

Abstract
Background
Mesenchymal/stromal stem cells (MSCs) are favorably regarded in anti-cancer cytotherapies for their spontaneous chemotaxis toward inflammatory and tumor environments associated with an intrinsic cytotoxicity against tumor cells. Placenta-derived or TRAIL-engineered adipose MSCs have been shown to exert anti-tumor activity in both in-vitro and in-vivo models of multiple myeloma (MM) while TRAIL-transduced umbilical cord (UC)-MSCs appear efficient inducers of apoptosis in a few solid tumors. However, apoptosis is not selective for cancer cells since specific TRAIL receptors are also expressed by a number of normal cells. To overcome this drawback, we propose to transduce UC-MSCs with a bicistronic vector including the TRAIL sequence under the control of IL-6 promoter (pIL6) whose transcriptional activation is promoted by the MM milieu.
Methods
UC-MSCs were transduced with a bicistronic retroviral vector (pMIGR1) encoding for green fluorescent protein (GFP) and modified to include the pIL6 sequence upstream of the full-length human TRAIL cDNA. TRAIL expression after stimulation with U-266 cell conditioned medium, or IL-1α/IL-1β, was evaluated by flow cytometry, confocal microscopy, real-time PCR, western blot analysis, and ELISA. Apoptosis in MM cells was assayed by Annexin V staining and by caspase-8 activation. The cytotoxic effect of pIL6-TRAIL + -GFP +-UC-MSCs on MM growth was evaluated in SCID mice by bioluminescence and ex vivo by caspase-3 activation and X-ray imaging. Statistical analyses were performed by Student’s t test, ANOVA, and logrank test for survival curves.
Results
pIL6-TRAIL + -GFP +-UC-MSCs significantly expressed TRAIL after stimulation by either conditioned medium or by IL-1α/IL-1β, and induced apoptosis in U-266 cells. Moreover, when systemically injected in SCID mice intratibially xenografted with U-266, those cells underwent within MM tibia lesions and significantly reduced the tumor burden by specific induction of apoptosis in MM cells as revealed by caspase-3 activation.
Conclusions
Our tumor microenvironment-sensitive model of anti-MM cytotherapy is regulated by the axis pIL6/IL-1α/IL-1β and appears suitable for further preclinical investigation not only in myeloma bone disease in which UC-MSCs would even participate to bone healing as described, but also in other osteotropic tumors whose milieu is enriched of cytokines triggering the pIL6.
Keywords: Multiple myeloma, Umbilical cord mesenchymal stromal/stem cells, TRAIL, Apoptosis
Background
Mesenchymal stem cells (MSCs) are presently under intensive investigation aimed not only at elucidating their nature and propensity to generate skeletal-related tissues [1, 2], but also to develop possible cell-based therapies against a number of diseases including cancer [3–5]. In this regard, within a few gene-therapy approaches, MSCs from bone marrow (BM) as well as from adipose tissue (AT) have been modified to enhance their secretory functions in a more targeted fashion either by releasing specific cytokines [6, 7] or as pro-apoptogen molecule producers to reverse the cell growth in both in vitro and in vivo models of solid tumors [8–10]. Consistent work by different groups of investigators showed indeed that both BM-derived and AT-derived MSCs are capable of inducing the tumor shrinkage in xenografted human glioma [11], gastric [12] or pancreatic [13] cancers, as well as in melanoma [14], and that this native anti-tumor cell growth activity may be definitely enhanced with MSCs transduced to express the tumor necrosis factor related apoptosis inducing ligand (TRAIL), namely a pro-apoptogen molecule linking the death receptors (DR) 4 and 5 on cancer cells [15]. In this context, fetal MSCs from umbilical cord (UC) engineered to produce a membrane-TRAIL protein have been reported to efficiently restrain the intracranial glioblastoma growth in mice as an effect of their innate chemotactic tendency to migrate toward the tumor microenvironment while exposing the death ligand to the tumor cells [16].
Based on their fetal tissue derivation, UC-MSCs have thus been focused with high interest to design novel cell-based therapeutic strategies. Besides their native restraining activity on Burkitt’s lymphoma cell proliferation [17], these cells have been demonstrated by ourselves to express a peculiar molecular profile of inhibitory properties on malignant plasma cells in vitro in relation to their secretome which significantly differs from both BM-derived and AT-derived MSCs [18]. On the other hand, placenta-derived MSCs, as a further fetal tissue source, are also capable of exerting spontaneous killing of multiple myeloma (MM) cells both in vitro and in a severe combined immunodeficient (SCID)-rab mice MM model developing osteolytic lesions [19]. In these studies, in addition to the anti-myeloma activity, MSCs were also found capable of repairing the bone loss within the bone lytic lesions in relation to their bone-regenerating constitutive capability [20].
However, since MSCs, particularly BM-MSCs, physiologically support hematopoiesis, it has also been debated whether a MSC-based model of cytotherapy for MM would sustain, rather than suppress, the proliferation of malignant plasma cells [21]. Contrarily to BM-derived and AT-derived MSCs, those from fetal tissues appear resistant to the genomic conditioning by MM cells that drives the molecular potential to support tumor cell proliferation [22], whereas UC-MSCs apparently show a genomic profile of a definite anti-MM killing secretome [18].
Previous work with TRAIL-engineered BM-MSCs or UC-MSCs, however, included transduction of cells with viral vectors allowing the constant expression of the apoptogen molecule by the cell membranes with the potential risk of cell death induction even in normal cells exposing DR4/DR5 receptors. Unselective binding of target cells thus represents a major drawback of this cytotherapy model since the occurrence of liver as well as of other parenchymal damage has already been reported in preliminary studies by Kim et al. [16] when treating glioma in mice with TRAIL-transduced UC-MSCs.
To overcome such a major drawback and generate UC-MSCs transduced to kill MM cells, we thus used a bicistronic vector to regulate TRAIL expression only after their molecular cross-talk with soluble factors of the MM tumor microenvironment. During the tumor progression of myeloma within the bone marrow, indeed, both interleukin (IL)-1α and IL-1β secreted by MM cells stimulate the stroma to produce IL-6 [23] through the linkage of the early growth response (EGR)-1 protein to the promoter of IL-6 (pIL6) [24]. Therefore, we transduced the UC-MSCs with a vector containing the full-length cDNA sequence of TRAIL under the control of the pIL6 and we evaluated the potential of pIL6-TRAIL + -GFP +-UC-MSCs to eradicate MM cells both in vitro and in SCID mice bearing intratibial human myeloma.
Results from our study support the effectiveness of an anti-MM cytotherapy approach in terms of selective killing of malignant plasma cells.
Methods
Cell cultures
UCs were obtained from parturients at the Obstetrician and Gynecology Department after informed consent approved by the local Ethical Committee of the University of Bari. UC-MSCs were isolated and maintained in alpha-Modified Eagle’s Medium (MEM) (Gibco, Life Technol., Lofer, Austria) and, at the second passage, were used for retroviral transduction. The U-266 MM cell line (DSMZ, Braunschweig, Germany) was grown in complete Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco), whereas HEK293T, a human embryonic kidney cell line (Sigma Aldrich, St. Louis, MO, USA), was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco).
