

Abstract
A plastic anemia is a rare life-threatening disease. However, since the introduction of immunosuppressive therapy and allogeneic stem cell transplantation, the outcome has improved considerably, and the 5-year survival is reported to be 70–80% in selected patient cohorts. Yet, contemporary population-based data on incidence and survival are lacking. We performed a national retrospective study to determine the incidence, treatment, and survival of patients with aplastic anemia diagnosed in Sweden from 2000–2011. Patients were included via the National Patient Registry, and diagnosed according to the Camitta criteria. In total, 257 confirmed cases were identified, with an overall incidence of 2.35 (95% CI: 2.06–2.64) cases per million inhabitants per year. Median age was 60 years (range: 2–92), and median follow up was 76 (0–193) months. Primary treatments included immunosuppressive therapy (63%), allogenic stem cell transplantation (10%), or single-agent cyclosporine/no specific therapy (27%). The 5-year survival was 90.7% in patients aged 0–18 years, 90.5% in patients aged 19–39 years, 70.7% in patients aged 40–59 years, and 38.1% in patients aged ≥60 years. Multivariate analysis showed that age (both 40–59 and ≥60 age groups), very severe aplastic anemia and single-agent cyclosporine/no specific therapy were independent risk factors for inferior survival. In conclusion, younger aplastic anemia patients experience a very good long-term survival, while that of patients ≥60 years in particular remains poor. Apparently, the challenge today is to improve the management of older aplastic anemia patients, and prospective studies to address this medical need are warranted.
Introduction
Aplastic anemia (AA) is a rare life-threatening disease. The incidence and median age at diagnosis, which varies according to geography, ranges from 1.5 to about seven cases per million inhabitants/year, and from 25–60 years, respectively.1–7 Following the introduction of immunosuppressive therapy (IST) with antithymocyte globulin (ATG), and allogeneic stem cell transplantation (SCT) in the 1980–90s, several studies have reported an improved outcome with a 5-year overall survival of approximately 70–80%.8–16 However, most of these studies were performed in selected patient cohorts, and included randomized trials, transplantation registry studies, or single center experiences, in which the majority of included patients were younger. In addition, there are several epidemiological studies on AA from Europe, the United States, South America, and Asia. Most of them reported on patients diagnosed from 1970 until the 1990s, and only a few presented outcome data, with a relatively short follow up.2,4,6,17,18 The most recent epidemiological study in Scandinavia was published in 1996, and focused on AA in the pediatric population.19 Therefore, it appears as if contemporary population-based (or real-world) data on treatment and survival in patients with AA are missing. Genuine population-based studies are difficult to perform because of the lack of nationwide AA registries on incidence and outcome. Additionally, given that many patients with AA are referred to a regional or academic center, followed by a transplant unit, many cases are lost to follow up at the referring center, and their long-term outcome is not reported. The importance of complete longitudinal population-based data was highlighted at the International Working Group on Severe Aplastic Anemia meeting in 2010, where the establishment of a population-based registry for longitudinal collection of data on patients from diagnosis, during and after treatment was proposed.20 In Sweden, where disease codes of all patients in the community-based health care system are centrally registered, there is the unique possibility to identify patients with AA, and collect detailed data on incidence, treatment, and outcome of the entire population. Such a study, while waiting for mature data from the proposed population-based AA registry, could allow for the investigation into several important issues (e.g., possible changes in incidence, potential etiological factors, the proportion of patients undergoing potentially curative treatment, and if the reported improvement of outcome is translatable to a population-based cohort, especially in older patients).
Therefore, the aim of the present national retrospective population-based study was to determine the incidence, treatment type, and survival of patients with AA diagnosed in Sweden during the years 2000–2011.
Methods
Identification of patients
We first aimed to identify all patients with AA (both children and adults) diagnosed in Sweden between January 2000 and December 2011. In the year 2000, Sweden had 8,882,792 inhabitants, while in 2011 the figure was 9,482,855. Patients were identified in the National Patient Registry held by the Swedish National Board of Health and Welfare, which collects patient, geographical, administrative, and medical data. The registry includes data on all patients treated as in- or outpatients in the national health care system regarding primary and secondary diagnoses and procedures. In the registry, patients are identified by a unique national social security number. For the registry search, the 10th version of International Classification of Diseases was used, and for a complete search, the disease codes D61.0–D61.9 were initially applied. For a detailed description of the identification of patients from the registry, see Online Supplementary Figure S1. The study was approved by the Regional Ethical Review Board in Gothenburg.
