

Abstract
Cardiosiderosis is a leading cause of mortality in transfusion-dependent thalassemias. Plasma non-transferrin-bound iron and its redox-active component, labile plasma iron, are key sources of iron loading in cardiosiderosis. Risk factors were identified in 73 patients with or without cardiosiderosis. Soluble transferrin receptor-1 levels were significantly lower in patients with cardiosiderosis (odds ratio 21). This risk increased when transfusion-iron loading rates exceeded the erythroid transferrin uptake rate (derived from soluble transferrin receptor-1) by >0.21 mg/kg/day (odds ratio 48). Labile plasma iron was >3-fold higher when this uptake rate threshold was exceeded, but non-transferrin-bound iron and transferrin saturation were comparable. The risk of cardiosiderosis was decreased in patients with low liver iron, ferritin and labile plasma iron, or high bilirubin, reticulocyte counts or hepcidin. We hypothesized that high erythroid transferrin uptake rate decreases cardiosiderosis through increased erythroid re-generation of apotransferrin. To test this, iron uptake and intracellular reactive oxygen species were examined in HL-1 cardiomyocytes under conditions modeling transferrin effects on non-transferrin-bound iron speciation with ferric citrate. Intracellular iron and reactive oxygen species increased with ferric citrate concentrations especially when iron-to-citrate ratios exceeded 1:100, i.e. conditions favoring kinetically labile monoferric rather than oligomer species. Excess iron-binding equivalents of apotransferrin inhibited iron uptake and decreased both intracellular reactive oxygen species and labile plasma iron under conditions favoring monoferric species. In conclusion, high transferrin iron utilization, relative to the transfusion-iron load rate, decreases the risk of cardiosiderosis. A putative mechanism is the transient re-generation of apotransferrin by an active erythron, rapidly binding labile plasma iron-detectable ferric monocitrate species.
Introduction
Cardiosiderosis or myocardial hemosiderosis (MH) is a leading cause of mortality in transfusion-dependent thalassemias.1 Suggested risk factors for MH and/or consequent cardiomyopathy have included: sustained high serum ferritin levels,2 high liver iron concentrations (LIC),3 poor adherence to chelation therapy,4 as well as genetic susceptibility factors.5 However, while improved monitoring by magnetic resonance imaging and better use of iron chelators6 have led to falling frequencies of MH, as judged by cardiac magnetic resonance studies, there remains a variable prevalence between populations7 and between individuals that is not fully understood.
The conduit through which MH develops is plasma non-transferrin-bound iron (NTBI), as transferrin-mediated iron uptake by cardiomyocytes is relatively ‘insignificant’.8 NTBI is detectable as chelatable iron9,10 or redox-active iron (labile plasma iron, LPI)11 at transferrin saturations >75% for NTBI12 or 100% in the case of LPI.13 Animal data suggest that NTBI is taken into the myocardium through L-type calcium channels,14 and are supported by recent clinical data indicating that MH is inhibited in transfusion-dependent thalassemia patients by the calcium channel blocker amlodipine.15 Plasma NTBI is not a single entity, however, being a heterogeneous, multispeciated pool of ferric iron (not bound to high-affinity transferrin iron-binding sites) containing monomeric, oligomeric, and polymeric iron citrate species16 with weak albumin binding,17 or stronger binding, where post-translational modifications to albumin occur.18 There are a number of potential non-protein ligands for NTBI, including phosphate, acetate, amino acids, pyrophosphate, and citrate. However, phosphate, acetate, and amino acids cannot compete with the hydroxyl ion for iron(III) at pH 7.4. Pyrophosphate is potentially a potent iron(III) ligand but has a vanishingly small concentration in plasma after accounting for the effect of magnesium and calcium binding, thus citrate is the dominant ligand. The iron-to-citrate ratio determines the mix of species present. As the plasma citrate concentration is roughly constant at 100 μM, the iron-to-citrate ratio is determined by the plasma NTBI concentration. At 1 μM NTBI the citrate excess is 100-fold and chelatable iron citrate species predominate, whereas, at ≥10 μM NTBI, the proportion of chelatable iron drops substantially.17,19 LPI refers to the redox-active fraction of NTBI, hence its term ‘labile’, which is chelatable,20 and has been implicated in organ hemosiderosis.21 Its chemical nature is not characterized, although it is predicted to comprise both monomeric and oligomeric ferric citrate, albumin iron complexes, and possibly partially coordinated iron chelates, e.g. of deferiprone,22 which are able to form hydroxyl radicals in the presence of ascorbate and hydrogen peroxide.
The balance between the rate of transferrin iron utilization by the erythron and the transfusion iron loading rate is key to determining levels of plasma NTBI.23 Blood transfusion delivers a mean of 0.4 mg/kg/day but with a wide range (0.2–0.6 mg/kg/day),24 exceeding by 10-fold the gut iron loading rate seen in non-transfusion-dependent thalassemia.25 Transfused red cells are ultimately catabolized within macrophages of the spleen, liver, and bone marrow, with iron released onto transferrin and - when the latter approaches saturation - forming plasma NTBI. Transferrin iron uptake by the erythron via transferrin receptor-1 (TfR1) liberates iron from transferrin during receptor-mediated endocytosis, whereupon iron-free apotransferrin is recycled back to plasma, as described in detail by Gkouvatsos et al.26 The extracellular domain of TfR1 is shed by red cell progenitors and circulates bound to holotransferrin (sTfR1).27 TfR1 expression in the erythron is transcriptionally regulated such that levels increase in iron deficiency.28 However, in thalassaemias, sTfR1 levels primarily reflect the degree of expansion of the erythron.29–31 Blood transfusion suppresses expansion and activity of the erythron, thereby decreasing sTfR1 proportionately to the pre-transfusion hemoglobin.32
In this study, we examined clinical factors associated with MH as well as the mechanisms underlying these associations. In particular, we focused on how transferrin-iron utilization by the erythron marked by sTfR1, relative to the transfusion-iron loading rate, affects the risk of MH. We considered how apotransferrin formed after endocytosis in the bone marrow may bind NTBI species, decreasing their availability for myocardial uptake.