TRAIL transduction of UC-MSCs
To generate TRAIL-transduced UC-MSCs, we adopted a bicistronic retroviral vector (pMIGR1) from a murine stem cell virus encoding for the green fluorescent protein (GFP) gene. This vector was modified to include the pIL6 sequence upstream of the full-length human TRAIL cDNA (Fig. 1a). Briefly, a 315-nucleotide fragment of human pIL6 (nucleotides –303 to +12, Ensembl ENSG00000136244), obtained from genomic DNA by cutting with restriction enzymes for BglII and XhoI sites, was amplified in polymerase chain reaction (PCR) by dedicated primers (forward, 5′-GAATTAGATCTTCAAGACATGCCAAAGTGC-3′; and reverse, 5′-GCCATCCTCGAGGGCAGAATGAGCCTCA-3′). Full-length human TRAIL gene (NM_003810.2) was amplified from cDNA using Expand High Fidelity Taq (Roche, Indianapolis, IN, USA) by primers containing XhoI (forward 5′-GCCCTCGAGGATGGCTATGATGGAGGTCCA-3′) and EcoRI (reverse 5′-GCGGAATTCCTTAGCCAACTAAAAAGGCCCC-3′) sites. Both PCR products were digested by XhoI and ligated with each other to generate a single insert. Thus, the pIL6-TRAIL was cloned into pMIGR1 at BglII and EcoRI sites, and defined as pIL6-TRAIL + -GFP +-pMIGR1 vector, whereas the empty GFP +-pMIGR1 vector was used as control.
Fig. 1.
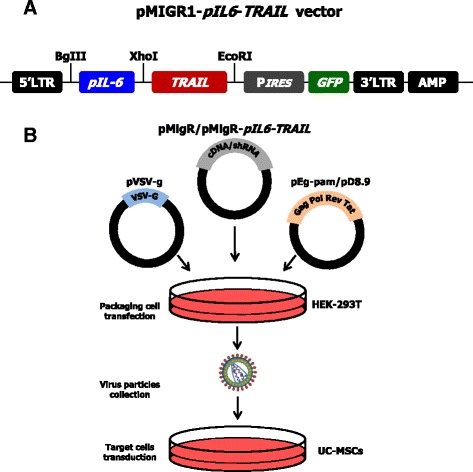
Structure of pMIGR1 vector and steps for UC-MSC transduction. a Structural construction of the bicistronic retroviral vector including both TRAIL and GFP sequences controlled by the IL-6 promoter. PIRES sequence was inserted to codify two different proteins from a single mRNA. b Sequential phases of multiple cell transfection, viral particle enrichment, and final transduction of UC-MSCs. GFP green fluorescent protein, MSC mesenchymal/stromal stem cell, pIL6 interleukin-6 promoter, PIRES poliovirus internal ribosome entry site, TRAIL tumor necrosis factor related apoptosis inducing ligand, UC umbilical cord
Retroviruses were produced by cotransfection of HEK293T cells with both pMIGR1 construct and the packaging plasmids, namely pΔ8.9 and pVSV-G, using XTreme Gene 9 DNA transfection Reagent (Roche). HEK293T retrovirus-enriched supernatants were collected 48 h after transfection and concentrated by ultracentrifuge at 17,000 rpm (SW28 rotor, Optima LE80K Ultracentrifuge; Beckman, Brea, CA, USA) for 2 h at 4 °C (Fig. 1b). Thus, UC-MSCs were transduced by virus-containing media from either pIL6-TRAIL + -GFP +-HEK293T or GFP +-HEK293T (both ∼ 1 × 106 transducing units/ml) and 4 μg/ml polybrene (Sigma-Aldrich). The transfected UC-MSCs were distinguished as pIL6-TRAIL + -GFP +-UC-MSCs and GFP +-UC-MSCs respectively, and expanded until the seventh passage.
TRAIL expression and modulation in transfected UC-MSCs
After transfection, UC-MSCs were investigated by flow cytometry (FACScanto; Becton-Dickinson, San Diego, CA, USA) for GFP fluorescence to define the efficiency of the vector insertion. The transfected UC-MSCs were then sorted for GFP expression by FACS Aria III (Becton Dickinson, Milan, Italy) to obtain homogeneous populations to be used in subsequent experiments.
Basal levels of TRAIL and further expression on pIL6-TRAIL + -GFP +-UC-MSCs by U-266 supernatant, and by 10 ng/ml of IL-1α and IL-1β, were investigated by flow cytometry, confocal microscopy, quantitative (q)PCR, and western blot (WB) analysis.
TRAIL flow cytometry analysis was assessed using specific phycoerythrin (PE)-conjugated monoclonal (Mo) antibody (Ab) (Abcam, Cambridge, UK) in triplicate with isotype control. Data were reported as both the percentage of TRAIL-positive cells and the mean fluorescence intensity (MFI) ratio as described previously [25].
Furthermore, TRAIL expression by pIL6-TRAIL + -GFP +-UC-MSCs was also analyzed by confocal microscope and NIS element software (C2plus; Nikon Instr., Lewisville, TX, USA). Transfected UC-MSCs were incubated with unconjugated anti-human TRAIL rabbit polyclonal Ab (Cell Signaling, Danvers, MA, USA) and then with anti-rabbit fluorescein isothiocyanate (FITC) (Sigma). The samples were counterstained by tetramethylrhodamine (TRITC)-conjugated phalloidin (Life Technologies) to visualize F-actin and by Hoechst 33342 (Sigma Aldrich) for nuclear staining.
In addition, to assess the TRAIL expression by qPCR, total RNA was extracted by RNeasy kit (Qiagen, Hilden, Germany), and 500 ng was reverse-transcribed by IScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). cDNA was then amplified by Fast SYBR Green Master Mix and the StepOne Plus Real Time PCR (Life Technologies Inc., Carlsbad, CA, USA) using the specific primers for TRAIL: forward, 5′-GCTCTGGGCCGCAAAAT-3′; and reverse, 5′-TGCAAGTTGCTCAGGAATGAA-3′. Data were normalized on glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels and TRAIL amounts were detected as fold change with respect to basal condition.
Also, the protein was evaluated by WB analysis using polyclonal anti-human TRAIL Ab (Abcam) and ECL reagent (Bio-Rad), and then visualized by the UVIchemi (UVItec, Cambridge, UK) imaging system using UVI-1D quantification software. Expression levels were calculated as mean ± 3 standard deviations (SDs) of the optical density (OD) ratio between TRAIL and housekeeping GAPDH in three different experiments. Finally, soluble TRAIL was also measured in supernatants of pIL6-TRAIL + -GFP +-UC-MSCs after treatment with U-266 conditioned medium, or IL-1α and IL-1β, by dedicated ELISA kit (Abcam).
In vitro apoptosis of U-266 cells
To investigate the pro-apoptogen potential of pIL6-TRAIL + -GFP +-UC-MSCs toward U-266 cells, we carried out cocultures at 1:2 ratio and evaluated the cytotoxicity at 24 h by Annexin V-FITC/propidium iodide (PI) staining (eBioscience, Bender MedSystems GmbH, Vienna, Austria) using the FACScanto. The U-266 population was gated based on forward scatter (FSC) and side scatter (SSC) parameters.
Specificity of apoptosis of U-266 cells following the TRAIL cross-talk was analyzed by caspase-8 activation using the CaspGLOW™ Fluorescein Active Caspase-8 Staining Kit (eBioscience, Bender MedSystems GmbH) and the active caspase-8 was evaluated by flow cytometry.
In vivo functional studies
To investigate the activity of transfected UC-MSCs against MM cells in vivo, we generated stably transduced bioluminescent U-266 cells (Red-Luc+U-266) using RediFect™ lentiviral particles containing red-shifted firefly luciferase (Luciola italica) transgene (Perkin Elmer, Waltham, MA, USA). Briefly, U-266 cells were seeded into a 24-well plate and were incubated with 106 viral particles for 24 h. Transduced cells were selected and expanded in medium containing 1 μg/ml puromycin for 4 weeks. Luciferase expression by transduced U-266 was assayed using IVIS Lumina SIII (Perkin Elmer) by adding d-luciferin potassium salt (Perkin Elmer) to the cultures.