Most of the patients were treated at one of the seven university hospitals. Some patients were treated at the regional or county hospitals (29 hospitals in total) without referral to a university hospital.
The diagnosis of AA was confirmed according to the Camitta criteria21: bone marrow biopsy cellularity <25% (or 25–50% with <30% residual hematopoietic cells) together with two of the following three criteria: hemoglobin <100 g/L, reticulocytes <50×109/L, or <1%; platelets <50×109/L; and neutrophil leucocytes (ANC) <1.5×109/L. Disease severity was classified as follows: severe aplastic anemia (SAA), with two of the following three characteristics: ANC <0.5×109/L, platelet count <20×109/L, or reticulocyte count <20×109/L; very severe aplastic anemia (VSAA) had the same characteristics as SAA, but with ANC <0.2×109/L; and non-severe aplastic anemia (NSAA). Patients with congenital disease, pancytopenia without a marrow biopsy performed, marrow fibrosis, or other signs of malignancy or dysplasia were excluded. A mild dysplasia in erythropoiesis was accepted.
All data were collected in a case report form, and unclear cases were re-evaluated. Follow-up information was obtained from medical charts and/or through matching the social security number in the Swedish Cause of Death Registry.
Statistics
Incidence and confidence intervals (CI) were calculated according to Rothmann.22 Rates and proportions were compared using Pearson’s χ2 test. The Kaplan-Meier method was used for estimation of overall survival, and comparisons were based on the log-rank test. Analysis of risk factors for survival were calculated using Cox regression proportional hazards, including age, sex, type of treatment and disease severity. Relative survival ratios were calculated using the Ederer II23 method by dividing the observed survival of patients with AA with the expected survival in a general Swedish population with corresponding age, sex, and calendar year. The statistical analyses were performed either by SPSS version 23 or Stata for Macintosh, version 13.1, and relative survival ratio was calculated by use of the strs module.
Results
Basic data
Between 1st January 2000 and 31st December 2011, we identified 257 confirmed cases of acquired AA among 1,362 potential cases. The remaining cases had malignant diseases, secondary anemia/pancytopenia, or other benign hematological disorders (Online Supplementary Figure S1).
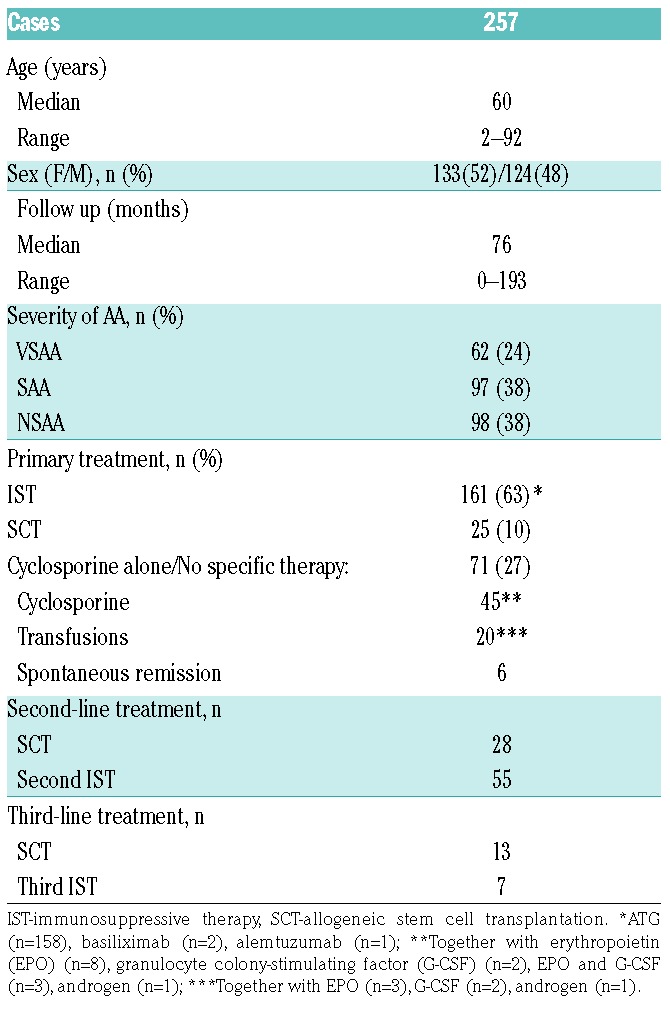
Clinical characteristics are shown in Table 1. The median age at diagnosis was 60 years (95% CI: 54–64, range: 2–92). A total of 133 patients (52%) were female, and the median age was 60 years for both sexes (females: 95% CI: 51–66, range: 2–90; males: 95% CI: 51–65, range: 7–92). At diagnosis, 38% had NSAA, 38% had SAA, and 24% had VSAA. There was no significant age-related distribution difference between patients with SAA and VSAA, however NSAA patients were older (P=0.028 and P=0.001, respectively).