Methods
Patients
A cohort of 73 transfusion-dependent thalassemia patients on deferasirox, with known transfusion-iron load rate,24 was divided into those with MH (n=24, cardiac T2*<20 ms, R2*>50 s−1) and those without MH (n=49, T2*>20 ms, R2* <50 s−1).33 Details are provided in the Online Supplement. Patients gave written informed consent to participate in the study and approval was obtained from The Joint University College London/University College London Hospitals Committees on Ethics of Human Research.
Biomarkers
Pre-transfusion samples were tested, after a 48-h washout from deferasirox chelation, for iron metabolism and routine biochemical variables, and compared in both groups (±MH). The tests included assays for NTBI,9 LPI,11 hepcidin, urea-gel transferrin saturation, sTfR1, and growth differentiation factor 15 (GDF-15), as published previously,23 as well as routine clinical variables (hemoglobin, absolute reticulocyte count, nucleated red cells, serum ferritin, bilirubin, and serum iron), as described in detail in the Online Supplement.
Cell-line experiments
Murine HL-1 cardiomyocytes (ATCC number CRL-12197) were grown in Claycomb medium (Sigma); the culture protocol is described in the Online Supplement. Buffered ferric citrate17,16 or ferric ammonium citrate (FAC) was used to model NTBI. Ferric nitrilotriacetate was used to saturate transferrin. Total cellular iron was assayed by ferrozine. The level of cytosolic reactive oxygen species, tested using 2,7-dichlorofluorescein diacetate, was calculated from slopes of fluorescence-versus-time data. Detailed methods are provided in the Online Supplement.
Statistics
Continuous data are presented as mean ± SD or median ± interquartile range and compared using the t-test or nonparametric tests, depending on assumed distribution, unless otherwise specified. Categorical variables are compared using the χ2-test. A P value <0.05 is considered statistically significant.
Results
Factors associated with myocardial hemosiderosis
To determine factors associated with MH, biomarkers were compared in transfusion-dependent thalassemia patients with (n=24) and without (n=49) MH. The sTfR1 emerged as the factor most significantly associated with MH (P<0.001, Figure 1A–C) being >3-fold higher in controls than in patients with MH (medians 4.06 versus 1.26 μg/mL, P=0.0005) with an area-under-curve for the receiver operating characteristic curve (AUCROC) of 0.8 (P=0.0004). A threshold of 1.77 μg/mL was predictive of MH with 89.7% sensitivity and 64.7% specificity (normal range for sTfR1: 0.3–1.65 μg/mL). Other biomarkers significantly associated with MH were bilirubin, reticulocyte count, hepcidin, LIC, and serum ferritin (Table 1). Although the median LPI differed insignificantly between patients with and without MH, a cut-off threshold of >0.31 μM was significantly associated with MH (χ2 test, P=0.04). Differences in NTBI, LPI, transferrin saturation (TfSat), and transfusion iron load rate (ILR) were insignificant (Figure 1D–G), and no such thresholds were found for NTBI or TfSat.
Figure 1.

Biomarkers in patients with and without myocardial siderosis. Comparison of means and medians in patients with and without cardiac iron, MH(+) and MH(-), respectively, shown as box and whisker (min-max) plots. (A) Soluble transferrin receptor-1, sTfR1: 4.06 vs. 1.26 μg/mL, MW P=0.0005. (B) Receiver operating characteristic (ROC) curve for sTfR1, AUCROC=0.8±0.07 (P=0.0004). (C) Plot of cardiac R2* vs. sTfR1, Spearman correlation coefficient r = –0.61, P<0.0001. (D) Non-transferrin-bound iron, NTBI: 2.86 vs. 2.7 μM, MW P=0.59. (E) Transferrin saturation, TfSat: 100 vs. 100%, MW P=0.94. (F) Labile plasma iron, LPI: 0.1 vs. 0.27 μM, MW P=0.13. (G) Transfusion-iron load rate, ILR: 0.32 vs. 0.35 mg Fe/kg/day, t-test P=0.19. (H) Growth-differentiation factor-15, GDF-15: 6702 vs. 4430 pg/mL, MW P=0.4 (I) Plasma hepcidin 6.8 vs. 14.3 nM, MW P=0.04. MW, Mann-Whitney test; AUC, area under curve, MH, myocardial hemosiderosis.
Table 1.
Significant clinical and laboratory variables associated with myocardial hemosiderosis in transfusion-dependent thalassemias.
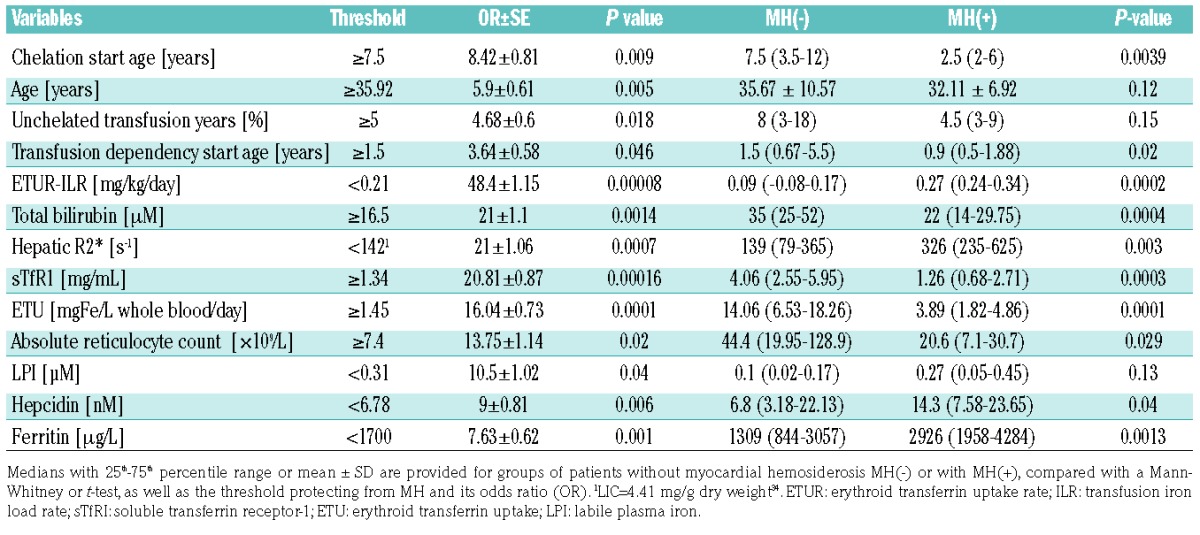
High LIC and, to a lesser extent, serum ferritin were also significant risk factors for MH in our study, which is of interest, as this was not seen in earlier studies.33 However, determination of liver iron in earlier studies was not optimal34 and many patients had recently undergone intesive iron chelation which removes iron faster from the liver than from the heart,35 thereby obscuring such a relationship.36 A further significant, though weak, risk factor for MH was high plasma hepcidin. Hepcidin did not correlate with markers of iron overload such as LIC (as hepatic R2*, P=0.4), or ferritin (P=0.52), but was inversely and significantly correlated with sTfR1 (Spearman r=−0.54, P<0.0001). sTfR1 did not correlate with LIC or ferritin (P=0.4 and P=0.52, respectively; Online Supplementary Figure S1). Associations between MH an nucleated red cells, total serum iron, age, weight, ILR, TfSat, and GDF-15 were insignificant.