Anti-MM activity of pIL6-TRAIL + -GFP +-UC-MSCs was investigated in 6–8-week-old NOD.CB17-Prkdcscid/J mice (Charles River, Milan, Italy) in line with the rules and institutional guidelines of the Italian Ministry of the Health. For this, 42 mice were intratibially (IT) injected with 2 × 105 Red-Luc+U-266 cells and the tumor engraftment was evaluated by luminescence imaging 3 days after inoculation. Briefly, anesthetized mice were intraperitoneally administered with luciferin (150 mg/kg) and the tumor luminescence was captured 10 min after injection. Regions of interest encompassing the area of signal were defined using Living Image software and the total photons per second (photons/s) were recorded. The MM-bearing mice were then randomly divided into three groups and injected intracardially (IC) as follows: phosphate buffered saline (PBS) for the control group (N = 14); 2.5 × 105 GFP +-UC-MSCs (N = 14); and 2.5 × 105 pIL6-TRAIL + -GFP +-UC-MSCs (N = 14). Three mice from each group were sacrificed after 12 and 48 h to evaluate the distribution of UC-MSCs and MM cell apoptosis within tibiae. In addition, the tumor burden was evaluated by luminescence imaging in six mice of each condition at different times up to 30 days after the treatment with pIL6-TRAIL + -GFP +-UC-MSCs and GFP +-UC-MSCs as control. The tumor growth rate in each group of MM-bearing mice was expressed as the relative fold increase of tumor volume calculated as the ratio of total photon flux at various time points with respect to basal condition. All animals were ultimately sacrificed at 30 days for ethical reasons.
Ex vivo measurement of pIL6-TRAIL+-GFP+-UC-MSC activity against MM
Several tissue samples, including heart, lung, spleen, liver, and kidney from MM-bearing mice sacrificed at 12 and 48 h, were fixed in formaldehyde and embedded in paraffin, whereas explanted tibiae were decalcified in formic acid and included in paraffin.
PCR for the GFP insert was performed to reveal the pIL6-TRAIL + -GFP +-UC-MSC distribution in mice. DNA was extracted from all tissues using the AllPrep DNA/RNA FFPE Kit (Qiagen). Briefly, 100 ng template DNA was used to perform PCR using the HotstarTaq Master Mix (Qiagen) and 0.3 μM of primers, for 35 cycles with annealing temperatures of 60 °C (GFP) or 55 °C (β-actin). Primer sequences for GFP were forward 5′GTGCTTCAGCCGCTACCC-3′ and reverse 5′-TGTCGGCCATGATATAGACGTTG-3′, whereas for β-actin they were forward 5′-ACGGGGTCACCCACACTGTGC-3′ and reverse 5′-CCGCTCGTTGCCAATAGTGATGA-3′.
To evaluate the intratibiae MM cell apoptosis, sections 3 μm thick were stained with hematoxylin–eosin and in parallel for active caspase-3 by a specific anti-human mouse MoAb (MyBiosource, San Diego, CA, USA). The test was completed by EnvisionFlex kit (DakoCytomation, Santa Clara, CA, USA) according to the manufacturer’s instructions. All samples were then examined under light microscopy (Olympus Bx61; Shinjuku, Tokyo, Japan). To visualize the macroscopic effect of our model, we completed radiography evaluations of tibiae. Briefly, animals were euthanized by carbon dioxide and X-ray scans were taken at 20 kV and 25 mAs for 5 s using a mammographic device (Model Flat E; Metaltronica, Rome). Films from the three groups were inspected comparatively for visible bone lesions that were carefully measured for their bone devastation size (mm2) (ImageJ software, version 1.45; NIH, Bethesda, MD, USA).
Statistical analysis
Results were shown as mean ± SD of experimental triplicates. Statistical analyses were completed by Microsoft® Excel (Microsoft, Inc., Redmond, WA, USA) and GraphPad Software (GraphPad Software, San Diego, CA, USA). Significance between differences in Kaplan–Meier survival curves were generated using MedCalc 12.7.0.0 software. For the Kaplan–Meier analyses, survival curves were compared using the logrank test. Student’s t test was used to compare two groups while comparisons between multiple groups (n > 2) were performed by ANOVA and differences were considered significant with p < 0.05.
Results
pIL6-TRAIL+-GFP+-pMIGR1 vector construction and UC-MSC transduction
pMIGR1 bicistronic retroviral vector containing poliovirus internal ribosome entry site (PIRES) and GFP sequences was modified to express full-length TRAIL under the control of pIL6 (Fig. 1a). pIL6-TRAIL construct was obtained by ligation of the relative PCR products in the XhoI restriction site and subsequently cloned between the BglII and EcoRI sites on pMIGR1. Thus, modified pMIGR1 construct and viral packaging plasmids were transfected in HEK293T competent cells and the culture supernatant containing viral particles bearing pIL6-TRAIL + -GFP +-pMIGR1 vector was used to infect UC-MSCs (Fig. 1b). pMIGR1 wildtype construct was used as control empty vector.
TRAIL expression in transduced UC-MSCs
The efficiency of transfection was evaluated by GFP expression in flow cytometry. As shown in Fig. 2a (left), GFP was largely detected in 83.5% of GFP +-UC-MSCs and 87.6% of pIL6-TRAIL + -GFP +-UC-MSCs, whereas wildtype UC-MSCs were negative as expected. After cell sorting we obtained GFP +-UC-MSCs and pIL6-TRAIL + -GFP +-UC-MSCs with purity of 99.0% and 99.5% respectively. These last cell populations were expanded and used for subsequent experiments.
Fig. 2.

TRAIL expression and modulation in pIL6-TRAIL + -GFP +-UC-MSCs. a Left: transfection efficiency of UC-MSCs evaluated by flow cytometry for GFP fluorescence. Wildtype UC-MSCs were negative as compared to GFP +-UC-MSCs (red histogram: 83.5%) in a similar amount as for pIL6-TRAIL + -GFP +-UC-MSCs (blue histogram: 87.6%). Right: FACS analysis revealed constitutive TRAIL expression in transduced UC-MSCs (green histogram: 62.5%) that was significantly enhanced by the conditioned medium from U-266 cells (blue histogram: 80.3%) as well as in response to IL-1α/IL-1β (red histogram: 91.2%). b Confocal microscopy of pIL6-TRAIL + -GFP +-UC-MSCs confirmed the upregulation of TRAIL (FITC, green) induced by either U-266 supernatant or IL-1α/IL-1β treatment. Phalloidin staining was used to show actin (TRITC-red); nuclei were counterstained with Hoechst 33342 (blue). Magnification: 100×. c Evaluation of TRAIL expression in transduced cells by RT-qPCR, western blot analysis, and ELISA. (i) After either U-266 or IL-1α/IL-1β treatment, TRAIL mRNA levels in pIL6-TRAIL + -GFP +-UC-MSCs increased up to 2.2-fold and 2.5-fold compared to basal condition. Data are relative amount of mRNA expression normalized to GAPDH and are presented as mean ± SD of three independent experiments. *p < 0.05. (ii) Western blot analysis of lysates from transduced cells showing increased levels of TRAIL protein after U-266 medium or IL-1α/IL-1β conditioning. GAPDH was used as loading control. (iii) ELISA measurement of soluble TRAIL in supernatants of pIL6-TRAIL +-GFP +-UC-MSCs after 24 h of treatment with both U-266 conditioned medium and IL-1α/IL-1β. In both instances the increase was significant as compared to control cells (p < 0.05). GFP green fluorescent protein, MSC mesenchymal/stromal stem cell, pIL6 interleukin-6 promoter, TRAIL tumor necrosis factor related apoptosis inducing ligand, UC umbilical cord, WT wildtype
The expression of TRAIL, both constitutively and following the variation by U-266 conditioned medium, or by IL-1α and IL-1β, in pIL6-TRAIL + -GFP + -UC-MSCs is depicted by flow cytometry histograms in Fig. 2a (right). As shown, TRAIL was expressed in 62.5% transduced cells in basal conditions suggesting that pIL6-TRAIL + -GFP + -UC-MSCs were able to maintain the constitutive protein production including cytokines that activate pIL6 [18]. However, when treated with the U-266 conditioned medium, pIL6-TRAIL + -GFP +-UC-MSCs significantly enhanced their TRAIL expression up to 80.3% of positive cells (blue histogram), which was confirmed up to 91.2% of positive population by IL-1α and IL-1β (red histogram). Moreover, TRAIL MFI was 74.6 on pIL6-TRAIL + -GFP +-UC-MSCs after stimulation with the U-266 conditioned medium that was significantly increased compared to basal condition MFI (44.7). The TRAIL upregulation was also confirmed after stimulation by IL-1α and IL-1β (MFI = 70.7). The increased TRAIL expression was shown on representative pictures by confocal microscopy. Figure 2b depicts the enriched TRAIL fluorescent signal on pIL6-TRAIL + -GFP +-UC-MSCs stimulated with U-266 conditioned medium (middle), or in response to IL-1α/IL-1β (right) with respect to basal condition (left).
