

Abstract
Long non-coding RNAs are defined as transcripts larger than 200 nucleotides but without protein-coding potential. There is growing evidence of the important role of long non-coding RNAs in cancer initiation, development and progression. In this study, we sought to evaluate the long non-coding RNA expression profile of patients with cytogenetically normal acute myeloid leukemia (AML). RNA-sequencing of 40 cytogenetically normal AML patients allowed us to quantify 11,036 long non-coding RNAs. Among these, more than 8000 were previously undescribed long non-coding RNAs. Using unsupervised analysis, we observed a specific long non-coding RNA expression profile dependent on the mutational status of the NPM1 gene. Statistical analysis allowed us to identify a minimal set of 12 long non-coding RNAs capable of discriminating NPM1-mutated from NPM1-wild-type patients. These results were validated by qRT-PCR on an independent cohort composed of 134 cytogenetically normal AML patients. Furthermore, we have identified one putative biomarker, the long non-coding RNA XLOC_109948 whose expression pattern predicts clinical outcome. Interestingly, low XLOC_109948 expression indicates a good prognosis especially for NPM1-mutated patients. Transient transfection of GapmeR against XLOC_109948 in NPM1-mutated OCI-AML3 cell line treated with Ara-C or ATRA enhances apoptosis suggesting XLOC_109948 plays a role in drug sensitivity. This study improves our knowledge of the long non-coding RNA transcriptome in cytogenetically normal AML patients. We observed a distinct long non-coding RNA expression profile in patients with the NPM1 mutation. The newly identified XLOC_109948 long non-coding RNA emerged as a strong prognostic factor able to better stratify NPM1-mutated patients.
Introduction
Acute myeloid leukemia (AML) is a clinically and biologically heterogeneous disease with marked differences in survival following intensive chemotherapy. These differences are based on age, blast cell count, cytogenetic abnormalities, and gene mutations.1 Patients with cytogenetically normal AML (CN-AML) account for approximately 50% of all AML and are currently categorized in the intermediate-risk group.2 However, this large group of AML is clinically and molecularly heterogeneous. In recent years, the identification of several gene mutations, deregulated coding genes, and the aberrant expression of several microRNAs have provided important prognostic tools and a more complete understanding of the molecular basis of AML.3 However, important questions about the molecular mechanisms underlying AML development and progression remain unanswered. To date, most efforts have been focused on genetic alterations that affect protein-coding genes. Therefore, the discovery of genes that code for long non-coding RNAs (lncRNAs) could reveal a new set of players that participate in AML development. LncRNAs are defined as RNA transcripts that are larger than 200nt but do not appear to have protein-coding potential.4 Recent studies have demonstrated that lncRNAs regulate many processes such as transcription,5 translation,6 epigenetic modification,5,7 cellular differentiation,8 and cell cycle regulation.9 There is growing evidence that also points towards an important role for lncRNAs in cancer initiation, development, and progression.10 Recently, Garzon et al. showed that some deregulated lncRNAs are associated with recurrent mutations and clinical outcome in AML.11 However, our overall knowledge of lncRNA expression patterns in hematologic malignancies remains very limited.12,13 The aim of our study was to better decipher the lncRNA transcriptome and to assess their prognostic role in patients with CN-AML.
Methods
AML samples
Cohort 1 was composed of 40 AML samples collected from patients registered at the HIMIP (Hémopathies INSERM Midi-Pyrénées, France) collection (Online Supplementary Table S1). For the validation cohort (Cohort 2, n=134), 34 AML samples were obtained from the HIMIP collection, 7 AML samples from the Hematology Institute of Perugia University, Italy, and 93 AML samples from the FILOtheque AML, Paris, France (Online Supplementary Table S1). The study was approved by local ethics committees.
RNA-Sequencing Library Preparation, Read Generation and Mapping
RNA sequencing was performed at the BGI, Hong Kong, for samples from Cohort 1 (n=40). rRNA depletion was performed from total RNA with Ribo-Zero™ rRNA Removal Kits (Epicentre, Madison, WI, USA). Paired-end, strand-specific reads of 91 nt were generated on an Illumina HiSeqTM2000. Alignment and mapping were performed using Tophat against the hg19 genome, and the mapped reads were assembled by Cufflinks v.2.0.2.
Fluidigm 96.96 dynamic array integrated fluidic circuits
LncRNA expression analysis was performed using the BioMark 96×96 gene expression platform (Fluidigm) according to the manufacturer’s instructions. Online Supplementary Table S2 shows the list of primers used in the experiment. RNAseP, 5S rRNA and MLN51 were used as reference genes for RT-qPCR data normalization. The Biomark System was used to run the chips and data were collected with Fluidigm Real-Time PCR analysis software.
Statistical analysis
Differential expression analysis was performed with the EdgeR package on R and cut offs were established at a fold change of 4 or more, and False Discovery Rate (FDR) less than 0.001. Fisher’s exact test and the Mann Whitney test were used to assess the statistical significance of the differences between groups. Survival curves were generated by the Kaplan-Meier method and P values were calculated by the log-rank test.
Cell culture
The OCI-AML3 cells were grown in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin and 100 U/mL streptomycin at 37°C in humidified atmosphere containing 5% CO2.
Transient transfection of GapmeRs: the antisense LNA GapmeRs were designed and supplied by Exiqon (Online Supplementary Table S2). OCI-AML3 cells were transiently transfected with 50 nM (final concentration) of GapmeR mediated by Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions.
Apoptosis assay
Apoptosis was induced one day post transfection with Cytarabine (Ara-C, Sigma) or Retinoic acid (ATRA, Sigma) at a final concentration of 10 μM and 1 μM, respectively. The cells were harvested at day 1 after induction with Ara-C and day 2 after induction with ATRA and stained using Pacific Blue™ Annexin V Apoptosis Detection Kit with PI (Biolegend) according to the manufacturer’s instructions. AnnexinV/PI stained samples were analyzed using a MACSQuant-10 flow cytometer (Miltenyi Biotec) and further analysis was performed using FlowJo.
Additional methods are described in the Online Supplementary Materials and Methods.
Results
Identification and quantification of lncRNAs expressed in CN-AML by RNA-sequencing
In this study, we sought to determine the lncRNA expression profiles of CN-AML samples. Our first goal was to determine whether specific signatures could be associated with common AML molecular characteristics. We focused our attention on the well-known AML mutations in NPM1,14 FLT3 (FLT3-ITD),15 CEBPa,16 DNMT3a,17 and IDH (IDH1R132 and IDH2R140).18 Forty CN-AML patients were selected such that around 30% of patients carried each mutation (Figure 1, Cohort 1, and Online Supplementary Table S1). We then used RNA sequencing to identify and determine the lncRNA transcriptome (method described in the Online Supplementary Appendix). RNA sequencing was performed on rRNA-depleted total bone marrow samples in order to evaluate the entire lncRNA landscape (polyadenylated and non-polyadenylated transcripts). This approach allowed us to identify 11036 lncRNAs expressed amongst our CN-AML patients. Among them, 8526 were new lncRNAs that had not been previously reported in the RefSeq, UCSC or ENCODE databases (GRCh37/hg19).
Figure 1.