Relationship of transferrin-iron utilization to myocardial hemosiderosis in transfusion-dependent thalassemia
To gain insight into the relationship between MH and sTfR1, we utilized understandings from ferrokinetic studies29,30 which established the erythron transferrin uptake (ETU) as proportional to plasma sTfR1. The regression equation (r2=0.84) for ETU was calculated from published data:29
Assuming that the relationship remains temporally stable, sTfR1 represents erythropoiesis quantitatively, given that plasma levels are proportionate to tissue transferrin receptors of the whole organism,31 of which erythroid cells are the main dynamic component. ETU can be expressed as a rate corrected for estimated blood volume (BV) [70 mL/kg (males), 65 mL/kg (females)] and the patient’s weight (erythroid transferrin uptake rate, ETUR).
Although transfusion does not directly relate to cardiac iron, when plasma sTfR1 levels were expressed as ETUR (i.e. transferrin-iron utilization rate) and compared to the transfusion ILR (Figure 2A–C), MH was present only when the ‘net ILR’ (ILR-ETUR) exceeded 0.21 mg/kg/day (P<0.001). Above and below this threshold, LIC,34 TfSat, and NTBI were similar (9.7 versus 10.1 mg/g dry weight, 96 versus 98%, 3 versus 2.6 μM, P=ns), while LPI was >3-fold higher (0.35 versus 0.1 μM, P=0.01). Thus, LPI was high only in patients in whom the net ILR exceeded 0.21 mg/kg/day, whereas no differences were seen in LIC, TfSat, and NTBI between patients with and without MH, even when adjusted for the net ILR (Figure 2C).
Figure 2.

Balance between transfusion-iron load rate and erythroid transferrin uptake rate derived from soluble transferrin receptor-1 relates to cardiac iron. (A) Cardiac R2* plotted against transfusion-iron load rate (ILR), gray circles mark patients with cardiac iron (cR2*>50 s−1), open circles – patients without cardiac iron; no relationship overall. (B) Cardiac R2* (cR2*) plotted against ILR, open circles mark patients with cardiac iron (cR2*>50 s−1), gray circles – patients without cardiac iron; no relationship overall, relationship present in patients with cardiac iron only, r2=0.61; points differ in size according to sTfR1 level [μg/mL], see inset. (C) same as in panel B but the x-axis shows ILR corrected for utilization rate derived from sTfR1 according to Beguin et al. 199329 (ETU[μmol/L/day]=0.013×sTfR1 [μg/L]+2.25; ETUR[mg/kg/day]=ETU×55.845[g/mol]/1000×blood volume[mL]/1000/body weight[kg]). Highly discriminant threshold 0.21mg/kg/day P<0.0001, 100% sensitive, 83% specific for myocardial hemosiderosis (positive predictive value 71%, negative predictive value 100%) above and below which the liver iron concentration (LIC), transferrin saturation (TfSat) and non-transferrin-bound iron (NTBI) do not differ (P=ns) but labile plasma iron is 0.35 μM vs. 0.1 μM P=0.01, respectively. (D) same as panel C but points differ in size according to total body iron derived from LIC values.
This suggests a link between iron clearance from transferrin - a process implying the formation of transient levels of apotransferrin - and the low propensity to MH in transfusion-dependent thalassemia. We hypothesized that increased transient local concentrations of apotransferrin in the marrow and circulating reticulocytes37 could decrease the uptake of NTBI into the myocardium. With similar TfSat and NTBI in MH-positive and MH-negative patients, this effect of recycled apotransferrin was expected to influence the speciation of NTBI.
Effects of iron-to-citrate ratio on iron detection in the labile plasma iron assay
As LPI was found to differ between MH-positive and MH-negative patients, we wished to characterize which fraction of NTBI was detected by the LPI assay. In particular, we wished to determine whether LPI was most detectable under conditions in which either monomer or oligomer ferric citrate species predominated. As shown in Figure 3A, at constant citrate concentrations (100 μM), the proportion of iron detectable in the LPI assay decreases as iron concentration increases. Conversely, LPI is proportionately most detectable at 1000-fold citrate excesses. Under these clinically relevant conditions, the citrate excess favors the formation of monomer rather than oligomer species. As LPI was associated with an increased risk of MH in our clinical observations, these findings suggest that the monomer LPI might be the species most readily taken into myocardial cells.
Figure 3.