Furthermore, as shown in Fig. 2c, qPCR of TRAIL mRNA confirmed the stimulation of pIL6-TRAIL + -GFP +-UC-MSCs by either U-266 conditioned medium or IL-1α and IL-1β, since the TRAIL gene expression significantly increased up to 2.2-fold and 2.5-fold respectively as compared to basal condition (Fig. 2ci). This was also confirmed by WB analysis that revealed the increase of TRAIL protein after stimulation. The OD ratio of pIL6-TRAIL + -GFP +-UC-MSCs treated with both U-266 conditioned medium and IL-1α/IL-1β, indeed, was 2.67 ± 1.4 and 3.72 ± 2.1 respectively, compared to untreated cells (p < 0.005 in all instances) (Fig. 2cii).
Finally, we also demonstrated the production of soluble TRAIL by transduced UC-MSCs. In fact, ELISA detected minimal mean levels of the ligand (25 pg/ml) in basal condition which, however, was significantly increased in 24-h treated cells up to 140 pg/ml after stimulation with both conditioned medium and IL-1α/IL-1β (p < 0.005 in all instances) (Fig. 2ciii).
These results suggested that following the transduction of UC-MSCs with the pIL6-TRAIL + -GFP + -pMigr vector, these cells significantly enhanced their TRAIL expression as both membrane-bound and soluble protein after their activation by pIL6.
pIL6-TRAIL+-GFP+-UC-MSCs are apoptosis effectors in U-266 cells
The capability of pIL6-TRAIL + -GFP +-UC-MSCs to kill MM cells was evaluated in vitro by Annexin V/PI staining. In 24-h coculture, the apoptosis extent of U-266 cells was 50.4% ± 6.6 as compared to control wildtype UC-MSCs (19.4% ± 3.4) or to GFP +-UC-MSCs (20.2% ± 2.3). When IL-1α and IL-1β were added to the cocultures, we observed an increase of the programmed cell death associated to the additional expression of TRAIL by pIL6 (70.8% ± 6.5) (p < 0.01). Figure 3a shows a representative pattern of U-266 apoptosis induced by TRAIL-transduced UC-MSCs.
Fig. 3.
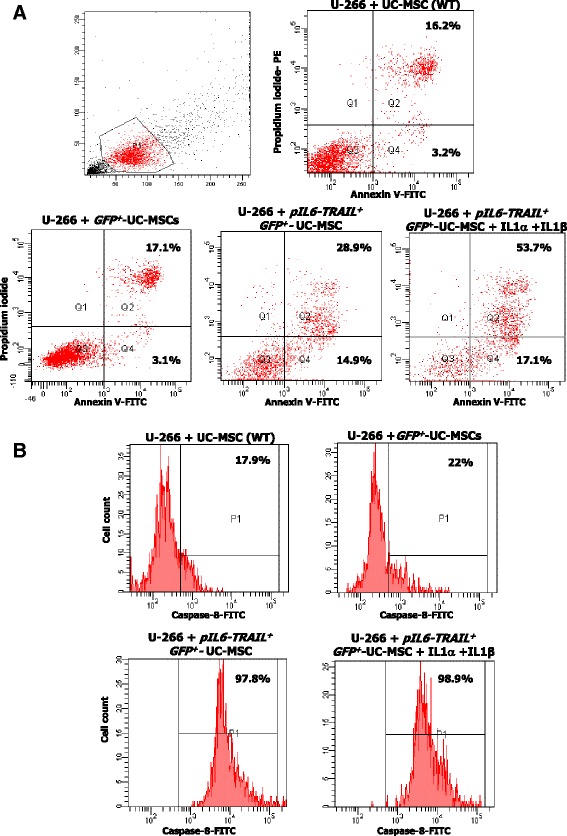
In vitro apoptosis of U-266 cells induced by pIL6-TRAIL + -GFP +-UC-MSCs. a Apoptosis in U-266 cells by transduced UC-MSCs was measured by Annexin V/PI staining using flow cytometry. Representative dot plots revealed that the apoptosis extent was significantly increased (43.8%) after 24 h of coculture with pIL6-TRAIL + -GFP +-UC-MSCs with respect to control UC-MSCs (19.4%) and GFP +-UC-MSCs (20.2%). The effect was also enhanced when IL-1α and IL-1β were added to the cocultures (70.9% of U-266 cell apoptosis). b Active caspase-8 in U-266 cells as signature of TRAIL-induced apoptosis was measured by flow cytometry after 24 h of coculture with UC-MSCs, GFP +-UC-MSCs, and pIL6-TRAIL + -GFP +-UC-MSCs. This representative experiment depicts the activity of caspase-8 in 97.8% of U266 cells cocultured with pIL6-TRAIL + -GFP +-UC-MSCs as compared to 17.9% of control UC-MSCs, and 22% of GFP +-UC-MSCs. Such high levels of active caspase-8 were not further modified by supplementing the cultures with IL-1α/IL-1β (98.9% of positive cells). GFP green fluorescent protein, MSC mesenchymal/stromal stem cell, pIL6 interleukin-6 promoter, TRAIL tumor necrosis factor related apoptosis inducing ligand, UC umbilical cord, WT wildtype
To assess the molecular pathway of apoptosis in U-266 cells we measured their levels of active caspase-8. Figure 3b shows these results. As depicted, the flow cytometric analysis of U-266 cells cocultured by pIL6-TRAIL + -GFP +-UC-MSCs suggested a significant enrichment of active caspase-8 up to 97.8% ± 0.7 of this population (p < 0.05). The addition of IL-1α and IL-1β was ineffective in changing the active caspase-8 levels (98.9% ± 0.6) as compared to the same cells cultured with UC-MSCs or GFP +-UC-MSCs (17.9% ± 3.3 and 22.0% ± 2.7 respectively) (p < 0.01).
pIL6-TRAIL+-GFP+-UC-MSCs exert anti-MM activity in vivo
We generated stably transduced bioluminescent Red-Luc+U-266 to monitor the tumor growth in mice. The timeline of in vivo experiments is reported in Fig. 4a. Forty-two mice were implanted IT with Red-Luc+U-266 cells and, after 3 days, the tumor engraftment was evaluated by bioluminescence imaging. Recorded total photon flux at day 3 in all animals was variable, ranging from 3.5 × 106 to 5 × 107 (photon/s) with a mean of 2.64 × 107 ± 2.16 × 107. Thus, mice distributed in the groups were injected IC with PBS (group A), GFP +-UC-MSCs (group B), or pIL6-TRAIL + -GFP +-UC-MSCs (Group C). Their drop-out [26], representing the rate of dead animals following the ventricle injection failure, was approximately 14%.