Table 1.
Clinical characteristics.
Incidence
The overall incidence was estimated to be 2.35 (95% CI: 2.06–2.64) cases per million inhabitants per year. For all patients, a biphasic age distribution was observed; one peak in patients aged 15–20 years, 2.87 (95% CI: 1.72–4.03), and one in patients >60 years old, 4.36 (95% CI: 3.55–5.18). The incidence according to age groups and sex are shown in Online Supplementary Table S1. The biphasic distribution was predominantly observed in male patients, while the incidence among females was more evenly distributed and increased steadily with a peak above the age of 60 years. The incidence in children <10 years old was lower: 1.8 per million per year. Furthermore, no difference in incidence was observed when grouping the patients according to two different time periods (2000–2005 and 2006–2011); 2.19 (95% CI: 1.80–2.29) and 2.5 (95% CI: 2.08–2.91), respectively.
Treatment
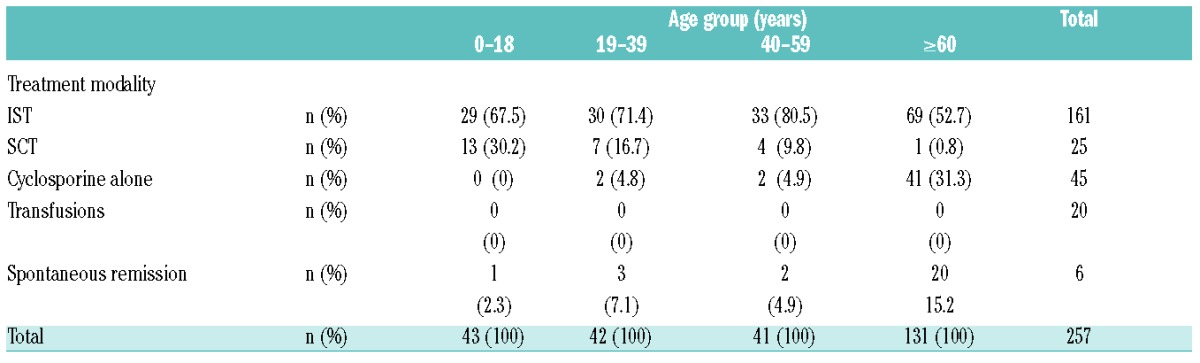
Primary treatment included IST (defined as treatment with ATG or other anti-T-cell monoclonal antibody together with cyclosporine, n=161, 63%), a total of 25 patients (10%) underwent allogeneic SCT, and 71 patients (27%) were treated with single-agent cyclosporine (CSA alone) or no specific therapy. Spontaneous remission was observed in six cases (five cases of NSAA and one of SAA). Treatment data are shown in Table 1, and data on type of initial treatment in the different age groups are shown in Table 2. The median age in the ≥60 years group who received IST, CSA alone, and no specific therapy was 67 (range: 60–85), 79 (range: 60–90), and 82.5 (range: 62–92) years, respectively.
Table 2.
Primary treatment in different age groups.