Effect of ferric citrate speciation on detectability of labile plasma iron, total cellular iron and transferrin binding. (A) Labile plasma iron (LPI) as a percentage of ferric citrate with constant citrate (100 μM) and variable iron (0–10 μM, shown next to data points in μM), as a function of excess citrate (fold), representative of three experiments. This illustrates what proportion of a given ratio species is LPI-detectable. (B) Total cellular iron dose response in confluent HL-1 cells to 24 h incubation in Claycomb medium (CM) with ferric citrate in MOPS pH=7.4 at variable Fe:citrate ratios and constant citrate at 100 μM, r2=0.95, EC50=0.31 μM, EC90=1.09 μM; shown as mean±SD, n=6. (C) Percentage of ferric citrate iron that binds to 35.5 μM apotransferrin (ApoTf) to form ferrotransferrin (over 2 h incubation at 37°C) as a function of citrate:Fe ratio. 0–30 μM iron and 100 μM citrate were increased 25.5-fold to keep the same ratio (see legend: 0–765 μM and 2550 μM f.c. respectively) in order to better resolve absorbance peaks of holotransferrin formation (at 465 nm, inset), and compared to 35.5 μM 100% saturated ferrotransferrin absorbance of 1.69. Percentage of iron bound to transferrin was calculated from transferrin iron content/nominal concentrations prepared (see inset, mean±SD, n=2). (D) Apotransferrin-dependent inhibition of LPI in ferric citrate. Apotransferrin (ApoTf) with 25 mM bicarbonate was added at 0–10 μM (f.c.) together with ferric citrate, subsequent DHR oxidation was followed up for 1 h. LPI values are interpolated from the standard curve, mean±SD, n=3. (E) HL-1 cells grown to confluence and incubated for 24 h in CM with 1 μM ferric ammonium citrate (FAC) ±0–2.5 μM ApoTf; ANOVA P<0.001, mean±SD, n=2, Bonferroni post-test significant for ApoTf effect in the FAC group. Global fit r2=0.91, ApoTf IC50=0.81 μM, curves significantly different (P<0.0001). (F) Transferrin saturation-dependent model of NTBI uptake (as FAC) into HL-1 cells grown to confluence then incubated in CM with apotransferrin and 100% saturated holotransferrin at relevant ratios to obtain a 37.5 μM TfSat model ±5 μM FAC; difference vs. control (no NTBI) only detectable for TfSat=100%, i.e. when no apotransferrin is present (multiple t-test with Holm-Sidak correction for multiple comparisons, P=0.004, mean±SD, n=6).
Effect of iron-to-citrate ratio on iron uptake into HL-1 cardiomyocytes
To confirm this interpretation, we examined iron uptake in HL-1 cardiomyocytes at varying iron-to-citrate ratios. Figure 3B shows the effect of increasing ferric iron at constant citrate concentration (100 μM) on iron uptake into HL-1 cells at 24 h. A clear plateau effect is seen whereby iron uptake does not increase further when citrate exceeds iron by less than 100-fold. Under these conditions oligomer rather than monomer ferric citrate species are favored.17 This suggests that oligomer iron citrate species are less available than the monomer species for uptake by cardiomyocytes.
Effect of iron-to-citrate ratio on iron binding to transferrin
We also wished to determine how the iron-to-citrate ratio affected the availability of ferric iron to bind transferrin. Apotransferrin (at 35.5 μM or 71 μM iron binding equivalents, IBE) was incubated with buffered ferric citrate at 0 to 4000-fold citrate excess (Figure 3C). Iron binding to transferrin, determined by spectroscopy, increased to a maximum of 40% as the citrate excess increased. Thus binding of iron to transferrin is more rapid under conditions favoring monomer ferric citrate species than under conditions in which oligomers predominate.
Low (nanomolar) concentrations of apotransferrin substantially decrease labile plasma iron
The preferential binding of the monomer ferric citrate species by transferrin was predicted to decrease those same species responsible for activity in the LPI assay. We tested this; the results are shown in Figure 3D. The control without transferrin showed increasing LPI values with increasing ferric iron concentration (at a constant 100 μM concentration of citrate). When apotransferrin was added at remarkably low concentrations (30 nM or 60 nM IBE), LPI detectability decreased substantially across all ratios. This effect exceeded results expected from simple stoichiometric binding. A higher concentration of apotransferrin (10 μM) completely abrogated LPI detectability up to 10 μM iron. These findings show that the redox-active iron species responsible for the majority of LPI detectability is present at a very low concentration (nanomolar). This species is most likely to be ferric monocitrate because apotransferrin is known to bind ferric monocitrate most avidly.
Effect of apotransferrin on HL-1 cardiomyocyte iron uptake from ferric citrate species
Having established the conditions favoring LPI detectability and apotransferrin binding of ferric citrate species, we wanted to test whether these same conditions are those that most inhibit iron uptake in HL-1 cardiomyocyte cells.
We first examined the effect of transferrin on uptake from FAC, which is a stable form of monomeric ferric iron coordinated by two citrate molecules.38 Figure 3E shows that iron uptake from 1 μM FAC was almost completely inhibited by apotransferrin at 1 μM (2 μM IBE). Figure 3F illustrates the effect of a constant transferrin concentration with varying percentage saturations on iron uptake from 5 μM FAC at 24 h. Uptake was completely inhibited by physiologically relevant transferrin concentrations of 37 μM (74 μM IBE) at saturations below 99%.
We then wished to examine the inhibitory effect of apotransferrin on iron uptake at varying iron-to-citrate ratios. In order to study short-time intervals we developed an intracellular reactive oxygen species assay and validated this against total cellular iron at 24 h (Figure 4A). The control iron uptake marked by intracellular reactive oxygen species at 60 min in Figure 4B (closed circles) showed a plateau, as previously noted for total iron uptake at 24 h (Figure 3B). Thus, under conditions in which oligomer species increasingly predominate, uptake into HL-1 cells forms a plateau. In the same experiment (Figure 4B, triangles), the addition of 5 μM apotransferrin inhibited uptake up to the point where ferric citrate approached 10 μM, at which concentration iron uptake exceeded that from control. Importantly, this contrasts with the effects of apotransferrin on iron uptake from FAC (Figure 3F) where uptake was inhibited at every apotransferrin concentration. The key difference between Figure 3B,E and F is the form of iron citrate presented to the cells. Freshly prepared FAC is a fully coordinated monomeric iron dicitrate,38 whereas at the 1:10 iron-to-citrate ratio used in the experiments shown in Figure 3B, mixtures of oligomer and monomer iron species were present.17,16
Figure 4.

