The strains were cultured on 2% Ogawa egg slants for 3 weeks at 37 °C. Genomic DNA was purified as described (Parish and Stoker 1998). The complete genome sequence of four strains (OCU464, HP17, S2, and P7) and the draft genome sequences of eight other strains were determined (supplementary table S1, Supplementary Material online).
Determination of Genome Sequences
Draft genome sequences for eight MAH strains were determined by shotgun sequencing, using Illumina technology (GAIIx, Miseq, and Hiseq systems) for multiplexed paired-end libraries. The reads were trimmed and filtered using Trimmomatic ver. 0.20 software (Bolger etal. 2014) and contigs were assembled using Velvet ver. 1.2.03 software (Zerbino and Birney 2008) after optimizing the k-mer value from 17–199 depending on the read length. Complete genome sequences were determined using the PacBio RSII platform (Pacific Biosciences, Menlo Park, CA), a 20-kb insert size library, and P4C2 chemistry. De novo assembly was conducted using HGAP ver. 2.0 and Sprai ver. 0.9.5.1.3. software for strain OCU464, or Canu 1.3 and Falcon 0.6.4 software for strains S2, P7, and HP17. The genomes were annotated using the Rapid Annotations Subsystem Technology pipeline (Aziz etal. 2008).
MLVA Data Linked with Isolation Source Information
Epidemiological analyses of Mycobacteria routinely include a microsatellite copy number analysis (MLVA) to investigate the genetic relatedness of clinical and environmental isolates (Supply etal. 2006; Iakhiaeva etal. 2016). In the present study, we first retrieved 14-locus MLVA data for 560 MAH isolates from three previous studies (Iwamoto etal. 2012, 2014; Ichikawa etal. 2015). Next, we generated novel MLVA data for 135 MAH isolates using a previously established experimental method (Iwamoto etal. 2012) or whole genome sequence data. A part of the MLVA data (i.e. comprising 11 of the 14 loci) for 90 MAH isolates from Russia were published previously (Starkova etal. 2014). In summary, we collected 14-locus MLVA data for 692 MAH isolates from six countries: 346 from Japan, 175 from Korea, 43 from USA, 90 from Russia, 27 from the Netherlands, and 11 from Germany. Of these, 614 MAH strains were from humans, 75 from pigs, 2 from animals other than pigs, and 1 from a bathroom. MLVA data profiles and source details are summarized in supplementary data S2 in Supplementary Material online. Of these 692 isolates, 25 isolates were included in the core-genome SNP-based population structure analysis.
Basic Data Analysis
We used EMBOSS or the Bioconductor package Biostrings during sequence handling, and NCBI BLAST+ to conduct homology searches (Rice etal. 2000; Camacho etal. 2009; Pagès etal. 2016). Unless otherwise specified, all statistical analysis, distance calculations, hierarchical clustering, model fitting, and data visualizations were performed on R (R Core Team 2016).
Genome data were retrieved from the PATRIC database (Wattam etal. 2017) for MAH and MAP, and from NCBI RefSeq for MTB.
Both the average pairwise nucleotide diversity (pi), and Tajima D statistic were calculated for nonoverlapping 10-kb sliding window of core genome per species/subspecies (MAH, n= 36; MAP, n = 30; MTB, n = 35) using the R package PopGenome (Pfeifer etal. 2014). Genome alignments were generated using the Parsnp program with –c option (Treangen etal. 2014) for the listed MAH strains (supplementary data S1, Supplementary Material online). The sites containing gaps and N were removed.
Gene Content Analysis
The Pan and core genomes were defined using Roary software (Page etal. 2015). Complete and draft genome sequences (supplementary data S1, Supplementary Material online) were reannotated to generate gff3 files using PROKKA ver. 1.1.12 software (Seemann 2014). Homologous proteins (i.e. protein families) were clustered using the CD-Hit and MCL algorithms. The BLASTp cut-off value was set at 95%. The number of core- and pan-genome protein families were estimated via genome sampling up to the number of input genomes at the default setting in Roary. Genes overrepresented in East Asian lineages are screened based on p.value of one-tailed Fisher’s exact test. Chromosomal locations of those genes were manually inspected as to whether they were located in the recombination tracts predicted by fastGEAR (see below). Pairwise BLASTn analysis for chromosomal regions containing East Asian alleles was performed using GenomeMatcher software (Ohtsubo etal. 2008).
Population Structure and Recombination Analysis
Population structure was inferred using BAPS6 software, assuming that MAH is a recombinogenic bacterium. For this analysis, core genome SNPs were first detected by Parsnp genome aligner software using the complete genome of TH135 as a reference (Uchiya etal. 2013). SNPs on locally collinear blocks (LCB) <200 bp, poor alignment regions identified by the Harvesttools software (Treangen etal. 2014), and sites containing gap and N were removed using a custom R script. In the BAPS, iteration in sampling steps was set to 100. The optimal number of population (K) was estimated to be five based on the likelihood scores for a 36-strain core-genome data set. When the BAPS analysis was applied to the 14-loci MLVA data (supplementary data S2, Supplementary Material online), the upper limit of K was manually set to six, such that members in a single population group defined using core-genome SNP data were allocated into 1–2 result population group.
Chromosome recombination was inferred using fastGEAR software (Mostowy etal. 2017). For each genome, the filtered polymorphic sites used for BAPS analysis were combined with intervening reference genome sequences, such that the SNP position was matched to the reference genome position. The resulting string was combined as a multi-FASTA file, and used as the input for fastGEAR. The iteration number was set to 15 (default). fastGEAR infers two types of recombination (Mostowy etal. 2017). “Recent recombination” refers to interlineage recombination for which that donor-recipient relation can be inferred, whereas “ancestral recombination” refers to the recombination for which donor-recipient relation cannot be inferred due to high conservation level of SNP tract in two lineages. Recent recombinations detected with a Bayesian factor (BF) > 1 and ancestral recombinations with a BF >10 were taken into account. The relationships between strains were represented using a PSA tree (Mostowy etal. 2017) generated by hierarchical clustering for the distance matrix, where distance is defined as of 1−PSA (the proportion of the genome fragment length for which two strains share ancestry). Both ancestral and recent recombination were taken into account during construction of the PSA tree. In our data set, members of each of the five lineages did not change before and after construction of the final PSA tree. We conducted the same analysis for the three genome alignments generated using the different reference genomes (Mah104, TH135, HP17) to validate the median import length (supplementary fig. S2, Supplementary Material online), lineage member, and the presence of gene flux between lineages.