Survival
The median follow up was 76 months (95% CI: 66–86, range: 0–193). During follow up, 121 (47.1%) patients died. Twenty-six died within 3 months, and a further 48 died within the first 24 months. The most common causes of death during the first 24 months were infections (n=41), bleeding (n=14), and unspecified conditions related to AA (n=8). The 5-year survival for all patients with AA was 60.7% (95% CI: 57.7–63.7) (Figure 1A), and median survival was 150 months. The 5-year survival, irrespective of treatment modality, varied according to the different age groups, and was significantly lower in patients aged 40–59 years and ≥60 years: 90.7% (95% CI: 77.1–96.4) in patients aged 0–18 years, 90.5% (76.6–96.3) in patients 19–39 years (P=0.95), 70.7% (54.3–82.2) in patients 40–59 years (P=0.029), and 38.1% (29.8–46.4) in patients ≥60 years (P=0.001) (Figure 1B). When dividing the ≥60 years group into two further groups, 60–69 years and ≥70 years, the 5-year survival was 57.5% (41.8–70.5) and 27.9 (18.9–37.6) (P=0.001), respectively. The age-related survival difference was obvious early after diagnosis: patients ≥60 years had a 3-month survival of 84.0% compared with 97.7% for patients aged 0–18 years, 97.6 % for patients 19–39 years, and 92.7% for patients 40–59 years (P=0.021, P=0.024, and P=0.155, respectively). When grouping patients according to disease severity, the 5-year survival was lower in patients with VSAA compared with those with SAA (P=0.025), but not compared with NSAA (P=0.13) (Figure 2). However, during follow up, almost 39% of patients with NSAA developed SAA or VSAA and the majority (84.7%) were treated: IST (n=61), CSA alone (n=18), or SCT (n=4). Early mortality rate (at 3 months) was significantly higher in VSAA compared with SAA and NSAA: 22.6% versus 8.2% and 4.1% (P=0.009 and P<0.0001), respectively.
Figure 1.

Overall survival. A. For all patients. B. In different age groups: 0–18 years, 19–39 years, 40–59 years, and ≥60 years.
Figure 2.

Overall survival according to disease severity at diagnosis (very severe, severe, and non-severe AA).
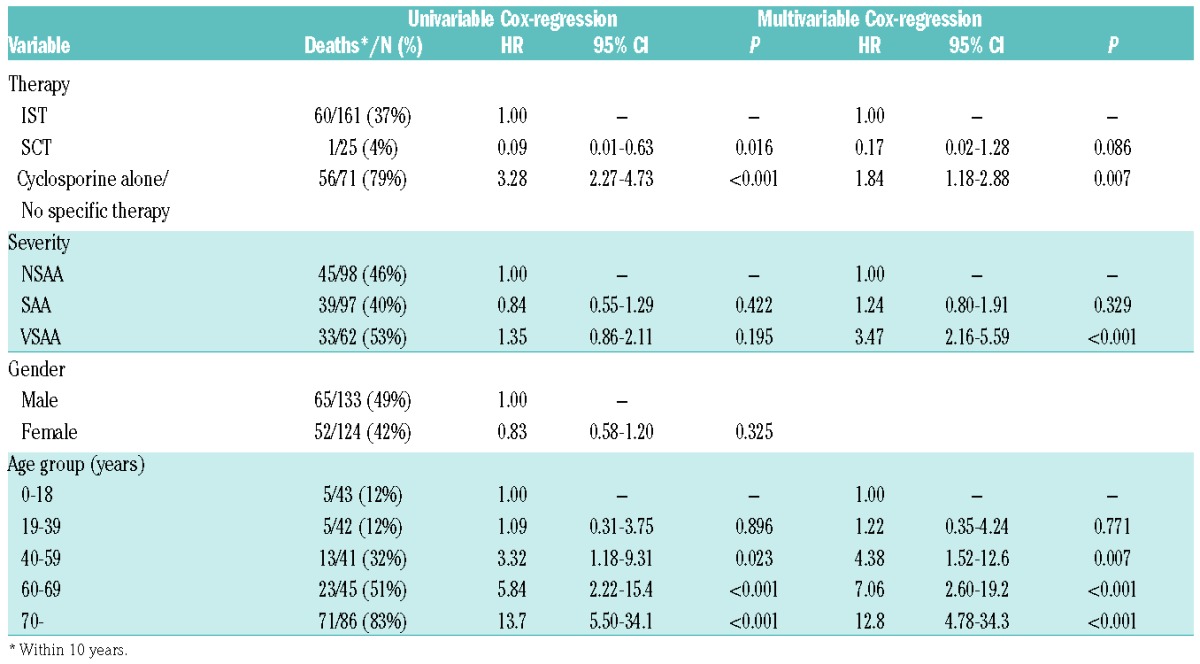
The 5-year survival rate was 96.0% (74.8–99.4) in patients who underwent SCT, 68.9% (61.2–75.5) in the IST group (P=0.009), and 29.6% (19.5–40.4) in patients who received CSA alone or no specific therapy (P<0.0001) (Figure 3A). In younger patients (0–18 and 19–39 years), there was no significant difference in survival, whether they were primarily treated with IST or if they underwent SCT: 0–18 years, 86.2% vs. 100% (P=0.169); and 19–39 years, 90% vs. 100% (P=0.395). Furthermore, when grouping these patients together, the corresponding values were 88.1% vs. 100% (P=0.113). Regarding the group of patients treated with IST (n=161), there was no survival difference at 5 years between the age groups, 0–18 years: 86.2% (67.3–94.6), 19–39 years: 90% (72.1–96.7), and 40– 59 years: 69.7% (51.0–82.4) (P=0.67, P=0.12, and P=0.056, respectively). Patients ≥60 years had a significantly worse survival rate of 52.2% (39.8–63.2) compared with patients aged 0–18 years (P=0.003) and 19–39 years (P=0.001), but not with patients aged 40–59 years (P=0.15) (Figure 3B). The Cox regression analysis revealed that age (40–59 and ≥60 years age groups), VSAA and treatment with CSA alone/no therapy were independent risk factors for inferior survival (Table 3). The hazard ratio for SCT compared to IST was 0.17, but due to the small number of patients this was not statistically significant.