Effect of apotransferrin and citrate speciation on the iron uptake marked by cytosolic reactive oxygen species. (A) The relationship of intracellular reactive oxygene species (ROS) levels by the DCF method and total cellular iron by the ferrozine method, r=0.91, P<0.0001; Deming regression slope 1.14×10−5±1.68×10−6, intercept 2.69×10−5±5.08×10−6, mean±SD, n=6. (B) Cytosolic ROS levels in HL-1 cardiomyocytes as a function of control ferric citrate (iron 0–30 μM, citrate 100 μM) in MOPS pH=7.4 with (triangles) and without (circles) 5 μM apotransferrin (ApoTf) in 25 mM bicarbonate; mean±SD, n=5. (C) Dose response of cytosolic ROS levels in HL-1 cardiomyocytes to 95% saturated transferrin (0.37–15 μM) ±100 μM iron-binding equivalents of CP40, an extracellular chelator, mean±SD, n=4. (D) Cytosolic ROS formation in HL-1 cardiomyocytes showing recent uptake of iron from predominantly oligomer species (10 μM iron, 100 μM citrate) or control (citrate only) under dose-response effect of 0–16 μM apotransferrin in 30 mM bicarbonate/phosphate-buffered saline; mean±SD, n=4.
The increased uptake for apotransferrin shown in Figure 4B might be interpreted superficially as increased uptake from holotransferrin, formed from the addition of apotransferrin to iron citrate. However control experiments, including those illustrated in Figure 4C and Online Supplementary Figure S2, showed that incubation of HL-1 cardiomyocytes with holotransferrin does not increase iron uptake: increasing concentrations of 95%-saturated transferrin actually inhibited iron accumulation.
An alternative explanation for the findings presented in Figure 4B is that the interaction of apotransferrin with iron citrate alters the proportions of citrate species. To test this hypothesis further, we examined the iron uptake at increasing concentrations of apotransferrin but constant iron citrate concentrations that favor oligomer species (10 μM ferric iron and 100 μM citrate) (Figure 4D). Apotransferrin concentrations between 1 and 8 μM increased iron uptake but at higher apotransferrin concentrations (8 μM or 16 μM) uptake was inhibited. This is more consistent with catalytic depolymerization of oligomers to monomers as elaborated in the discussion.
We therefore suggest that the presence of apotransferrin favors the formation of citrate species that are more rapidly taken into cells, namely monomeric iron species. Only when the iron binding capacity of apotransferrin exceeds the iron content of the ferric citrate (here above about 10 μM), is iron uptake inhibited. We conclude that speciation of iron citrate is critical to cardiomyocyte iron uptake and that apotransferrin alters this speciation.
Discussion
In this stduy we sought to identfy risk factors for MH in transfused thalassemia patients. A key novel finding is that low levels of sTfR1 appear to be a powerful predictor for MH with an odds ratio of 21. It is unlikely that sTfR1 has a direct mechanistic role in iron distribution, as mice transfected with sTfR1 showed similar iron absorption and hepcidin to controls.39 Furthermore, the circulating concentration of sTfR1 (nanomolar) is three logs less than that of diferric transferrin and thus unlikely to be a significant receptor trap for transferrin iron utilization. We also found a clear relationship between the transfusion-iron loading rate and myocardial iron, as determined by magnetic resonance imaging (mR2*), in MH-positive patients (Figure 2B). This suggests that the balance between the transfusion iron-loading rate and erythron iron utilization might be key to MH. We therefore developed a model building on data linking sTfR1 to quantitate iron utilization by the erythron (the ETUR),29,30 and found that the difference between the ILR and the ETUR predicted MH with an odds ratio of 48. We identified a threshold for this difference of 0.21 mg/kg/day, above which MH was more likely and below which MH was not seen (Figure 2C), even in the presence of high total body iron (Figure 2D). Hence a high transfusion iron intake relative to endogenous erythropoiesis puts patients at increased risk of MH.
The high transfusion iron intake relative to endogenous erythropoiesis as a risk for MH is supported by other clinical observations. Additional factors that we found correlated with lower MH risk, such as high bilirubin concentration and high reticulocyte count, are also consistent with pronounced activity of the erythron. Others have shown that a reticulocyte count below 5% in sickle cell disease actually predicted premature development of cardiac iron deposition.40 Erythropoietin, although not measured in this cohort, would be predicted to negatively correlate with MH because it drives sTfR1 levels29 and correlates with them closely in transfusion-dependent thalassemia. The relationship of age at onset of transfusion dependence to MH suggests that an initial expansion of the bone marrow, by under-transfusion or late introduction of transfusion, is hard to suppress even after many subsequent years of hypertransfusion (Online Supplementary Figure S3). Others have indirectly linked the absence of MH with extramedullary hematopoiesis in transfused thalassemia patients, using extramedullary masses by magnetic resonance imaging as surrogate markers of erythroid mass,41 which also proves the point that even life-long hypertransfusion does not suppress erythropoiesis completely. A high risk of MH has also been identified in another transfusion-dependent condition with low iron utilization and extremely low sTfR1, namely Diamond-Blackfan anemia.23,42 Conversely, non-transfusion-dependent thalassemia patients have very high sTfR1 levels13 with high ineffective erythropoiesis but a low risk of MH,43,44 despite substantial iron overload. Factors in addition to iron uptake may influence net MH. For example, our findings of increased plasma hepcidin in MH-positive patients are consistent with recent work in animal models of iron overload implicating iron exit through cardiac ferroportin channels as important to myocardial iron retention.45,46 There was a significant relationship between hepcidin and sTfR1, but not with GDF-15, nonetheless it would be of value to look at this relationship with erythroferrone when the assay becomes available.