Recombination hot regions were inferred using OrderedPainting software (Yahara etal. 2014). Filtered polymorphic sites were used for this analysis. Recombination intensity was represented as a recombination hotness index Hi (also referred to as the realized recombination rate) (Yahara etal. 2016). Sites with Hi > 3.0 (top 0.1%) were discussed. Genes in the recombination hot regions are listed in supplementary table S2 in Supplementary Material online.
Phylogenetic Analysis
The phylogenetic network was generated using SplitTree 4 software (Huson and Bryant 2006) with filtered polymorphic sites (see above). The gene tree was generated using PhyML software (Guindon etal. 2010) after identifying an optimal substitution model using ModelGenerator software (Keane etal. 2006). The amino acid sequence alignment was generated using MAFFT software (Katoh and Standley 2016). Trees were visualized using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/).
Nucleotide Sequence Accession Numbers
Complete genome sequences were submitted under the following DDBJ/EMBL/GenBank accession numbers: strain OCU464 chromosome, CP009360; OCU464 plasmid p78Kb, CP009405; OCU464 plasmid p18Kb, CP009406; strain P7 chromosome, CP018020; strain S2 chromosome, CP018014; strain HP17 chromosome, CP016818. Whole Genome Shotgun project for strains OCU462, OCU404, OCU466, OCU556, OCU491, CAM57, CAM177, and CAM78 were deposited under the accession numbers MBFQ00000000, MKCW00000000, LBGZ00000000, MKCX00000000, MKCY00000000, MBFN00000000, MBFO00000000, and MBFP00000000, respectively.
Results
Basic Genomic Features of MAH
High Genetic Diversity in MAH
Prior to the present study, only one Asian-isolate MAH genome was available in the Genbank database. To overcome this geographical bias of present genomic sequence data, we determined eight draft and four complete genome sequences of Japanese MAH strains (supplementary table S1, Supplementary Material online), selected to cover a wide variety of microsatellite polymorphisms revealed by our previous molecular epidemiological study (Iwamoto etal. 2012). By combining these 12 with the 24 previously determined genome sequences in the PATRIC database, we generated a genetically diverse MAH database consisting of 36 strains (supplementary data S1, Supplementary Material online). Since the present study represents the first population-wide MAH genome analysis, we compared the basic MAH genomic features with those of more host-specialized Mycobacteria, including cattle- or sheep-adapted MAP, and human-adapted M. tuberculosis (MTB). The mean chromosome size of the whole (completely sequenced) MAH genome was 5.0 Mb (fig. 1A), larger than the sizes of either MAP (4.8 Mb) or MTB (4.4 Mb). The GC content of MAH was 69.0%, which was higher than that of MTB (65.0%), and slightly lower than that of MAP (69.2%). The mean sequence diversity (pi) of the genome alignment was 20-fold greater in MAH (0.00562) compared with MAP (0.00028) or MTB (0.00027). Tajima’s D statistics for MAH comprised positive values (median 0.65), whereas those for MAP and MTB comprised negative values, (median −0.56 and −1.87, respectively). The data suggest that evolution of the MAH genome has been influenced predominantly by balancing selection, whereas the MTB and MAP genomes have predominantly been subject to purifying selection. Together, the basic MAH genome features that were identified clearly suggest that MAH is a genetically diverse mycobacterial subspecies, as compared with more host-specialized mycobacterial groups, such as MAH and MTB.
Fig. 1.

—Basic genomic features of MAH. (A) Chromosome size, G+C content, average sequence diversity (pi), and Tajima D statistics of MTB, MAP, and MAH. N=35 (MTB), 4 (MAP), and 8 (MAH) for chromosome size. N=35 (MTB), 30 (MAP), and 36 (MAH) for all other categories. Pi and Tajima D statistics were calculated for 10-kb nonoverlapping sliding windows of the genome alignments. Chromosome size was shown only for completely sequenced strains. Except for chromosome size, the statistical significance between MAH and MTB or MAP was analyzed via a two-sided Wilcoxon rank sum test: *P=0.00404; **P=0.00025; ***P<0.00001. (B) Comparison of pan genome size among the three Mycobacterium groups. Protein families were identified using Roary software (Page etal. 2015). γ denotes the parameter estimate obtained by data fitting to a power model n=κ×N^γ, where n is the number of protein families, N is the number of sampled genomes, and κ and γ are coefficients.
MAH Acquires Genes More Frequently Than Either MAP or MTB
Previous reports suggest that MAH horizontally acquires genes as genomic islands (Lahiri etal. 2014b; Sanchini etal. 2016). To address the extent of new gene acquisition of MAH, we conducted pan genome analysis (Medini etal. 2005; Mira etal. 2010). This method has been used to infer the species capacity to acquire gene from outside the defined species or lineages by mathematical extrapolation. We estimated the number of MAH pan genomic genes compared with those of MAP and MTB. Sampling of 30 genomes revealed the mean number of homologous protein families in the core genome to be 3,344 (± 21) for MTB, 2,130 (± 0) for MAP, and 1,988 (± 35) for MAH. The mean number of protein families in the pan genome was 5,323 (± 45) for MTB, 10,450 (± 0) for MAP, and 16,227 (± 262) for MAH (fig. 1B). Together, these data indicate that comparatively, the MAH gene repertoire is larger than that of either MTB or MAP. The rate of new gene discovery is expected to be the highest in MAH among the three species groups according to the gamma coefficient obtained by data fitting to a power model (fig. 1B). Thus, MAH acquires genes more frequently than either MAP or MTB.
Genetic Population Structure of MAH
To evaluate potential genetic linkage of MAH strains with their geographic distribution, we first constructed a phylogenetic network of the 36 MAH strains based on the similarity in 62,210 polymorphic sites in the core genome. This resulting network was very complex, suggesting that recombination may frequently occur between different lineages (fig. 2A). Therefore, we inferred genetic population structure using Bayesian population structure analysis software (BAPS), assuming that MAH is a sexual organism (Corander and Marttinen 2006). BAPS partitions the entire population into smaller populations that are equivalent to random mating units. Five major populations were detected by BAPS mixture analysis (fig. 2B). Japanese human isolates were assigned to two populations, designated MahEastAsia1 and MahEastAsia2 (since the origin of most strains in these populations was Japan). This assignment was supported by the high genetic relatedness observed between Japanese and Korean isolates at the MLVA level (see the analysis below). Notably, strain 5581, which was assigned to MahEastAsia2, was isolated in USA; however, the host of the strain isolated was an Asian elephant. The USA and European isolates, as well as the Japanese Animal isolates, were assigned to one of three other populations, designated sequence cluster 1 (SC1), SC2, and SC3. Strains DT78, CAM78, 2495, and 3388 were inferred to be admixed strains. Except for MahEastAsia1, each genetic population group contained both human and nonhuman isolates. This observation suggests that most MAH genetic population groups behave as zoonotic, broad host–range pathogens, and that MahEastAsia1, consisting of only Japanese human isolates, is specialized to infect only human hosts. We tested this hypothesis by extending the genetic population structure analysis to include a MLVA (i.e. a type of MLST) data set that comprised a much larger set of population data.