Figure 3.

Overall survival. A. According to primary treatment. B. For patients treated with IST divided into different age groups.
Table 3.
Uni-and multivariable Cox-regression.
Forty-three (27%) patients in the entire IST group were allografted (related donor, n=11; unrelated donor, n=32) after not responding to/relapsing following one or two cycles of IST. Only two (3%) patients ≥60 years underwent SCT compared with 14 (48%) patients in the group aged 0–18 years, 17 (57%) in the group aged 19–39 years, and 10 (30%) in the group aged 40–59 years.
For patients receiving CSA alone/no specific therapy, we observed no survival difference between patients with a more palliative approach (“Transfusions”; n=20) and CSA alone patients (n=45), 20.0% vs. 24.4% (P=0.172).
When grouping the entire patient cohort according to two different time-periods (from 2000–2005 or 2006– 2011), we found no difference in 5-year survival for all patients (61.0% vs. 61.5%), or in the different age groups (data not shown).
The relative 5-year survival (i.e., excess mortality: the difference between observed mortality and expected mortality) for all patients was 65.4% (95% CI: 58.6–71.5) (Figure 4A). When grouping all patients according to the median age at diagnosis, the relative 5-year survival was 84.6% (76.9–90.0) in patients less than 60 years, while the corresponding figure for patients ≥60 years was significantly worse, 45.3% (35.4–55.1) (Figure 4B). When grouping the patients into a younger group, 0–39 years, and a group ≥40 years, the relative survival at 5 years was significantly higher in the younger group: 90.8% (82.2–95.4) and 52.1% (43.4–60.3), respectively. When dividing the ≥60 years group into 60–69 years and ≥70 years, there was a numerical difference however without statistical significance: 60.7% (44.1–74.5) compared to 37.1% (25.1–49.9).
Figure 4.

Relative survival. A. For all patients. B. Divided into two age groups according to median age at diagnosis: < and ≥60 years.
Discussion
In this comprehensive population-based study on patients with AA patients in Sweden, diagnosed from 2000–2011, we found that the 5-year survival among younger patients (up to the age of 40) was about 90%, which is similar to the reported survival rates from modern clinical trials with ATG, or from SCT registry data.7,9,13–16,24 Furthermore, there was no survival difference between IST and SCT as the primary treatment in the younger patients, which corroborate with recently published data from the European Society for Blood and Marrow Transplantation (EBMT) registry of the SAA Working Party.25 Additionally, patients from 40–59 years old experienced a 5-year survival of about 70%. Together, these figures appear to be superior in comparison to data from the latest population-based AA study, on Spanish patients diagnosed between 1980 and 2003, where the reported 2-year survival was about 80% and 40% in the comparable patient groups.7 Thus, also in a population-based cohort, the outcome for patients with AA below the median age seems to have improved during the last decade. There could be several possible reasons for this: a higher percentage of these patients are treated with SCT, earlier onset of treatment, and better supportive care including infection prophylaxis.
However, even though AA patients 40–59 years also had an inferior survival, patients aged ≥60 years do not seem to have gained any survival improvement in the last few decades. Less than 40% of these patients were alive after 5 years, and the relative 5-year survival was 45%, which indicated considerable excess mortality. In addition, patients ≥60 years had a significantly higher risk of early death compared with younger patients; 16% were deceased 3 months after diagnosis. On the other hand, patients ≥60 years old treated with IST (52.7 %) had a 5-year survival of around 50%, which was similar to the results of IST reported from EBMT AA registry data.26 In their study, although survival was worse in older patients than in younger ones, the response to IST was independent of age. This finding, together with our results, may imply that more older patients should be treated with IST.