Fig. 4.

In vivo effect of pIL6-TRAIL + -GFP + -UC-MSCs on MM growth. a Timeline elucidating the experimental design of SCID mice xenografted intratibially (IT) with 2 × 105 Red-Luc+U266 cells (D0), followed after 3 days (D3) by injection intracardially (IC) with PBS (group A), 2.5 × 105 GFP +-UC-MSCs (group B), or 2.5 × 105 pIL6-TRAIL + -GFP +-UC-MSCs (group C) (N = 14 for each group). Three mice for each group were sacrificed randomly, respectively at 12 h (h12) and 48 h (h48) after injection IC for ex vivo evaluation of UC-MSC distribution and tumor apoptosis. The remaining mice were investigated at defined time points (D3, D10, D20 and D30) by bioluminescence imaging with IVIS Lumina. b The biodistribution of pIL6-TRAIL + -GFP +-UC-MSC, revealed by PCR analysis for GFP after injection IC, confirmed the presence of these cells in tibiae as well as in lung, heart, and kidney, while transduced cells were not observed in spleen and liver. Actin was used as loading control. c Representative bioluminescence images at different time points of MM-bearing mice, injected IC with PBS (group A), GFP +-UC-MSCs (group B), or pIL6-TRAIL + -GFP +-UC-MSCs (group C). The color scale ranged from blue (just higher than background noise; set to 1 × 107 photons/s/cm2/sr) to red (at least 2.5 × 108 photons/s/cm2/sr). Quantitative analysis of tumor growth in mice was assessed by Living Image Software. Data represent the relative increase of median photon flux (photon/s) within ROI areas in each group at different time points. Tumor growth was timely reduced in mice treated with pIL6-TRAIL + -GFP + -UC-MSCs as compared to both PBS and GFP +-UC-MSC treated groups (p < 0.03). Error bars represent the standard error of the mean (SEM); *p value calculated by Student’s t test. d Hindlimb radiographs of MM bearing mice was performed 27 days after injection IC of transduced UC-MSCs or PBS. Representative images of the three experimental groups show a minor expansion of the osteolytic lesions in mice systemically injected with pIL6-TRAIL +-GFP +- UC-MSCs compared to those of groups A and B. GFP green fluorescent protein, MM multiple myeloma, MSC mesenchymal/stromal stem cell, PBS phosphate buffered saline, UC umbilical cord, NTC no template control
To investigate the organ distribution of transduced UC-MSCs in mice, we analyzed tissues isolated at 12 h after injection IC. PCR analysis for GFP confirmed the presence of pIL6-TRAIL + -GFP +-UC-MSCs in tibiae as well as in lung, heart, and renal glomeruli as described previously [27], while transduced cells were not detected in spleen and liver (Fig. 4b). However, there was no evidence of toxicity on healthy tissues evaluated at the experimental endpoint (data not shown).
The MM burden was evaluated by bioluminescence in mice of each group at experimental time points (10, 20, 30 days), as depicted by representative images in Fig. 4c (upper). The quantitative analysis (Fig. 4c, lower), calculated as the ratio of total photon flux at various time points with respect to basal condition, showed a decrease of tumor growth, although not significant, already on D10 in group C compared to groups A and B (relative fold increase: group C = 185 ± 80; group A = 778 ± 131; group B = 595 ± 221). At D20 the differences between groups were increased, becoming significant (relative fold increase: group C = 360 ± 96; group A = 1442 ± 288; group B = 1264 ± 182), and this effect was maintained at the endpoint (relative fold increase: group C = 844 ± 167; group A = 2076 ± 332; group B = 2136 ± 325) (p < 0.03 in each instance).
Bone devastations induced in vivo by MM cells were also evaluated by X-ray imaging. The average size of the osteolytic areas measured on X-ray films of tibiae in group A and B mice was apparently larger and displaying blown cortical bone (mean value 2.5 ± 0.35 mm2), as compared to that observed in pIL6-TRAIL + -GFP +-UC-MSC-treated mice (0.96 ± 0.39 mm2) (p < 0.02) (Fig. 4c).
Finally, to demonstrate MM cell apoptosis in tibia samples, we measured the active caspase-3 by immunohistochemistry. Figure 5a shows the enrichment of plasma cells evidenced by hematoxylin–eosin staining in the bone matrix of mice tibiae in each group. As shown, plasma cells are accumulated within resorptive lacunae where they appear strictly adjacent to the resorbed bone. Also, the active isoform of caspase-3 was detected already after 12 h from injection IC of pIL6-TRAIL + -GFP +-UC-MSCs and this effect persisted up to 48 h (Fig. 5b). On the contrary, the sections of mice inoculated with GFP +-UC-MSCs were negative for active caspase-3.
Fig. 5.

Ex vivo immunohistochemical detection of MM cell apoptosis. a Hematoxylin–eosin staining of tibia sections in mice xenografted IT shows the plasma cell bone expansion. As can be seen, U-266 cells are accumulated within resorptive lacunae where they appear strictly to the resorbed bone. b Immunostaining of active caspase-3 showed a high extent of apoptosis in tibiae sections of mice injected systemically with pIL6-TRAIL + -GFP +-UC-MSCs after 12 and 48 h. Active caspase-3 was undetectable in control mice treated with GFP +-UC-MSCs. GFP green fluorescent protein, MSC mesenchymal/stromal stem cell, pIL6 interleukin-6 promoter, TRAIL tumor necrosis factor related apoptosis inducing ligand, UC umbilical cord
These data suggest that pIL6-TRAIL + -GFP +-UC-MSCs were able to remarkably inhibit the tumor burden and ultimately restrain the bone devastation by U-266 cells.
Discussion
Novel anti-cancer cytotherapies are presently under intensive investigation and MSCs have potential as cell vehicles for targeted delivery and/or local production of cytotoxic molecules for tumor cells. Besides their large bioavailability and easy recruitment from the human body, the suitability of these cells in fighting cancers is based on their tropism toward inflamed sites including the tumor microenvironment, as well as on their proclivity to be genetically engineered with gene sequences encoding for anti-tumor biological agents [28, 29]. In this context, we have demonstrated previously that UC-MSCs constitutively migrate in vitro toward myeloma cells and express a defined anti-MM secretome [18]. Here, we provide further evidence that, once engineered to express TRAIL in the presence of malignant plasma cells, UC-MSCs specifically induce apoptosis in these cells both in vitro and in vivo in SCID mice bearing human MM. The selective activity against MM cells is related to the control of TRAIL production by pIL6 which is inserted before the TRAIL sequence within the vector, thus rendering suitable this cell-based approach of anti-MM treatment for future translation in human studies.