Fig. 2.

—Population structure of MAH. (A) Phylogenetic network of MAH isolates based on core genome SNPs at 62,210 polymorphic sites. The network was generated via the NeigborNet method and p-distance in Split tree 4 software. The geographic locations of isolates are indicated by the color of the dot of the leaf. Isolation sources are indicated by illustration of the strain name. (B) Population structure and admixed strains inferred by BAPS. Polymorphic sites used are the same as those used in (A). Colors indicate population groups defined according to BAPS mixture analysis results.
To evaluate the worldwide distribution pattern of the MAH genetic population groups, the BAPS analysis was performed using a 14-loci MLVA data set containing information for 692 isolates from six different countries (supplementary data S2, Supplementary Material online). This MLVA data was obtained by combining clinical isolate data obtained during the present study with previously published data, and with sequence information used in the core genome-based analysis (again, generated during the present study). To determine population partitions, the upper limit of population (K) was manually set to six, such that the members in a single population group defined using core-genome SNP data were allocated into 1–2 resultant populations. 692 isolates were derived from one of seven isolation origins: “European human,” “Japanese human,” “Japanese pig,” “Korean human,” “USA human,” “USA animal,” or “Japanese environment.” The Japanese human and Korean human categories consisted predominantly of two population groups that contain strains assigned into MahEastAsia1 or MahEastAsia2 in core genome SNP-based population structure analysis (supplementary fig. S2, Supplementary Material online). Three other populations detected in the core genome SNP-based analysis were found in various proportions in the European human, USA human, and Japanese pig categories. Genetic population groups equivalent to SC2 (Pop2 and Pop3 in supplementary fig. S2, Supplementary Material online) were predominantly categorized as European human and Japanese pig in origin. The Japanese pig category did not contain genetic population groups equivalent to either MahEastAsia1 or MahEastAsia2. Despite the presence of the SC2 population in Japan, strains designated as MahEastAsia1 and MahEastAsia2 by BAPS together comprised 93.3% (252/270) of Japanese human isolates. These distribution patterns of the various genetic population groups suggest that, firstly, MAH transmission from pigs to humans in Japan is very rare. Secondly, MahEastAsia1 and MahEastAsia2 are more specialized for infection of Asian humans than that by the various other genetic population groups.
Extensive interlineage recombination in M. avium
To investigate potential genetic flux between MAH populations, we analyzed the MAH genomes using recently developed fastGEAR software to infer imported genomic fragments and their origins (Mostowy etal. 2017). fastGEAR infers genome lineage using Hidden Markov Model approach based on clustering pattern given by BAPS, and then infers the coordinates of imported genomic fragments for each genome and their donor lineage. Recently imported fragment, (i.e. those not shared between two lineages) and their predicted donor lineage were visualized in figure 3. Ancestral recombination was visualized in supplementary fig. S2 panel A in Supplementary Material online. The results of this analysis showed the imported genomic fragments to be distributed throughout the MAH genome, and the median import length to be 4,724 bp. This held true even when an alternative reference genome was used to detect core-genome single nucleotide polymorphisms (SNP) (supplementary fig. S2, panel B, Supplementary Material online). The genealogical relationships between the various isolates were represented using a proportion of shared ancestry (PSA) tree, in which the pairwise distance was defined as 1−the proportion of the total fragment length that exhibited shared ancestry (Mostowy etal. 2017). Five major lineages in the PSA tree were equivalent to the five genetic population groups detected by BAPS (fig. 1B); therefore, we used the same names (MahEastAsia1, MahEastAsia2, SC1, SC2, and SC3) to refer to each lineage. As suggested by the phylogenetic network, strains CAM78 and DT78 received a large proportion of their genome from the MahEastAsia2 and SC2 lineages, respectively. Furthermore, gene flux from the MahEastAsia1 population to USA isolate 2495 (SC3) and USA isolate 3388 (SC2) became evident (fig. 3). When SC1 (comprising only two members) was omitted, genetic flux was observed between any pair of the four lineages. MahEastAsia1 predominantly received imports from MahEastAsia2, and vice versa.
Fig. 3.

—Genome-wide occurrence of interlineage recombinations. Left: Hierarchical clustering showing a proportion of shared ancestry (PSA) tree. Clustering was achieved using the complete linkage method. Middle: Visualization of recent genomic imports. Right: Proportion of recent genomic import. Colors indicates donor lineage (population group) of the recent genomic imports. Gray color indicates imports from sources external to the five population lineages. MAH lineage and recombination were inferred using fastGEAR software (Mostowy etal. 2017).
Even within the same lineage, there were strains comprising a relatively large proportion (∼47%) of recently imported fragments, and strains that comprised a very limited proportion (∼5%) of imported fragments. For example, in the SC2 lineage, MAH strain 3388 comprised 42.7% imported fragments, whereas strain 104 comprised only 4.8% imported fragments. A similar phenomenon was observed in MahEastAsia2, were imported fragments represented 21.7% of the genome of strain TH135, but conversely represented 3.7% and 1.2% of the genome of strains P7 and S2, respectively. Strains exhibiting a low proportion of imports were all human isolates (fig. 3, right). Together, these results suggest ecological segregation of the MAH sublineages, and/or the presence of a genetic barrier to horizontal gene transfer. The proportion of imported fragments was generally largest in the MahEastAsia1 and SC3 populations (median proportions 28.0% and 25.2%, respectively). These observed trends held true when the number of polymorphic sites within the imported genomic fragments was calculated (supplementary fig. S2, panel C, Supplementary Material online).
Genome mosaicism can be generated through natural transformation, generalized transduction, or conjugation. Mycobacterium smegmatis and Mycobacterium canetti are known to be capable of conducting intraspecies chromosome exchange via conjugation, which generates progenies with mosaic genomes, each carrying up to >200 kb long multiple imported fragments distributed over the genome. The specific mechanism underlying this phenomenon was termed “distributive conjugative transfer” (DCT) (Derbyshire and Gray 2014; Mortimer and Pepperell 2014; Boritsch etal. 2016; Gray etal. 2016). In genus Mycobacterium, the occurrence of natural transformation has not been reported. Generalized transduction depends on the presence of bacteriophage, for which the size of transferable DNA is limited. We observed large import size (4.7 kb in median, 157 kb in maximum) and genome-wide distribution of the imports (fig. 3). This pattern is most consistent with DCT among the three possible gene transfer mechanisms.