In our cohort of patients ≥60 years old, a relatively large number (n=45) were treated with single-agent therapy of cyclosporine. In a phase III trial comparing cyclosporine alone versus the combination of ATG and cyclosporine in patients with NSAA, the overall response rate of cyclosporine alone was 46% compared with 74% in the combination arm.27 After a median follow up of 1 year, no difference in overall survival was observed. In contrast, our cohort of patients treated with cyclosporine had no survival benefit compared with patients treated with a stricter transfusion policy, i.e., with a palliative approach. In our cohort that received cyclosporine alone, only 38% had NSAA, whereas 62% had SAA or VSAA. In retrospective studies, obtaining reliable information on therapeutic intent and endurance can be difficult. However, since patients starting treatment with cyclosporine alone likely require sufficient renal function to tolerate therapeutic doses, it is plausible to assume that at least some of the patients in this cohort would have been eligible for IST.
Furthermore, only three patients ≥60 years received an allograft (one upfront, and two after failing IST). In the first comprehensive treatment guidelines for severe AA published in 1994,28 the Working Party on SAA recommended SCT from a HLA-identical sibling as upfront treatment for patients up to the age of 40. The first British AA guidelines (published in 2003) had similar recommendations, although patients from 30–40 years old could receive either IST or SCT.29 In the guidelines from 2009, the age limit was set at 40 years,30 and in the most recent guidelines from 2016,31 patients up to the age of 50 were considered eligible for upfront SCT. Regarding SCT as salvage for patients failing IST, the guidelines from 2003 advocated a matched unrelated donor SCT up to the age of 40. In the subsequent update, this age limit was changed to 50 years, but was also considered as an option for patients between 50–60 years if they had good performance status. Finally, in the 2016 version, the authors indicated that there is no strict upper age limit. At present, one of the most frequently allotransplanted groups, with a reduced intensity conditioning regimen, are those with acute myelogenous leukemia (AML) between 50–70 years old. This procedure is now considered standard of care for older patients with AML.32 Therefore, given our poor outcome data for patients ≥60 years old, this treatment option should likely be discussed more often. A possible alternative for older patients who are not eligible for IST with ATG, or for SCT, is treatment (preferably within a multicenter prospective clinical trial) with the thrombopoietin receptor antagonist, eltrombopag, which has shown promising results in refractory AA.33,34 In addition, very promising results with the combination of eltrombopag and IST was most recently reported,35 which could potentially become a future treatment option for all patients considered eligible for IST.
We found that the incidence of AA during the study period was 2.35 per million inhabitants per year, and showed a biphasic age distribution. Previous epidemiological studies have shown a broad variation in incidence depending on time and geographical location. Reports from Europe and the United States in the 1960–70s presented a very high incidence: from six to ten cases of AA per million per year. In addition, in some studies, there was an association with toxic agricultural substances.3,4,6,17 However, different diagnostic criteria were used, and many cases likely represented other diseases.4 The International Agranulocytosis and Aplastic Anemia Study was published in 1987, and established a well-accepted overall incidence of around two cases of AA per million.6 This was later confirmed by data from Spain published in 2008, with an overall incidence of 2.34 per million per year.7 In contrast, studies performed in Asia showed a higher incidence: over four patients with AA per million per year.5,36 A higher incidence in younger people in Asia has been suggested to be associated with environmental factors related to occupation.36 Our incidence data correspond to the aforementioned later reports from Europe, the United States, and South America. In some studies, the incidence has also been reported to be slightly higher among females,2,6 while data from Turkey and Bangkok have instead shown a male predominance.5,17 We found no differences between female and male incidence rate (1.07:1), which was consistent with data from the Spanish study from 2008.
The majority of patient deaths that occurred within two years from diagnosis were from serious infections (50%) or bleeding (15%), indicating the importance of early treatment and prevention of infections with antibacterial and antifungal therapy. It has been reported that despite the lack of progress in achieving higher response rates for patients with AA in the last two decades, both infection-related and overall mortality have been reduced in patients with SAA who are unresponsive to initial IST, mainly because of prompt empirical antifungal therapy.37 Patients in our cohort who died later had refractory disease, complications from transplantation, worsening of heart failure because of chronic anemia, or secondary cancer.