Previous models of cytotherapies with MSCs in MM included placenta-derived MSCs [19] that showed a native activity against MM cells in vitro in parallel with a moderate osteogenic potential in vivo in healing the MM bone lesions in SCID-rab mice. These studies, however, supported the general cytostatic property of MSCs which was apparently mild and unselective for MM cells while the bone-repairing capacity was also related to the constitutive osteoblast differentiation of cells belonging to the mesenchymal lineage as placenta-derived MSCs [19]. Further work by ourselves adopted AT-MSCs which were transduced with TRAIL and definitely promoted apoptosis in U-266 cells by the caspase-3 activation [30] in a similar manner as for other tumor models using these engineered MSCs [31–34]. In the present study, we preferred to transduce UC-MSCs with TRAIL for several reasons: large availability of MSCs from UCs; spontaneous chemotactic activity toward the tumor; and constitutive anti-MM activity by their secretome. We also used UC-MSCs since fetal MSCs are very poorly immunogenic for the minor expression of costimulatory molecules [18] while showing multipotent plasticity [35] and no tumorigenic potential [36]. In addition, UC-MSCs have been reported to show higher kinetics of proliferation and karyotypic stability in culture than AT-MSCs and BM-MSCs [37, 38].
Animal models of xenogenic tumors treated with TRAIL-transduced MSCs provided evidence on the effectiveness of this cell-based approach in relation to the expression of DR4/DR5 by the target tumors [16, 33, 34, 39], although several concerns regard the defective specificity since the engineered MSCs also deliver TRAIL to both tumor and normal cells, and therefore normal tissues including liver and kidney are recurrently damaged following the systemic injections of these cells [27]. On the other hand, it has been reported that, particularly in hematological malignancy including MM, based on the high expression of DR4/DR5 molecules, the soluble TRAIL isoform is also capable of inducing apoptosis in a similar manner, although less effective, than in the membrane bound form as presented by the transduced cells [40]. Thus, differentially structured viral vectors were engineered for TRAIL gene insertion to transduce MSCs and the full-length gene sequence codifying its soluble form has been variably inserted. However, some evidence demonstrated that the cancer cell-killing potential induced by the full-length form expressed on MSC membrane is more efficacious than that obtained by the soluble molecule [15].
Therefore, to maintain the full-length structure of the apoptotic protein and to selectively kill MM cells, we designed a vector inducing the expression of TRAIL only in the presence of cytokines secreted locally within the MM microenvironment. To this, we adopted the retroviral bicistronic vector pMIGR1 incorporating the full-length TRAIL gene whose expression is regulated by pIL6. Several cytokines such as IL-1α and IL-1β are largely secreted by MM cells within the marrow microenvironment to activate the IL-6 secretion by BM MSCs [41–43] through the stimulation of its own promoter by EGR-1 [23, 24, 44]. We reasoned that the pIL6 insertion just before TRAIL within the pMIGR1 would have been able to upregulate the expression of the protein by pIL6-TRAIL + -GFP +-UC-MSCs in response to IL-1α and IL-1β. Although, in the presence of a basal expression of TRAIL which was probably driven by an autocrine loop of cytokine secretion regulated by the UC-MSCs secretome [18], in our experiments we found that this structural variant of the vector significantly increased the production of TRAIL as both membrane-bound and soluble protein also in response to the conditioned medium from U-266 cells. On the other hand, the minor expansion of the osteolytic lesions in tibiae of mice treated with those cells confirmed the capacity of our vector to trigger the secretion of the apoptosis inducer in the presence of MM cells resulting in tumor cell death by activation of caspase-8 [31, 39]. This result was enough to support the efficacy of our cell-based approach against MM in mice.
The anti-MM killing of these cells was tested in our orthotopic in vivo model of MM. SCID mice were injected IT with U-266 cells to resemble the human MM model in which malignant plasma cells expand within the bone marrow and promote the bone resorption inducing osteolytic lesions [45]. Then, after developing the bone lesions, MM-bearing mice were treated with injections IC of pIL6-TRAIL + -GFP + -UC-MSCs and periodically investigated for the anti-MM effect as well as for their tissue distribution. By evaluating the GFP expression in several ex vivo organs, we found that the accumulation of pIL6-TRAIL + -GFP +-UC-MSCs occurred in tibiae as well as in lung, heart, and renal glomeruli, thus supporting the typical tropism also to the tumor sites ascribed to MSCs [27]. However, these cells did not produce local damage in healthy tissues and they had no toxicity that could compromise the quality of life in mice. This is probably due to the low levels of both membrane and soluble TRAIL basally expressed by pIL6-TRAIL + -GFP +-UC-MSCs, since the transcription of TRAIL is reinforced by both IL-1α and IL-1β usually abundant within the tumor sites. Such a spontaneous homing of UC-MSCs toward the myeloma tumor milieu within tibiae is apparently related to the overexpression of a number of genes such as growth factor receptor-bound 2 (GRB2), which are activated to promote the cell migration toward inflamed sites in response to the cell stimulation by tumor-derived chemokines [18]. Thus, in line with previous studies in different tumor models [6, 46], we interpreted the accumulation of pIL6-TRAIL + -GFP +-UC-MSCs within tibiae of MM-bearing mice as an effect of their typical attraction toward the cytokine-enriched MM environment. Our result is consistent when considering that these cells migrated toward the tumor sites after the injections IC that were adopted in our animal model to avoid the potential entrapment of transduced UC-MSCs within lungs after intravenous administration.
In our model, as pIL6-TRAIL + -GFP + -UC-MSCs were induced in vitro to express TRAIL in response to the U-266 conditioned medium and IL-1α and IL-1β stimulation, we observed in vivo the selective pressure of the MM microenvironment to induce TRAIL at high levels. In fact, after 12 h post inoculation we observed on tibiae sections an extended apoptosis of U-266 cells by caspase-3 activation. This effect persisted up to 48 h, suggesting that the pIL6-TRAIL + -GFP +-UC-MSCs exhibit a higher survival rate in bone tissue compared to other organs in which these cells are cleared faster [20], while the reduction of the bioluminescent signal of the tumor burden at different time points and even 30 days after injection supported their MM cytotoxic activity.
It has been described that placenta-derived MSCs are capable of rebuilding the MM bone lesions in mice [19]. In our model the transduced UC-MSCs also induced a partial restoration of the bone structure as shown by X-ray analysis of tibiae. Although this aspect needs to be confirmed by further work, UC-MSCs also appear capable of restoring the balance between osteoblasts and osteoclasts altered by intratibial MM expansion. The accumulation of functional MSCs within tibiae was apparently functional in interacting and stimulating the bone marrow osteoblast precursors by secreted factors that induce their differentiation into bone-building osteoblasts [20]. At the same time, UC-MSCs also restrain the osteoclast activity by secreting specific molecules [19].
Conclusions
Our model of cytotherapy appears suitable in overcoming the drawback of the high soluble TRAIL amounts injected to induce tumor suppression, which have a short half-life when systemically infused and for which previous clinical trials failed to obtain the expected results [47]. By contrast, since pIL6-TRAIL + -GFP +-UC-MSCs are committed to overexpress TRAIL only in the presence of specific cytokines secreted within the bone MM microenvironment, this cell-based therapy model would be suitable for human studies not only in controlling the marrow MM progression, but also in other osteotropic tumors since preclinical observation confirmed the biosafety of viral-transduced MSCs for TRAIL expression [48].
Acknowledgements
The authors are grateful to Marco Moschetta for X-ray analysis, and to Annalisa Savonarola and Morena d’Avenia for technical assistance.
Funding
The work was funded by a grant from the Italian Association for Cancer Research (IG grant 17536) and from the Apulia Region (‘Oncogenomic Project’ and ‘Jonico-Salentino Project’).
Availability of data and materials
Not applicable.