Genes at Recombination Hot Region
To gain insight into the phenotypic traits most frequently affected by recombination in the core genome, we inferred recombination hot regions using OrderedPainting (Yahara etal. 2014). This method ranks polymorphic sites according to the recombination intensity normalized to the genome average. Recombination intensity reflects both mechanistic bias for DNA recombination and natural selection for the alleles. The recombination hot regions often contain genes linked to cell surface structure (Yahara etal. 2016). Top 0.1% of high intensity sites in MAH core genome spanned four genomic regions (fig. 4). These regions included genes encoding mammalian cell entry (MCE) proteins of MCE operon (mceC, mceD) at locus 1 (see the following sections for locus definition), trehalose phosphate synthase (otsA), transcription-repair coupling factor (mfd), anti-anti-sigma phosphoserine phosphatase (rsbP), and acyl-CoA dehydrogenase (fadE). Except for fadE, multiple nonsynonymous substitutions were observed in each of these genes. Both the MCE operon and otsA encode proteins predicted to influence cell surface structure (Zhang and Xie 2011; Nobre etal. 2014).
Fig. 4.

—Recombination intensity in the MAH genome. Recombination hotness index at genome position i (Hi) was inferred using OrderedPainting software (Yahara etal. 2014). Genes harboring sites with a top 0.1% Hi values are highlighted. MCE locus 1 denotes mammalian cell entry protein operon locus 1.
Emergence of East Asia-Type Alleles and Their Introgressions
High new gene discovery rate in the MAH pan genome and extensive interlineage recombination suggests that gene acquisition from the mycobacterial pan genome and its local transmission via mutual homologous recombination is a mechanism to facilitate MAH evolution. To test this model, we investigated whether genes representing East Asian lineages are embedded in the fragments introduced via ancestral or recent recombinations. This revealed, for example, host infection-related proteins (MCE proteins), proteins consisting of Type I restriction and modification systems, and maltokinase as the proteins that represent East Asian populations (fig. 5). Among these protein families, seven (rsmD and six hypothetical proteins in fig. 5) were encoded in the fragment introduced via ancestral recombinations, and 26 were in the fragment imported via recent recombination between MahEastAsia1 and MahEastAsia2 (fig. 5). Thus, new gene acquisition and its transmission in East Asia had occurred at least for several genomic loci in evolutionary history.
Fig. 5.

—Protein families overrepresented in two East Asian lineages. Colored cells indicate the presence of a given protein family. The dendrogram indicates hierarchical clustering of protein families or strains, based on the Jaccard distance and complete linkage method. Protein families exhibiting a P value <0.005 in one-tailed Fisher’s exact test were used for clustering. Proteins encoded near the MCE operon locus 3 are indicated by the vertical blue line. Cells of coding sequence embedded in the fragments imported via recombination were boxed by solid line (recent recombination) or broken line (ancestral recombination) in magenta. Protein families without function annotation are all hypothetical proteins. Numbers in maltokinase indicate allele subtype: 1, EA1; 2, EA2. Protein names in orange are discussed in main text.
Inspection of the coding regions revealed that some of these East Asia-specific protein families were produced from allelic variants of the major alleles from other lineages. One chromosomal locus with allelic variation was the trehalose synthase (TreS)-maltokinase (Mak, also referred to as Pep2) operon (fig. 6A), which was previously discovered to influence alpha-glucan synthesis in MTB studies (Roy etal. 2013; Koliwer-Brandl etal. 2016). Another such locus was the MCE protein operon (tentatively named MCE operon locus 3). To obtain additional insights into recombination patterns around these East Asian alleles and their origins, we conducted comparative analysis using representative genomes below.
Fig. 6.

—Occurrence of East Asia-type alleles in the treharose synthase operon. (A) Nucleotide similarity around treS-mak locus among representative strains from MahEastAsia1, MahEastAsia2 and SC2, SC3. Similarity was visualized using GenomeMatcher software (Ohtsubo etal. 2008). TH135 coordinate 4,750,441–4,823,085 and its equivalent region of five other strains are shown. Rectangles indicate coding sequence. The 5.1-kb differentiated region in OCU464 is indicated by a white line, whereas the 53.8-kb differentiated region in TH135, OCU404, and 2344 was indicated by distinct filling pattern of coding sequence. (B) Maximum likelihood tree of Mak (left), TreS (middle). Protein sequence was aligned using MAFFT software. The substitution model used was JTT+G for TreS and Mak. Values on the nodes denote bootstrapping values obtained from 100 runs. The color of strain names indicates lineage. (C) One parsimonious explanation for the transmission history of the East Asia-type alleles. The dendrogram indicates PSA tree. Arrow indicates the direction of introgression of the alleles. The arrow color indicates the donor lineage of the allele.
treS-maK Operon
The trehalose synthase operon of East Asian strains showed reduced sequence identity with the corresponding region of other lineages compared with neighboring regions (see supplementary fig. S3, Supplementary Material online, for alignments). Strains in MahEastAsia2 lineage carry about a 53.8-kb segment that shows a highly differentiated sequence compared with four MahEastAsia1 strains (e.g. OCU464) and SC2 strains (broken lines in coding regions of TH135 map in fig. 6A). This region contains the treS-mak operon. The equivalent regions in a MahEastAsia1 strain OCU464 are mostly identical to the equivalent regions of other MAH lineages, however, they harbor about a 5.1-kb differentiated region just containing the treS-mak operon (white line below OCU464 map in fig. 6A). MahEastAsia1 strain OCU404 carries the 53.8-kb MahEastAsia2-type segment, but its 5.8-kb region containing the treS-mak operon was identical to the MahEastAsia1-type sequence (red line over OCU404 map in fig. 6A). Recombinations implicated in generating these SNPs patterns could not be detected by systematic recombination inference by fastGEAR due to poor alignment for 36 strains.