What is the role of the LPI fraction of NTBI in MH risk? A notable feature of non-transfusion-dependent thalassemias, found elsewhere in a large cohort of 155 patients, is that, even in the presence of high levels of body iron and raised NTBI, LPI is typically within the normal range, while the few cases with increased LPI have no apotransferrin.13 This is consistent with this study’s findings in trasfusion-dependent patients in whom a >3-fold difference in LPI was seen across the ILR-ETUR threshold, while plasma NTBI levels and transferrin saturations were indistinguishable (Figure 2C). This suggests that LPI-detectable species are those taken up by cardiomyocytes, consistent with previous clinical observations linking LPI-detectable iron to MH.21
As the LPI assay detects only the redox-active sub-fraction of NTBI, we wished to determine whether LPI-detectable NTBI species were those particularly prone to cardiomyocyte uptake. We used buffered ferric citrate as a model for NTBI at a constant citrate concentration of 100 μM to represent plasma concentration. Detectability of iron by the LPI assay was less using iron citrate as the source of iron than ferric nitrilotriacetate (Online Supplementary Figure S4), indicating that only a proportion of ferric citrate species is detectable by the LPI assay. Furthermore, LPI was proportionately most detectable when citrate exceeded ferric iron concentrations by >100-fold; ratios known to be associated with the appearance of monoferric citrate species.16,17 We, therefore, hypothesized that monoferric species are taken into cardiomyocytes most readily since, in our study, MH associated with LPI, rather than with total NTBI. Monoferric species include ferric monocitrate and ferric dicitrate16,17 (Figure 5, compounds A and C). Redox-cycling of iron species is dependent on the ability of reductants and oxidants to gain access to the iron center by displacing the monodentate water molecules which occupy the ‘free’ coordination sites of iron complex that is only partially coordinated by ligands such as one tridentate citrate molecule or one bidentate deferiprone molecule22 (Figure 5, coordination sites in bold). Ferric dicitrate, with its iron center fully hexa-coordinated by two tridentate citrate molecules, has a high stability constant for iron(III),16 is less redox-active,22 and thus less of a candidate for uptake (a process requiring reduction14,47,48). We therefore predicted that it is ferric monocitrate which is most available for cardiomyocyte uptake as well as transferrin binding, as discussed below.
Figure 5.

Model of apotransferrin-dependent re-speciation of polymeric ferric citrate. The paradoxical effect of apotransferrin seen in Figure 4D, in which uptake from 10 μM ferric citrate is increased before it is abolished by apotransferrin, is consistent with the sub-equivalent concentration of apotransferrin disrupting ferric citrate oligomers and releasing from them ferric monocitrate species. As apotransferrin binds a portion of iron in high ratio ferric citrate, this decreases the amount of iron per citrate, so effectively changes the iron-to-citrate ratio, i.e. re-speciates it. In the presence of apotransferrin and bicarbonate, which forms a ternary complex with citrate, oligomer complexes of ferric citrate become a source of ferric monocitrate species. These are subject to competition with uptake mechanisms for cellular entry (dotted line), with apotransferrin for the formation of ferrotransferrin and with citrate for the formation of ferric dicitrate, citrate also competing with apotransferrin for ferric monocitrate. Kinetic differences between ferrotransferrin formation and cellular uptake from ferric citrate may explain the additional iron uptake from newly released mononuclear species before apotransferrin can chelate them altogether. The coordination sites on the iron (shown in bold) are typically occupied by water, but they are labile sites and can also bind oxygen and H2O2, rendering the species susceptible to redox chemistry (marked in bold cursive on the right). They are also the sites of condensation with other iron complexes.
We found that cardiomyocyte iron uptake from citrate and its inhibition by apotransferrin are speciation-dependent. Cardiomyocyte uptake of ferric citrate did indeed occur most rapidly when citrate excess was high (Figure 3B), under conditions favoring monomer rather than oligomer species. This contrasts with previous findings for hepatocytes and cardiomyocytes8,47,49 using ferric nitrilotriacetate, a non-physiological but typically monomeric iron source, where the speciation dependence of uptake was not studied.
Very low concentrations of apotransferrin, and transferrin saturations ≤99% inhibited cardiomyocyte iron uptake from FAC (Figure 3E,F). This suggests that the NTBI species partaking in cardiac uptake constitute only a small fraction of the total NTBI. The small magnitude of this fraction is consistent with monocitrate being the predominant species for cardiomyocyte iron uptake. Furthermore, because apotransferrin decreased LPI in our assays, and iron monocitrate was the species most rapidly chelated by apotransferrin, as described above, ferric monocitrate is most likely the predominant LPI species. Very low transferrin concentrations (nanomolar) were all that were necessary to inhibit LPI-detectable iron citrate species (Figure 3D), which we identified above as being monoferric citrate. This suggests that this LPI-detectable species is present at a very low concentration but is kinetically labile.
Surprisingly, we found that at very high ferric citrate concentrations (10 μM, Figure 4D), under conditions that favor oligomer species, rarely observed clinically, sub-stoichiometric concentrations of apotransferrin, insufficient to bind all the ferric citrate, actually increased cellular iron uptake. This was not due to increased uptake by holotransferrin, as negligible iron was delivered to cardiomyocytes by holotransferrin (Figure 4C), consistent with previous observations in cardiomyocytes.8 We deduced therefore that increased uptake from ferric citrate in the presence of apotransferrin under these conditions may occur by accelerating the dissociation of oligomer to monomer citrate species (Figure 5). This deduction is consistent with the observation that the transition from absence to excess of transferrin binding capacity is associated with the generation of kinetically labile iron citrate species that are rapidly taken into HL-1 cardiomyocytes (Figure 4D). This effect is notable only when apotransferrin is present at intermediate concentrations allowing depolymerization of oligomers to their constituent monomers (i.e. ferric monocitrate, see Figure 5). Previous work, consistent with this model, showed that the rate-limiting step for exchange of polymeric ferric citrate iron with apotransferrin was the depolymerization and release of monomeric (mononuclear) ferric citrate.50 Ferric dicitrate was unreactive towards apotransferrin unless converted to an active intermediate, which the authors had supposed to be the monocitrate.50 Importantly, such conditions of very high NTBI are unlikely to occur clinically so that inhibitory effects of apotransferrin on NTBI uptake will predominate.