Although our study included all identifiable patients with AA diagnosed in Sweden between 2000 and 2011, and therefore reasonably reflects the true incidence and survival, it had limitations. First, there could be additional patients with AA who were not listed in the national diagnosis registry (e.g., patients with a mild form and subsequent spontaneous remission or patients who died before established diagnosis). However, based on the pilot study, when the search criteria were established, we estimated that the number of missing patients was going to be at most 2%, i.e., only four to five patients. Furthermore, other cases that could have been missed because of our search criteria of at least two medical contacts with D61.0–D61.9 would likely have been older individuals unsuited for active treatment, with a short life expectancy. Such patients would worsen the survival data of the entire ≥60 years old patient group. Second, the patient data were collected retrospectively. However, all of the charts were thoroughly examined, and our follow up data are almost complete (only one patient was lost during follow up). Third, we did not re-evaluate the bone marrow biopsies. Nevertheless, we scrutinized the biopsy reports from the hematopathologist for all patients, and even though the possibility of solitary cases of hypoplastic myelodysplastic syndromes cannot be ruled out, it seems unlikely that such cases would have significantly impacted our results.
In conclusion, apart from the limitations, incidence data obtained from this contemporary population-based study are consistent with previously established figures. Furthermore, younger patients, regardless of initial therapy, experienced a very good long-term survival. However, for patients above the median age at diagnosis (≥60 years), excess mortality was still substantial. Therefore, the management of older patients with AA should be improved. Prospective studies to address this medical need are warranted.
Supplementary Material
Acknowledgments
We would also like to thank Erik Holmberg, Regional Cancer Centrum Väst, for expert statistical assistance.
Footnotes
Funding
This study was supported by grants from ALF Västra Götaland, Gothenburg Medical Society, and a scholarship from Alexion Sweden.
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/10/1683
References
- 1.Camitta BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48(1):63–70. [PubMed] [Google Scholar]
- 2.Cartwright RA, McKinney PA, Williams L, et al. Aplastic anaemia incidence in parts of the United Kingdom in 1985. Leuk Res. 1988;12(6):459–463. [DOI] [PubMed] [Google Scholar]
- 3.Mary JY, Baumelou E, Guiguet M. Epidemiology of aplastic anemia in France: a prospective multicentric study. The French Cooperative Group for Epidemiological Study of Aplastic Anemia. Blood. 1990;75(8):1646–1653. [PubMed] [Google Scholar]
- 4.Szklo M, Sensenbrenner L, Markowitz J, Weida S, Warm S, Linet M. Incidence of aplastic anemia in metropolitan Baltimore: a population-based study. Blood. 1985;66(1):115–119. [PubMed] [Google Scholar]
- 5.Issaragrisil S, Kaufman DW, Anderson T, et al. The epidemiology of aplastic anemia in Thailand. Blood. 2006;107(4):1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.IAAS. Incidence of aplastic anemia: the rel evance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70(6):1718–1721. [PubMed] [Google Scholar]
- 7.Montane E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93(4):518–523. [DOI] [PubMed] [Google Scholar]
- 8.Speck B, Gratwohl A, Nissen C, et al. Treatment of severe aplastic anaemia with antilymphocyte globulin or bone-marrow transplantation. Br Med J (Clin Res Ed). 1981;282(6267):860–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica. 2007;92(1):11–18. [DOI] [PubMed] [Google Scholar]
- 10.Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al. Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. The German Aplastic Anemia Study Group. N Engl J Med. 1991;324(19):1297–1304. [DOI] [PubMed] [Google Scholar]
- 11.Bacigalupo A, Bruno B, Saracco P, et al. Antilymphocyte globulin, cyclosporine, prednisolone, and granulocyte colony-stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT study on 100 patients. European Group for Blood and Marrow Transplantation (EBMT) Working Party on Severe Aplastic Anemia and the Gruppo Italiano Trapianti di Midolio Osseo (GITMO). Blood. 2000;95(6):1931–1934. [PubMed] [Google Scholar]
- 12.Bacigalupo A, Brand R, Oneto R, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy–The European Group for Blood and Marrow Transplantation experience. Semin Hematol. 2000;37(1):69–80. [DOI] [PubMed] [Google Scholar]