Mak alleles in most MAH lineages were indistinguishable from MAP, MAA, and MAS Mak alleles at the amino-acid sequence level, thus we designated these as “M. avium-type alleles” (fig. 6B). The Mak and TreS alleles of MahEastAsia1 are different from those in MahEastAsia2 at both nucleotide and protein sequence levels (supplementary fig. S3, Supplementary Material online). We designated these alleles as East Asia-type alleles, “EA1 subtype,” and “EA2 subtype,” respectively (fig. 6B). At present, we could not find close relatives of East Asia-type alleles in the NCBI database except for the M. avium-type alleles. Thus, one parsimonious explanation for treS-mak allele transmission was as follows. First, the treS-mak operon was imported from an undiscovered mycobacterial lineage into two East Asian lineages (gray arrows in fig. 6C). Then, about a 53.8-kb MahEastAsia2-type segment was transferred into a sublineage in MahEastAsia1 (blue allow in fig. 6C). Finally, a part of the segment in the OCU404 ancestor was replaced with MahEastAsia1-derived segment (red arrow in fig. 6C). MahEastAsia1-type SNP tract was present in the SC3 strain 2495 (supplementary fig. S3, Supplementary Material online), suggesting another transmission through recombination. The Mak protein of OCU464 (EA1 subtype) shared 77% sequence identity with the protein encoded by the M. avium-type allele. The same pattern was observed for both TreS and the membrane protein encoded nearby, which were found to share 95%, and 76% sequence identity, respectively, with the protein encoded by the M. avium-type allele. These similarities corresponded to a divergence of 55 (Mak), 25 (TreS), and 31 (membrane protein) amino acids, respectively, with the product encoded by the M. avium-type allele.
MCE Operon Locus 3
The MCE operon consists of two genes encoding an ABC transporter permease domain (yrbEA yrbEB) and six genes encoding a membrane protein that contains the Mce domain (mceA-F). MCE operons in mycobacteria were deduced to play roles in interaction with eukaryotic cells (Zhang and Xie 2011). In the MAH genome we detected eight MCE operons, and tentatively designated those as locus 1 to locus 8. Locus 2, locus 6, and locus 7 are orthologous to Mce4, Mce2, and Mce1 of the MCE operon family in MTB, respectively, according to synteny analysis (supplementary fig. S4A, Supplementary Material online). The gene content analysis revealed 19 protein families that were common to MahEastAsia2 strains, as well as to strain OCU464 of MahEastAsia1 (vertical blue line in fig. 5), and were all included in a recently imported segment in OCU464 (TH135 coordinate 776,275 to 826,427 according to recombination inference). This segment contained one MCE operon named MCE operon locus 3 (supplementary fig. S4B, Supplementary Material online). The phylogenetic tree for MCE operon locus 3 in the import region suggests that the East Asian alleles diverged from the majority of the other M. avium-type alleles prior to the divergence of M. avium from M. intracellurae when M. kansassi was used as an outgroup (supplementary fig. S4C, Supplementary Material online). Thus, it is likely that MCE operon locus 3 was acquired by MahEastAsia2 from as yet undiscovered mycobacterial lineages, and then introgressed into a sublineage in MahEastAsia1.
Discussion
Genomic epidemiological studies have been previously used to elucidate the evolutionary background of pathogenic M. tuberculosis (Wlodarska etal. 2015; Stucki etal. 2016). Conversely, MAH is an emerging pathogen for which population-wide genome data is largely unavailable, restricting studies on its sources of infection, transmission route, and adaptive strategies. In the present study, we revealed, for the first time, the genetic linkage between global MAH strains based on genome scale data, and thus identified five major MAH lineages, including two that were specifically distributed in East Asia. A comprehensive population genomic analysis revealed that M. avium is a recombinogenic bacterium with a capacity to acquire new genes in the subspecies pan genome. Furthermore, we identified several MAH chromosome loci with allelic variations that may be potentially useful as clinical and/or epidemiological markers. Taken together, these results emphasize the importance of recombination in MAH local adaptation.
M. avium is ubiquitous in the environment and it causes infectious disease in both humans and animals. In particular, MAH is a major cause of antibiotic-resistant lung disease (Prevots and Marras 2015). Since the incidence of MAC lung disease is increasing worldwide, we postulated the emergence of human-adapted MAH lineages on both local and global scales. We found all Japanese human MAH isolates to fall within the MahEastAsia1 or MahEastAsia2 population groups (fig. 2A), and to be genetically distinct from SC2 Japanese pig isolates (figs. 2 and 3; supplementary fig. S2, Supplementary Material online). These results indicated repeated infection of East Asian human populations by MAH strains of the two lineages. Given that the incidence rate of NTM pulmonary disease in Japan is currently the highest for this disease worldwide (Namkoong etal. 2016), we hypothesize that MahEastAasia1 and MahEastAsia2 are likely carriers for adaptive alleles enabling infection of East Asian humans. We also found that the two strains isolated from a household (i.e. a bathroom for OCU462 in MahEastAsia2 and a vacuum cleaner for 27-1 in SC2) showed high similarity to human isolates at the core-genome level. This suggests that the living environment can be a reservoir of MAH clones capable of infecting humans (Nishiuchi etal. 2009; Iwamoto etal. 2012; Lahiri etal. 2014a; Iakhiaeva etal. 2016).
By investigating genetic variations of two East Asian lineages in detail, we observed a pattern of allelic transmission in MAH populations: acquisition of new alleles from as yet undiscovered mycobacterium lineage(s), and their transmission via recombination in population sharing geographic locations (fig. 6 and supplementary fig. S4, Supplementary Material online). We found the treS-mak operon to be a locus harboring East Asia-type alleles that predominated among two East Asia lineages (fig. 6) and otsA to be a recombination hot region (fig. 4). Another trehalose biosysthesis gene treT was included in a segment exchanged in ancestral recombination between MahEastAsia1 and MahEastAsia2 (data not shown). Trehalose is a component of both a glycolipid in the outer membrane and the alpha-glucan of capsules (Sambou etal. 2008; Nobre etal. 2014; Thanna and Sucheck 2016). Given that a large degree of amino-acid sequence dissimilarity was identified in the expected protein sequences (TreS, Mak) of East Asia-type alleles and the M. avium-type alleles, the biochemical nature of these proteins may vary between East Asian and other MAH populations. Genetic variations in the three glycosyltransferase genes (otsA, treS, mak), which could influence cell surface characteristics, may be considered to be candidates for clinical markers of M. avium.
Similarly, we identified MCE operon locus 1 as a recombination hot region, and MCE operon locus 3 as a locus harboring East Asia-type alleles. For MCE operon locus 3, we observed its transmission from MahEastAsia2 to MahEastAsia1 (supplementary fig. S4, Supplementary Material online). The MCE operon encodes cell surface proteins whose exact function is not well elucidated to date except for one orthologous family, mce1, for which its implication in reimport of mycolic acid of MTB has been proposed (Forrellad etal. 2014). Nonpathogenic E. coli strains that express mce1 or mce4 operon of M. tuberculosis (locus 7 and locus 2 in MAH) were shown to facilitate the strain’s invasion into mammalian cells (Arruda etal. 1993; Saini etal. 2008). Thus, MCE operons were deduced to play a role in host–bacteria interactions (Zhang and Xie 2011). MAH possesses as many as eight MCE loci, and allelic variants in one locus, as opposed to four loci (mce1 to mce4) in MTB (supplementary fig. S4, Supplementary Material online). The large repertoire of the MCE operon is consistent with a diverse niche (fig. 2) which MAH might encounter over evolutionary time.