In conclusion, we propose a mechanism of MH inhibition by the generation of apotransferrin during erythropoiesis. Taken together our clinical and in vitro data point to increased generation of apotransferrin by an active bone marrow (marked by high sTfR1) as a key mechanism for decreasing the risk of MH in transfusion-dependent thalassemias. We suggest that a local excess of apotransferrin in the bone marrow, around sinusoids and reticulocytes, chelates monomeric ferric citrate species, the same species most rapidly taken into the myocardium. The clinical implications of this are that a critical balance appears to exist between the transfusion rate, endogenous erythropoiesis, MH risk and NTBI speciation. We suggest that prospective longitudinal data collection, including sequential sTfR1 measurements, would be valuable in order that clear recommendations could be made about whether reducing transfusion exposure decreases the risk of MH. Furthermore, due to the marked geographic variability in MH risk,7 which cannot be related solely to chelation practices, cross-sectional studies on the impact of regional transfusion policies on ETUR and MH risk could be indicated.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Sukhvinder Bansal from the Department of Pharmacy at King’s College London for performing the hepcidin assay. MG would like to thank Dr. Farrukh Shah for Ph.D. co-supervision; the British Society for Haematology, Sickle Cell Society and UK Thalassaemia Society for the Haemoglobinopathy Fellowship Grant, as well as the Leukaemia and Blood Diseases Appeal for grant support. JP would like to thank UCL Biomedical Research Centre for Cardiometabolic Programme support. All authors would also like to thank the Wellcome Trust for grant support (WT093209MA).
References
- 1.Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Survival and complications in thalassemia. Ann NY Acad Sci. 2005;1054: 40–47. [DOI] [PubMed] [Google Scholar]
- 2.Olivieri NF, Nathan DG, MacMillan JH, et al. Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med. 1994;331(9):574–578. [DOI] [PubMed] [Google Scholar]
- 3.Telfer PT, Prestcott E, Holden S, Walker M, Hoffbrand AV, Wonke B. Hepatic iron concentration combined with long-term monitoring of serum ferritin to predict complications of iron overload in thalassaemia major. Br J Haematol. 2000;110(4):971–977. [DOI] [PubMed] [Google Scholar]
- 4.Gabutti V, Piga A. Results of long-term iron-chelating therapy. Acta Haematol. 1996;95(1):26–36. [DOI] [PubMed] [Google Scholar]
- 5.El Beshlawy A, El Tagui M, Hamdy M, et al. Low prevalence of cardiac siderosis in heavily iron loaded Egyptian thalassemia major patients. Ann Hematol. 2014;93(3):375–379. [DOI] [PubMed] [Google Scholar]
- 6.Pennell DJ, Berdoukas V, Karagiorga M, et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood. 2006;107(9):3738–3744. [DOI] [PubMed] [Google Scholar]
- 7.Aydinok Y, Porter JB, Piga A, et al. Prevalence and distribution of iron overload in patients with transfusion-dependent anemias differs across geographic regions: Results from the CORDELIA study. Eur J Haematol. 2015;95(3):244–253. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Parkes JG, Templeton DM. Differential accumulation of non-transferrin-bound iron by cardiac myocytes and fibroblasts. J Mol Cell Cardiol. 2003;35(5):505–514. [DOI] [PubMed] [Google Scholar]
- 9.Singh S, Hider RC, Porter JB. A direct method for quantification of non-transferrinbound iron. Anal Biochem. 1990;186(2):320–323. [DOI] [PubMed] [Google Scholar]
- 10.Garbowski MW, Ma Y, Fucharoen S, Srichairatanakool S, Hider R, Porter JB. Clinical and methodological factors affecting non-transferrin-bound iron values using a novel fluorescent bead assay. Transl Res. 2016;177:19–30.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood. 2003;102(7):2670–2677. [DOI] [PubMed] [Google Scholar]
- 12.Gosriwatana I, Loreal O, Lu S, Brissot P, Porter J, Hider RC. Quantification of non-transferrin-bound iron in the presence of unsaturated transferrin. Anal Biochem. 1999;273(2):212–220. [DOI] [PubMed] [Google Scholar]
- 13.Porter J, Cappellini M, Kattamis A, et al. Iron overload across the spectrum of non-transfusion-dependent thalassaemias: role of erythropoiesis, splenectomy and transfusion. Br J Haematol. 2017;176: 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oudit GY, Sun H, Trivieri MG, et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med. 2003;9(9):1187–1194. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes JL, Loggetto SR, Veríssimo MPA, et al. A randomized trial of amlodipine in addition to standard chelation therapy in patients with thalassemia major. Blood. 2016;128(12):1555–1561. [DOI] [PubMed] [Google Scholar]
- 16.Silva AM, Kong X, Parkin MC, Cammack R, Hider RC. Iron(III) citrate speciation in aqueous solution. Dalt Trans. 2009;(40):8616–8625. [DOI] [PubMed] [Google Scholar]
- 17.Evans RW, Rafique R, Zarea A, et al. Nature of non-transferrin-bound iron: studies on iron citrate complexes and thalassemic sera. J Biol Inorg Chem. 2008;13(1):57–74. [DOI] [PubMed] [Google Scholar]
- 18.Silva AM, Hider RC. Influence of non-enzymatic post-translation modifications on the ability of human serum albumin to bind iron. Implications for non-transferrin-bound iron speciation. Biochim Biophys Acta. 2009;1794(10):1449–1458. [DOI] [PubMed] [Google Scholar]
- 19.Evans P, Kayyali R, Hider RC, Eccleston J, Porter JB. Mechanisms for the shuttling of plasma non-transferrin-bound iron (NTBI) onto deferoxamine by deferiprone. Transl Res. 2010;156(2):55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanninelli G, Breuer W, Cabantchik ZI. Daily labile plasma iron as an indicator of chelator activity in thalassaemia major patients. Br J Haematol. 2009;147(5):744–751. [DOI] [PubMed] [Google Scholar]