Abbreviations
- Ab
Antibody
- AT
Adipose tissue
- BM
Bone marrow
- DMEM
Dulbecco’s modified Eagle’s medium
- DR
Death receptor
- EGR
Early growth response
- FITC
Fluorescein isothiocyanate
- FSC
Forward scatter
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GFP
Green fluorescent protein
- GRB
Growth factor receptor-bound
- IC
Intracardially
- IL
Interleukin
- IT
Intratibially
- MEM
Modified Eagle’s medium
- MFI
Mean fluorescence intensity
- MM
Multiple myeloma
- Mo
Monoclonal
- MSC
Mesenchymal/stromal stem cell
- OD
Optical density
- P
Promoter
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- PE
Phycoerythrin
- photons/s
Photons per second
- PI
Propidium iodide
- PIRES
Poliovirus internal ribosome entry site
- q
Quantitative
- RPMI
Roswell Park Memorial Institute
- SCID
Severe combined immunodeficient
- SD
Standard deviation
- SSC
Side scatter
- TRAIL
Tumor necrosis factor related apoptosis inducing ligand
- TRITC
Tetramethylrhodamine
- UC
Umbilical cord
- WB
Western blot
Authors’ contributions
PC planned the study design, performed experiments, collected data, and wrote the manuscript. LV designed and created the vector. FM performed the in vivo study and data analysis. EP performed in vitro experiments. CC performed histological sections and immunohistochemistry. EM planned immunohistochemistry experiments and microscopy analyses of sections. CF participated in vitro and in vivo experiments, and collected and analyzed data. FS provided the idea, and wrote and revised the final version of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Human UC samples from parturients were collected after obtaining the approval of the local independent Ethic Committee (Prot. 1409/CE). All patients provided their signed informed consent. All animal experiments in this study were approved by the Institutional Animal Care and Use Committee at University of Bari Aldo Moro (Prot. n.31414-X/10) and performed according to Italian Ministry of Health guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Paola Cafforio, Email: paola.cafforio@uniba.it.
Luigi Viggiano, Email: luigi.viggiano@uniba.it.
Francesco Mannavola, Email: francesco.mannavola@gmail.com.
Eleonora Pellè, Email: eleonora.pelle89@gmail.com.
Concetta Caporusso, Email: concetta.caporusso@policlinico.ba.it.
Eugenio Maiorano, Email: eugenio.maiorano@uniba.it.
Claudia Felici, Email: claudia.felici@uniba.it.
Francesco Silvestris, Email: francesco.silvestris@uniba.it.
References
- 1.Huang S, Xu L, Sun Y, Zhang Y, Li G. The fate of systemically administrated allogeneic mesenchymal stem cells in mouse femoral fracture healing. Stem Cell Res Ther. 2015;6:206. doi: 10.1186/s13287-015-0198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker N, Sohn J, Tuan RS. Promotion of human mesenchymal stem cell osteogenesis by PI3-kinase/Akt signaling, and the influence of caveolin-1/cholesterol homeostasis. Stem Cell Res Ther. 2015;6:238. doi: 10.1186/s13287-015-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang D, Zhang H, Cao M, Tang Y, Liang J, Feng X, et al. Efficacy of allogeneic mesenchymal stem cell transplantation in patients with drug-resistant polymyositis and dermatomyositis. Ann Rheum Dis. 2011;70:1285–8. doi: 10.1136/ard.2010.141804. [DOI] [PubMed] [Google Scholar]
- 4.Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E, et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. 2010;70:3718–29. doi: 10.1158/0008-5472.CAN-09-1865. [DOI] [PubMed] [Google Scholar]
- 5.Ciavarella S, Dominici M, Dammacco F, Silvestris F. Mesenchymal stem cells: a new promise in anticancer therapy. Stem Cells Dev. 2011;20:1–10. doi: 10.1089/scd.2010.0223. [DOI] [PubMed] [Google Scholar]
- 6.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–23. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Melo SM, Bittencourt S, Ferrazoli EG, da Silva CS, da Cunha FF, da Silva FH, et al. The anti-tumor effects of adipose tissue mesenchymal stem cell transduced with HSV-Tk gene on U-87-driven brain tumor. PLoS One. 2015;10:e0128922. doi: 10.1371/journal.pone.0128922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burdon TJ, Paul A, Noiseux N, Prakash S, Shum-Tim D. Bone marrow stem cell derived paracrine factors for regenerative medicine: current perspectives and therapeutic potential. Bone Marrow Res. 2011;2011:207326. doi: 10.1155/2011/207326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horwitz EM, Dominici M. How do mesenchymal stromal cells exert their therapeutic benefit? Cytotherapy. 2008;10:771–4. doi: 10.1080/14653240802618085. [DOI] [PubMed] [Google Scholar]
- 10.Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med. 2013;45:e54. doi: 10.1038/emm.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SM, Woo JS, Jeong CH, Ryu CH, Jang JD, Jeun SS. Potential application of temozolomide in mesenchymal stem cell-based TRAIL gene therapy against malignant glioma. Stem Cells Transl Med. 2014;3:172–82. doi: 10.5966/sctm.2013-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Su D, Xuan S, Ma G, Dai Z, Liu T, et al. Gene therapy of gastric cancer using LIGHT-secreting human umbilical cord blood-derived mesenchymal stem cells. Gastric Cancer. 2013;16:155–66. doi: 10.1007/s10120-012-0166-1. [DOI] [PubMed] [Google Scholar]
- 13.Kidd S, Caldwell L, Dietrich M, Samudio I, Spaeth EL, Watson K, et al. Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy. 2010;12:615–25. doi: 10.3109/14653241003631815. [DOI] [PubMed] [Google Scholar]
- 14.Krasikova LS, Karshieva SS, Cheglakov IB, Belyavsky AA. Mesenchymal stem cells expressing cytosine deaminase inhibit growth of murine melanoma B16F10 in vivo. Mol Biol (Mosk) 2015;49:1007–15. doi: 10.1134/S0026893315060126. [DOI] [PubMed] [Google Scholar]
- 15.Yuan Z, Kolluri KK, Sage EK, Gowers KH, Janes SM. Mesenchymal stromal cell delivery of full-length tumor necrosis factor-related apoptosis-inducing ligand is superior to soluble type for cancer therapy. Cytotherapy. 2015;17:885–96. doi: 10.1016/j.jcyt.2015.03.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SM, Lim JY, Park SI, Jeong CH, Oh JH, Jeong M, et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68:9614–23. doi: 10.1158/0008-5472.CAN-08-0451. [DOI] [PubMed] [Google Scholar]
- 17.Lin HD, Fong CY, Biswas A, Choolani M, Bongso A. Human Wharton’s jelly stem cells, its conditioned medium and cell-free lysate inhibit the growth of human lymphoma cells. Stem Cell Rev. 2014;10:573–86. doi: 10.1007/s12015-014-9514-3. [DOI] [PubMed] [Google Scholar]
- 18.Ciavarella S, Caselli A, Tamma AV, Savonarola A, Loverro G, Paganelli R, et al. A peculiar molecular profile of umbilical cord-mesenchymal stromal cells drives their inhibitory effects on multiple myeloma cell growth and tumor progression. Stem Cells Dev. 2015;24:1457–70. doi: 10.1089/scd.2014.0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Ling W, Pennisi A, Wang Y, Khan S, Heidaran M, et al. Human placenta-derived adherent cells prevent bone loss, stimulate bone formation, and suppress growth of multiple myeloma in bone. Stem Cells. 2011;29:263–73. doi: 10.1002/stem.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Ling W, Khan S, Yaccoby S. Therapeutic effects of intra bone and systemic mesenchymal stem cell cytotherapy on myeloma bone disease and tumor growth. J Bone Miner Res. 2012;27:1635–48. doi: 10.1002/jbmr.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–55. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeong JA, Hong SH, Gang EJ, Ahn C, Hwang SH, Yang IH, et al. Differential gene expression profiling of human umbilical cord blood-derived mesenchymal stem cells by DNA microarray. Stem Cells. 2005;23:584–93. doi: 10.1634/stemcells.2004-0304. [DOI] [PubMed] [Google Scholar]