DCT was first discovered in rapid-growing avirulent mycobacterium species M. smegmatis by conducting mating experiments and subsequent sequencing of transconjugant genomes (Nguyen etal. 2009; Gray etal. 2013; Boritsch etal. 2016). DCT allows the exchange of a large (33 kb in mean, up to 250 kb) chromosomal segment between donor and recipient cells, as opposed to recombination through natural transformation, which replaces fragments with lengths of a few hundred base pairs (Mortimer and Pepperell 2014). Among slow-growing mycobacterium species, MTB, which has evolved largely through clonal expansions, contains very few imports into the chromosome (Namouchi etal. 2012), whereas M. canetti, a progenitor species of MTB, contains clear recombination tracts in the chromosome (Gutierrez etal. 2005; Supply etal. 2013). DCT was predicted for M. canetti based on recombination inference on genome data of natural isolates (mean import size was 3.7 kb) (Mortimer and Pepperell 2014). Later, its DCT ability was demonstrated experimentally (Boritsch etal. 2016). As to MAC, a recombination tract was reported for an operon responsible for glycopeptidolipid (GPL) biosynthesis pathway in MAH (Krzywinska etal. 2004). As of yet a genome-wide survey of recombination has not been performed. Since the median predicted import size in MAH was 4.7 kb, and the unit of transmission of the treS-mak operon was estimated to be 5.1–5.8 kb (fig. 6A), we speculate that DCT is involved in gene transfer among M. avium. Together, except for MTB, Mycobacterium species sequenced for multiple strains showed a signature of DCT in the chromosome regardless of slow-growing or rapid-growing phenotype. Further functional studies for conjugation ability and the East Asia-type alleles would reveal whether DCT plays a critical role in adaptation of MAH.
Mycobacterium avium has been reported to reside in diverse ecological locations, particularly in close proximity to water and soil (Turenne etal. 2007; Lahiri etal. 2014a; Iakhiaeva etal. 2016). Given the genetic flux between MAH lineages revealed in this study, the ubiquity of M. avium residence no longer simply indicates the robustness of M. avium as an organism, but now supports the hypothesis that M. avium persists in the environment to increase opportunities of importing diverged alleles and new genes (fig. 7). Since human and animal isolates share genomic lineage (e.g., USA isolate 100 and USA isolate 5560 in SC3), we can assume that different animal hosts have acquired MAH from a common environmental pool. This in turn suggests that MAH from various hosts can be rereleased and merged again within the common environmental pool (fig. 7). When M. avium enters animal hosts, it is captured by macrophages, and subsequently segregated in granuloma (Bermudez etal. 1991; Smith etal. 1997). Thus, the environment is the easiest place to acquire genetic variation via recombination. This hypothesis is supported by the observed occurrence of distinct M. avium clones found in the same bathtub water or bathtub inlet (Nishiuchi etal. 2009; Iwamoto etal. 2012). Furthermore, recombination between different mycobacterial species is possible, as suggested by the occurrence of atypical alleles in East Asian strains (fig. 6). The animal infection stage subject genetic variants to natural selection, in that cells that survive the host immune system attack are able to proliferate in host cells, and subsequently rerelease clones to the environment. Furthermore, the variants may undergo microevolution during animal host infection, whereby they accumulate small genetic changes (e.g., point mutations and gene loss). Repetition of this cycle within a discrete geographic location would select adaptive alleles that are optimal for infection of local hosts, thus facilitating local adaption. Recent genomic analyses detected genome mosaicism in another NTM disease agent, Mycobacterium abscessus, which is also a species ubiquitous in water and soil (Brown-Elliott and Wallace 2002; Sapriel etal. 2016). Thus, M. abscessus may show a similar evolutionary pattern.
Fig. 7.
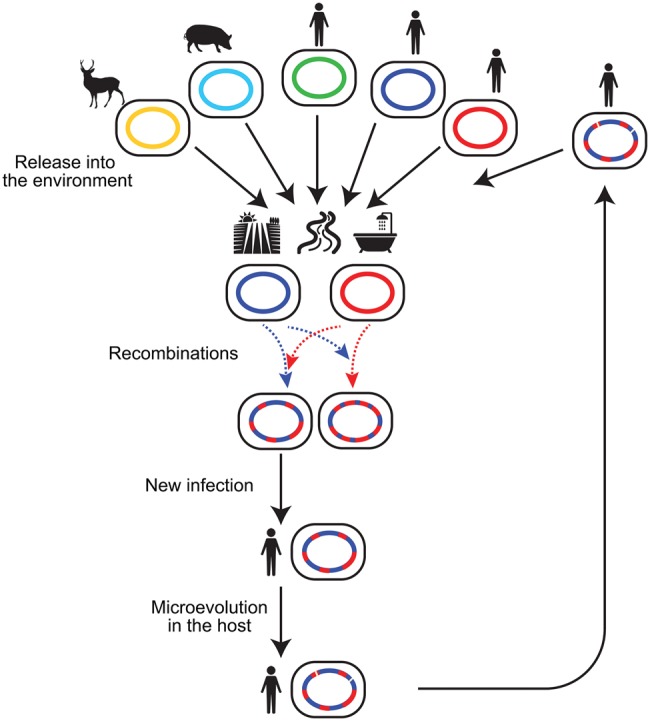
—A model for the flow of recombination-generated genetic variants in nature. Various hosts release MAH into a common environmental pool, where recombination occurs between MAH lineages or between MAH and other Mycobacterium species. This generates genetic variants that are then subject to natural selection. MAH clones with adaptive alleles that ensure survival in animal hosts, are rereleased into the environment, and then donate adaptive alleles to the environmental population. Solid line indicates flow of cells whereas broken line indicates flow of DNA.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Koji Yahara (National Institute for Infectious Diseases), Michael France (University of Idaho), Wen-Tso Liu (University of Illinois at Urbana–Champaign), Sayaka Mino (Hokkaido University), Hiroshi Mori (National Institute of Genetics), and Masataka Tsuda (Tohoku University) for the critical comments on this study. We thank Pekka Marttinen (Helsinki Institute for Information Technology), and Haruo Suzuki (Keio University) for their advice on optimizing the use of fastGEAR, and R, respectively. We also thank Gail E Deckert (Moses Lake Industries) and an anonymous reviewer for useful suggestions for improving this manuscript. This work was supported by the Institute of Medical Science, University of Tokyo, Human Genome Center (HGC) supercomputer system. This work is supported by grants from (1) Japan Society of Promotion of Science (JSPS) KAKENHI [grant number 15K08793 to T.I., 15K18665 to H.Y., 16H05830/16H0550/15K15675 to F.M.]; (2) Japan Agency for Medical Research and Development (AMED) [project number 17fk0108116h0401 to Y.N., T.I., M.A., and F.M.]; (3) Japan Science and Technology Agency (JST) ERATO [Grant Number JPMJER1502 to H.Y.]; (4) MEXT for the Joint Research Program of the Research Center for Zoonosis Control, Hokkaido University to T.I.