- 13.Bacigalupo A, Socie G, Lanino E, et al. Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants, in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica. 2010;95(6):976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Afable MG, 2nd, Shaik M, Sugimoto Y, et al. Efficacy of rabbit anti-thymocyte globulin in severe aplastic anemia. Haematologica. 2011;96(9):1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marsh JC, Pearce RM, Koh MB, et al. Retrospective study of alemtuzumab vs ATG-based conditioning without irradiation for unrelated and matched sibling donor transplants in acquired severe aplastic anemia: a study from the British Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2014;49(1):42–48. [DOI] [PubMed] [Google Scholar]
- 16.Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365(5):430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baslar Z, Aktuglu G, Bolaman Z, et al. Incidence of aplastic anemia in Turkey: a hospital-based prospective multicentre study. Leuk Res. 1997;21(11–12):1135–1139. [DOI] [PubMed] [Google Scholar]
- 18.Maluf E, Hamerschlak N, Cavalcanti AB, et al. Incidence and risk factors of aplastic anemia in Latin American countries: the LATIN case-control study. Haematologica. 2009;94(9):1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clausen N, Kreuger A, Salmi T, Storm-Mathisen I, Johannesson G. Severe aplastic anaemia in the Nordic countries: a population based study of incidence, presentation, course, and outcome. Arch Dis Child. 1996;74(4):319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pulsipher MA, Young NS, Tolar J, et al. Optimization of therapy for severe aplastic anemia based on clinical, biologic, and treatment response parameters: conclusions of an international working group on severe aplastic anemia convened by the Blood and Marrow Transplant Clinical Trials Network, March 2010. Biol Blood Marrow Transplant. 2011;17(3):291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975;45(3):355–363. [PubMed] [Google Scholar]
- 22.KJ R. Modern Epidemiology. Boston: Little Brown; 1986. [Google Scholar]
- 23.Ederer F, Axtell LM, Cutler SJ. The relative survival rate: a statistical methodology. Natl Cancer Inst Monogr. 1961;6:101–121. [PubMed] [Google Scholar]
- 24.Marsh JC, Bacigalupo A, Schrezenmeier H, et al. Prospective study of rabbit antithymocyte globulin and cyclosporine for aplastic anemia from the EBMT Severe Aplastic Anaemia Working Party. Blood. 2012; 119(23):5391–5396. [DOI] [PubMed] [Google Scholar]
- 25.Bacigalupo A, Giammarco S, Sica S. Bone marrow transplantation versus immunosuppressive therapy in patients with acquired severe aplastic anemia. Int J Hematol. 2016;104(2):168–174. [DOI] [PubMed] [Google Scholar]
- 26.Tichelli A, Socie G, Henry-Amar M, et al. Effectiveness of immunosuppressive therapy in older patients with aplastic anemia. European Group for Blood and Marrow Transplantation Severe Aplastic Anaemia Working Party. Ann Intern Med. 1999; 130(3):193–201. [DOI] [PubMed] [Google Scholar]
- 27.Marsh J, Schrezenmeier H, Marin P, et al. Prospective randomized multicenter study comparing cyclosporin alone versus the combination of antithymocyte globulin and cyclosporin for treatment of patients with nonsevere aplastic anemia: a report from the European Blood and Marrow Transplant (EBMT) Severe Aplastic Anaemia Working Party. Blood. 1999; 93(7):2191–2195. [PubMed] [Google Scholar]
- 28.Bacigalupo A. Guidelines for the treatment of severe aplastic anemia. Working Party on Severe Aplastic Anemia (WPSAA) of the European Group of Bone Marrow Transplantation (EBMT). Haematologica. 1994;79(5):438–444. [PubMed] [Google Scholar]
- 29.Marsh JC, Ball SE, Darbyshire P, et al. Guidelines for the diagnosis and management of acquired aplastic anaemia. Br J Haematol. 2003;123(5):782–801. [DOI] [PubMed] [Google Scholar]
- 30.Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. [DOI] [PubMed] [Google Scholar]
- 31.Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172(2):187–207. [DOI] [PubMed] [Google Scholar]
- 32.Champlin R. Reduced intensity allogeneic hematopoietic transplantation is an established standard of care for treatment of older patients with acute myeloid leukemia. Best Pract Res Clin Haematol. 2013;26(3):297–300. [DOI] [PubMed] [Google Scholar]
- 33.Desmond R, Townsley DM, Dumitriu B, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014;123(12):1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012;367(1):11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376(16):1540–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yong AS, Goh AS, Rahman M, Menon J, Purushothaman V. Epidemiology of aplastic anaemia in the state of Sabah, Malaysia. Med J Malaysia. 1998;53(1):59–62. [PubMed] [Google Scholar]
- 37.Valdez JM, Scheinberg P, Nunez O, Wu CO, Young NS, Walsh TJ. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011; 52(6):726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.