- 23.Carter A, Merchav S, Silvian-Draxler I, Tatarsky I. The role of interleukin-1 and tumour necrosis factor-alpha in human multiple myeloma. Br J Haematol. 1990;74:424–31. doi: 10.1111/j.1365-2141.1990.tb06330.x. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich-Breiholz O, Schneider H, et al. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun pathway. J Biol Chem. 2008;283:12120–8. doi: 10.1074/jbc.M800583200. [DOI] [PubMed] [Google Scholar]
- 25.Cives M, Quaresmini D, Rizzo FM, Felici C, D’Oronzo S, Simone V, et al. Osteotropism of neuroendocrine tumors: role of the CXCL12/CXCR4 pathway in promoting EMT in vitro. Oncotarget. 2017;8:22534–49. doi: 10.18632/oncotarget.15122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin EC, Aarons L, Yates JW. Accounting for dropout in xenografted tumour efficacy studies: integrated endpoint analysis, reduced bias and better use of animals. Cancer Chemother Pharmacol. 2016;78:131–41. doi: 10.1007/s00280-016-3059-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bentzon JF, Stenderup K, Hansen FD, Schroder HD, Abdallah BM, Jensen TG, et al. Tissue distribution and engraftment of human mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene. Biochem Biophys Res Commun. 2005;330:633–40. doi: 10.1016/j.bbrc.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Fan D, Xiong D. Mesenchymal stem cells as delivery vectors for anti-tumor therapy. Stem Cell Investig. 2015;2:6. doi: 10.3978/j.issn.2306-9759.2015.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramdasi S, Sarang S, Viswanathan C. Potential of mesenchymal stem cell based application in cancer. Int J Hematol Oncol Stem Cell Res. 2015;9:95–103. [PMC free article] [PubMed] [Google Scholar]
- 30.Ciavarella S, Grisendi G, Dominici M, Tucci M, Brunetti O, Dammacco F, et al. In vitro anti-myeloma activity of TRAIL-expressing adipose-derived mesenchymal stem cells. Br J Haematol. 2012;157:586–98. doi: 10.1111/j.1365-2141.2012.09082.x. [DOI] [PubMed] [Google Scholar]
- 31.Yan C, Yang M, Li Z, Li S, Hu X, Fan D, et al. Suppression of orthotopically implanted hepatocarcinoma in mice by umbilical cord-derived mesenchymal stem cells with sTRAIL gene expression driven by AFP promoter. Biomaterials. 2014;35:3035–43. doi: 10.1016/j.biomaterials.2013.12.037. [DOI] [PubMed] [Google Scholar]
- 32.Yan C, Li S, Li Z, Peng H, Yuan X, Jiang L, et al. Human umbilical cord mesenchymal stem cells as vehicles of CD20-specific TRAIL fusion protein delivery: a double-target therapy against non-Hodgkin’s lymphoma. Mol Pharm. 2013;10:142–51. doi: 10.1021/mp300261e. [DOI] [PubMed] [Google Scholar]
- 33.Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009;69:4134–42. doi: 10.1158/0008-5472.CAN-08-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guiho R, Biteau K, Grisendi G, Taurelle J, Chatelais M, Gantier M, et al. TRAIL delivered by mesenchymal stromal/stem cells counteracts tumor development in orthotopic Ewing sarcoma models. Int J Cancer. 2016;139:2802–11. doi: 10.1002/ijc.30402. [DOI] [PubMed] [Google Scholar]
- 35.Fong CY, Chak LL, Biswas A, Tan JH, Gauthaman K, Chan WK, et al. Human Wharton’s jelly stem cells have unique transcriptome profiles compared to human embryonic stem cells and other mesenchymal stem cells. Stem Cell Rev. 2011;7:1–16. doi: 10.1007/s12015-010-9166-x. [DOI] [PubMed] [Google Scholar]
- 36.Gauthaman K, Fong CY, Suganya CA, Subramanian A, Biswas A, Choolani M, et al. Extra-embryonic human Wharton’s jelly stem cells do not induce tumorigenesis, unlike human embryonic stem cells. Reprod Biomed Online. 2012;24:235–46. doi: 10.1016/j.rbmo.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Bai J, Ji X, Li R, Xuan Y, Wang Y. Comprehensive characterization of four different populations of human mesenchymal stem cells as regards their immune properties, proliferation and differentiation. Int J Mol Med. 2014;34:695–704. doi: 10.3892/ijmm.2014.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Hirai M, Cantero S, Ciubotariu R, Dobrila L, Hirsh A, et al. Isolation and characterization of mesenchymal stem cells from human umbilical cord blood: reevaluation of critical factors for successful isolation and high ability to proliferate and differentiate to chondrocytes as compared to mesenchymal stem cells from bone marrow and adipose tissue. J Cell Biochem. 2011;112:1206–18. doi: 10.1002/jcb.23042. [DOI] [PubMed] [Google Scholar]
- 39.Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA, Mohapatra G, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106:4822–7. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nowakowski A, Drela K, Rozycka J, Janowski M, Lukomska B. Engineered mesenchymal stem cells as an anti-cancer trojan horse. Stem Cells Dev. 2016;25:1513-31. [DOI] [PMC free article] [PubMed]
- 41.Cao Y, Luetkens T, Kobold S, Hildebrandt Y, Gordic M, Lajmi N, et al. The cytokine/chemokine pattern in the bone marrow environment of multiple myeloma patients. Exp Hematol. 2010;38:860–7. doi: 10.1016/j.exphem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Lacy MQ, Donovan KA, Heimbach JK, Ahmann GJ, Lust JA. Comparison of interleukin-1 beta expression by in situ hybridization in monoclonal gammopathy of undetermined significance and multiple myeloma. Blood. 1999;93:300–5. [PubMed] [Google Scholar]
- 43.Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84:114–22. doi: 10.4065/84.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donovan KA, Lacy MQ, Gertz MA, Lust JA. IL-1beta expression in IgM monoclonal gammopathy and its relationship to multiple myeloma. Leukemia. 2002;16:382–5. doi: 10.1038/sj.leu.2402374. [DOI] [PubMed] [Google Scholar]
- 45.Fryer RA, Graham TJ, Smith EM, Walker-Samuel S, Morgan GJ, Robinson SP, et al. Characterization of a novel mouse model of multiple myeloma and its use in preclinical therapeutic assessment. PLoS One. 2013;8:e57641. doi: 10.1371/journal.pone.0057641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menon LG, Picinich S, Koneru RM, Gao H, Lin SY, Koneru M, et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007;25:520–8. doi: 10.1634/stemcells.2006-0257. [DOI] [PubMed] [Google Scholar]
- 47.Hotte SJ, Hirte HW, Chen EX, Siu LL, Le LH, Corey A, et al. A phase 1 study of mapatumumab (fully human monoclonal antibody to TRAIL-R1) in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:3450–5. doi: 10.1158/1078-0432.CCR-07-1416. [DOI] [PubMed] [Google Scholar]
- 48.Choi SA, Yun JW, Joo KM, Lee JY, Kwak PA, Lee YE, et al. Preclinical biosafety evaluation of genetically modified human adipose tissue-derived mesenchymal stem cells for clinical applications to brainstem glioma. Stem Cells Dev. 2016;25:897–908. doi: 10.1089/scd.2015.0324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.