Literature Cited
- Adjemian J, et al. 2012. Spatial clusters of nontuberculous mycobacterial lung disease in the United States. Am J Respir Crit Care Med. 186(6):553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez J, et al. 2011. Epidemiological investigation of a Mycobacterium avium subsp. hominissuis outbreak in swine. Epidemiol Infect. 139(1):143–148. [DOI] [PubMed] [Google Scholar]
- Arruda S, Bomfim G, Knights R, Huima-Byron T, Riley LW.. 1993. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science 261(5127):1454–1457. [DOI] [PubMed] [Google Scholar]
- Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez LE, Young LS, Enkel H.. 1991. Interaction of Mycobacterium avium complex with human macrophages: roles of membrane receptors and serum proteins. Infect Immun. 59(5):1697–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boritsch EC, et al. 2016. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci U S A 113(35):9876–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Elliott BA, Wallace RJ.. 2002. Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev. 15(4):716–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Marttinen P.. 2006. Bayesian identification of admixture events using multilocus molecular markers. Mol Ecol. 15(10):2833–2843. [DOI] [PubMed] [Google Scholar]
- Derbyshire KM, Gray TA.. 2014. Distributive conjugal transfer: new insights into horizontal gene transfer and genetic exchange in mycobacteria. Microbiol Spectr. 2(1):MGM2-0022-2013. [DOI] [PubMed] [Google Scholar]
- Falkinham JO. 2015. Environmental sources of nontuberculous mycobacteria. Clin Chest Med. 36(1):35–41. [DOI] [PubMed] [Google Scholar]
- Field SK, Fisher D, Cowie RL.. 2004. Mycobacterium avium complex pulmonary disease in patients without HIV infection. Chest 126(2):566–581. [DOI] [PubMed] [Google Scholar]
- Forrellad MA, et al. 2014. Role of the Mce1 transporter in the lipid homeostasis of Mycobacterium tuberculosis. Tuberculosis (Edinb) 94(2):170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath K, Singh S.. 2010. Non-tuberculous mycobacteria in TB-endemic countries: are we neglecting the danger. PLoS Negl Trop Dis. 4(4):e615.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray TA, et al. 2016. Intercellular communication and conjugation are mediated by ESX secretion systems in mycobacteria. Science 354(6310):347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray TA, Krywy JA, Harold J, Palumbo MJ, Derbyshire KM.. 2013. Distributive conjugal transfer in mycobacteria generates progeny with meiotic-like genome-wide mosaicism, allowing mapping of a mating identity locus. PLoS Biol. 11(7):e1001602.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DE. 2007. Therapy of nontuberculous mycobacterial disease. Curr Opin Infect Dis. 20(2):198–203. [DOI] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Gutierrez MC, et al. 2005. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 1(1):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefsloot W, et al. 2013. The geographic diversity of nontuberculous mycobacteria isolated from pulmonary samples: an NTM-NET collaborative study. Eur Respir J. 42(6):1604–1613. [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 23(2):254–267. [DOI] [PubMed] [Google Scholar]
- Iakhiaeva E, et al. 2016. Variable-number tandem-repeat analysis of respiratory and household water biofilm isolates of “Mycobacterium avium subsp. hominissuis” with establishment of a PCR database. J Clin Microbiol. 54(4):891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa K, et al. 2015. Genetic diversity of clinical Mycobacterium avium subsp. hominissuis and Mycobacterium intracellulare isolates causing pulmonary diseases recovered from different geographical regions. Infect Genet Evol. 36:250–255. [DOI] [PubMed] [Google Scholar]
- Inagaki T, et al. 2009. Comparison of a variable-number tandem-repeat (VNTR) method for typing Mycobacterium avium with mycobacterial interspersed repetitive-unit-VNTR and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol. 47(7):2156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, et al. 2014. Intra-subspecies sequence variability of the MACPPE12 gene in Mycobacterium avium subsp. hominissuis. Infect Genet Evol. 21:479–483. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, et al. 2012. Genetic diversity of Mycobacterium avium subsp. hominissuis strains isolated from humans, pigs, and human living environment. Infect Genet Evol. 12(4):846–852. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2016. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 32(13):1933–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Creevey CJ, Pentony MM, Naughton TJ, Mclnerney JO.. 2006. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 6:29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh WJ, et al. 2013. Increasing recovery of nontuberculous mycobacteria from respiratory specimens over a 10-year period in a tertiary referral hospital in South Korea. Tuberc Respir Dis (Seoul) 75(5):199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliwer-Brandl H, et al. 2016. Metabolic network for the biosynthesis of intra- and extracellular α-glucans required for virulence of Mycobacterium tuberculosis. PLoS Pathog. 12(8):e1005768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinska E, Krzywinski J, Schorey JS.. 2004. Naturally occurring horizontal gene transfer and homologous recombination in Mycobacterium. Microbiology 150(Pt 6):1707–1712. [DOI] [PubMed] [Google Scholar]
- Lahiri A, Kneisel J, Kloster I, Kamal E, Lewin A.. 2014a. Abundance of Mycobacterium avium ssp. hominissuis in soil and dust in Germany – implications for the infection route. Lett Appl Microbiol. 59:65–70. [DOI] [PubMed] [Google Scholar]
- Lahiri A, Sanchini A, Semmler T, Schäfer H, Lewin A.. 2014b. Identification and comparative analysis of a genomic island in Mycobacterium avium subsp. hominissuis. FEBS Lett. 588(21):3906–3911. [DOI] [PubMed] [Google Scholar]
- Maekura R, et al. 2005. Clinical and prognostic importance of serotyping Mycobacterium avium-Mycobacterium intracellulare complex isolates in human immunodeficiency virus-negative patients. J Clin Microbiol. 43(7):3150–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R.. 2005. The microbial pan-genome. Curr Opin Genet Dev. 15(6):589–594. [DOI] [PubMed] [Google Scholar]
- Mira A, Martín-Cuadrado AB, D’Auria G, Rodríguez-Valera F.. 2010. The bacterial pan-genome: a new paradigm in microbiology. Int Microbiol. 13(2):45–57. [DOI] [PubMed] [Google Scholar]
- Mortimer TD, Pepperell CS.. 2014. Genomic signatures of distributive conjugal transfer among mycobacteria. Genome Biol Evol. 6(9):2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy R, et al. 2017. Efficient Inference of recent and ancestral recombination within bacterial populations. Mol Biol Evol. 34(5):1167–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkoong H, et al. 2016. Epidemiology of pulmonary nontuberculous mycobacterial disease, Japan(1). Emerg Infect Dis. 22(6):1116–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namouchi A, Didelot X, Schöck U, Gicquel B, Rocha EP.. 2012. After the bottleneck: genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res. 22(4):721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KT, Piastro K, Derbyshire KM.. 2009. LpqM, a mycobacterial lipoprotein-metalloproteinase, is required for conjugal DNA transfer in Mycobacterium smegmatis. J Bacteriol. 191(8):2721–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiuchi Y, et al. 2007. The recovery of Mycobacterium avium-intracellulare complex (MAC) from the residential bathrooms of patients with pulmonary MAC. Clin Infect Dis. 45(3):347–351. [DOI] [PubMed] [Google Scholar]