- 21.Wood JC, Glynos T, Thompson A, et al. Relationship between labile plasma iron, liver iron concentration and cardiac response in a deferasirox monotherapy trial. Haematologica. 2011;96(7):1055–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devanur LD, Neubert H, Hider RC. The Fenton activity of iron(III) in the presence of deferiprone. J Pharm Sci. 2008;97(4):1454–1467. [DOI] [PubMed] [Google Scholar]
- 23.Porter JB, Walter PB, Neumayr LD, et al. Mechanisms of plasma non-transferrin bound iron generation: insights from comparing transfused Diamond Blackfan anaemia with sickle cell and thalassaemia patients. Br J Haematol. 2014;167(5):692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen AR, Glimm E, Porter JB. Effect of transfusional iron intake on response to chelation therapy in β-thalassemia major. Blood. 2008;111(2):583–587. [DOI] [PubMed] [Google Scholar]
- 25.Taher AT, Porter J, Viprakasit V, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120(5):970–977. [DOI] [PubMed] [Google Scholar]
- 26.Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim Biophys Acta. 2012;1820(3):188–202. [DOI] [PubMed] [Google Scholar]
- 27.Hikawa A, Nomata Y, Suzuki T, Ozasa H, Yamada O. Soluble transferrin receptortransferrin complex in serum: measurement by latex agglutination nephelometric immunoassay. Clin Chim Acta. 1996;254(2): 159–172. [DOI] [PubMed] [Google Scholar]
- 28.Ponka P, Lok CN. The transferrin receptor: role in health and disease. Int J Biochem Cell Biol. 1999;31(10):1111–1137. [DOI] [PubMed] [Google Scholar]
- 29.Beguin Y, Clemons GK, Pootrakul P, Fillet G. Quantitative assessment of erythropoiesis and functional classification of anemia based on measurements of serum transferrin receptor and erythropoietin. Blood. 1993;81(4): 1067–1076. [PubMed] [Google Scholar]
- 30.Huebers HA, Beguin Y, Pootrakul P, Einspahr D, Finch CA. Intact transferrin receptors in human plasma and their relation to erythropoiesis. Blood. 1990;75(1):102–107. [PubMed] [Google Scholar]
- 31.R’zik S, Beguin Y. Serum soluble transferrin receptor concentration is an accurate estimate of the mass of tissue receptors. Exp Hematol. 2001;29(6):677–685. [DOI] [PubMed] [Google Scholar]
- 32.Cazzola M, De Stefano P, Ponchio L, et al. Relationship between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol. 1995;89(3):473–478. [DOI] [PubMed] [Google Scholar]
- 33.Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22(23):2171–2179. [DOI] [PubMed] [Google Scholar]
- 34.Garbowski MW, Carpenter JP, Smith G, et al. Biopsy-based calibration of T2* magnetic resonance for estimation of liver iron concentration and comparison with R2 Ferriscan. J Cardiovasc Magn Reson. 2014;16(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson LJ, Westwood MA, Holden S, et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. Br J Haematol. 2004;127(3):348–355. [DOI] [PubMed] [Google Scholar]
- 36.Noetzli LJ, Carson SM, Nord AS, Coates TD, Wood JC. Longitudinal analysis of heart and liver iron in thalassemia major. Blood. 2008;112(7):2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finch C, Huebers H, Eng M, Miller L. Effect of transfused reticulocytes on iron exchange. Blood. 1982;59(2):364–369. [PubMed] [Google Scholar]
- 38.Matzapetakis M, Raptopoulou CP, Tsohos A, Papaefthymiou V, Moon N, Salifoglou A. Synthesis, spectroscopic and structural characterization of the first mononuclear, water soluble iron−citrate complex, (NH4)5Fe(C6H4O7)2·2H2O. J Am Chem Soc. 1998;120(10):13266–13267. [Google Scholar]
- 39.Flanagan JM, Peng H, Wang L, et al. Soluble transferrin receptor-1 levels in mice do not affect iron absorption. Acta Haematol. 2006;116(4):249–254. [DOI] [PubMed] [Google Scholar]
- 40.Meloni A, Puliyel M, Pepe A, Berdoukas V, Coates TD, Wood JC. Cardiac iron overload in sickle-cell disease. Am J Hematol. 2014;89(7):678–683. [DOI] [PubMed] [Google Scholar]
- 41.Ricchi P, Meloni A, Spasiano A, et al. Extramedullary hematopoiesis is associated with lower cardiac iron loading in chronically transfused thalassemia patients. Am J Hematol. 2015;90(11):1008–1012. [DOI] [PubMed] [Google Scholar]
- 42.Wood JC. Cardiac iron across different transfusion-dependent diseases. Blood Rev. 2008;22(Suppl. 2):14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taher AT, Musallam KM, Wood JC, Cappellini MD. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am J Hematol. 2010;85(4):288–290. [DOI] [PubMed] [Google Scholar]
- 44.Roghi A, Cappellini MD, Wood JC, et al. Absence of cardiac siderosis despite hepatic iron overload in Italian patients with thalassemia intermedia: an MRI T2* study. Ann Hematol. 2010;89(6):585–589. [DOI] [PubMed] [Google Scholar]
- 45.Lakhal-Littleton S, Wolna M, Carr CA, et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc Natl Acad Sci USA. 2015;112(10):3164–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altamura S, Kessler R, Groene HJ, et al. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014;20(2):359–367. [DOI] [PubMed] [Google Scholar]
- 47.Parkes JG, Olivieri NF, Templeton DM. Characterization of Fe2+ and Fe3+ transport by iron-loaded cardiac myocytes. Toxicology. 1997;117(2–3):141–151. [DOI] [PubMed] [Google Scholar]
- 48.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci USA. 2006;103(37):13612–13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parkes JG, Randell EW, Olivieri NF, Templeton DM. Modulation by iron loading and chelation of the uptake of non-transferrin-bound iron by human liver cells. Biochim Biophys Acta. 1995;1243(3):373–380. [DOI] [PubMed] [Google Scholar]
- 50.Bates GW, Billups C, Saltman P. The kinetics and mechanism of iron(III) exchange between chelates and rransferrin. I. The complexes of citrate and nitrilotriacetic acid. J Biol Chem. 1967;242 (12):2810–2815. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.