- Nishiuchi Y, et al. 2009. Mycobacterium avium complex organisms predominantly colonize in the bathtub inlets of patients’ bathrooms. Jpn J Infect Dis. 62(3):182–186. [PubMed] [Google Scholar]
- Nobre A, Alarico S, Maranha A, Mendes V, Empadinhas N.. 2014. The molecular biology of mycobacterial trehalose in the quest for advanced tuberculosis therapies. Microbiology 160(8):1547–1570. [DOI] [PubMed] [Google Scholar]
- Ohtsubo Y, Ikeda-Ohtsubo W, Nagata Y, Tsuda M.. 2008. GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics 9:376.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, et al. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès H, Aboyoun PG, Gentleman R, DebRoy S.. 2016. Biostrings: string objects representing biological sequences, and matching algorithms. R Package version 2.42.1. https://bioconductor.org/packages/release/bioc/html/Biostrings.html.
- Parish T, Stoker NG.. 1998. Electroporation of mycobacteria. Methods Mol Biol. 101:129–144. [DOI] [PubMed] [Google Scholar]
- Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ.. 2014. PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol. 31(7):1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevots DR, Marras TK.. 2015. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin Chest Med. 36(1):13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. Available from: https://www.R-project.org/.
- Radomski N, et al. 2010. Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J Clin Microbiol. 48(4):1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16(6):276–277. [DOI] [PubMed] [Google Scholar]
- Rindi L, Garzelli C.. 2014. Genetic diversity and phylogeny of Mycobacterium avium. Infect Genet Evol. 21:375–383. [DOI] [PubMed] [Google Scholar]
- Roy R, et al. 2013. Synthesis of α-glucan in mycobacteria involves a hetero-octameric complex of trehalose synthase TreS and Maltokinase Pep2. ACS Chem Biol. 8(10):2245–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini NK, et al. 2008. Characterization of Mce4A protein of Mycobacterium tuberculosis: role in invasion and survival. BMC Microbiol. 8:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambou T, et al. 2008. Capsular glucan and intracellular glycogen of Mycobacterium tuberculosis: biosynthesis and impact on the persistence in mice. Mol Microbiol. 70(3):762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchini A, et al. 2016. A hypervariable genomic island identified in clinical and environmental Mycobacterium avium subsp. hominissuis isolates from Germany. Int J Med Microbiol. 306(7):495–503. [DOI] [PubMed] [Google Scholar]
- Sapriel G, et al. 2016. Genome-wide mosaicism within Mycobacterium abscessus: evolutionary and epidemiological implications. BMC Genomics 17:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. [DOI] [PubMed] [Google Scholar]
- Shah NM, et al. 2016. Pulmonary Mycobacterium avium-intracellulare is the main driver of the rise in non-tuberculous mycobacteria incidence in England, Wales and Northern Ireland, 2007-2012. BMC Infect Dis. 16:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons S, et al. 2011. Nontuberculous mycobacteria in respiratory tract infections, eastern Asia. Emerg Infect Dis. 17(3):343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Hänsch H, Bancroft G, Ehlers S.. 1997. T-cell-independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor-alpha and interferon-gamma. Immunology 92(4):413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkova DA, et al. 2014. The genome polymorphism of the Mycobacterium avium subsp. hominissuis strains. Mol Gen Mikrobiol Virusol. 4:14–19. [PubMed] [Google Scholar]
- Stucki D, et al. 2016. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 48(12):1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, et al. 2006. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 44(12):4498–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, et al. 2013. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. 45(2):172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanna S, Sucheck SJ.. 2016. Targeting the trehalose utilization pathways of Mycobacterium tuberculosis. Medchemcomm 7(1):69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson RM. 2010. Changing epidemiology of pulmonary nontuberculous mycobacteria infections. Emerg Infect Dis. 16(10):1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Ondov BD, Koren S, Phillippy AM.. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15(11):524.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turenne CY, Collins DM, Alexander DC, Behr MA.. 2008. Mycobacterium avium subsp. paratuberculosis and M. avium subsp. avium are independently evolved pathogenic clones of a much broader group of M. avium organisms. J Bacteriol. 190(7):2479–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turenne CY, Semret M, Cousins DV, Collins DM, Behr MA.. 2006. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J Clin Microbiol. 44(2):433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turenne CY, Wallace R, Behr MA.. 2007. Mycobacterium avium in the postgenomic era. Clin Microbiol Rev. 20(2):205–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiya K, et al. 2013. Comparative genome analysis of Mycobacterium avium revealed genetic diversity in strains that cause pulmonary and disseminated disease. PLoS One 8(8):e71831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattam AR, et al. 2017. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 45(D1):D535–D542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Johnston JC, Gardy JL, Tang P.. 2015. A microbiological revolution meets an ancient disease: improving the management of tuberculosis with genomics. Clin Microbiol Rev. 28(2):523–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HB, Jiang RH, Li L.. 2014. Treatment outcomes for Mycobacterium avium complex: a systematic review and meta-analysis. Eur J Clin Microbiol Infect Dis. 33(3):347–358. [DOI] [PubMed] [Google Scholar]
- Yahara K, Didelot X, Ansari MA, Sheppard SK, Falush D.. 2014. Efficient inference of recombination hot regions in bacterial genomes. Mol Biol Evol. 31(6):1593–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahara K, et al. 2016. The landscape of realized homologous recombination in pathogenic bacteria. Mol Biol Evol. 33(2):456–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E.. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Xie JP.. 2011. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol Cell Biochem. 352(1–2):1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.