Overview of the study design. RNA-sequencing was performed on rRNA-depleted total bone marrow of 40 cytogenetically normal acute myeloid leukemia (CN-AML) patients (Cohort 1) to determine the lncRNA transcriptome. A validation set composed of 134 new CN-AML patients (Cohort 2) was used to validate the results. Downregulation of XLOC_109948 lncRNA was performed in OCI-AML3 cells transfected with GapmeRs. sPLS-DA: Sparse partial least squares discriminant analysis.
Unsupervised analysis of the 11036 CN-AML lncRNAs using hierarchical clustering (Figure 2A) segregated the samples into two main groups. When different criteria [namely French-American-British classification (FAB) status, age and presence of mutations] were evaluated in order to define the two groups, the main observation was that NPM1-mutated patients were largely enriched in the first group (80%) compared to the second group (8%) (P=6×10−6). This result suggested there is an lncRNA expression profile that is specific to NPM1-mutated patients.
Figure 2.

Specific lncRNA expression profile for NPM1-mutated AML patients with normal cytogenetics. (A) Unsupervised hierarchical clustering analysis of 40 patients with cytogenetically normal acute myeloid leukemia (CN-AML) (Cohort 1: NPM1+, n=14; NPM1wt, n=26) using 11065 lncRNAs (RNA-seq data). (B) Hierarchical clustering and associated heatmap of Fluidigm data from the first cohort of CN-AML patients (n=40) with 31 lncRNAs differentially-expressed between NPM1-mutated (n=14) and NPM1-wild-type patients (n=26). (C) Hierarchical clustering and associated heatmap of Fluidigm data from the second cohort of 134 CN-AML patients [NPM1-mutated (n=80) and NPM1-wild type (n=54)]. The heatmap depicts high expression (red: +1) and low expression (blue: −1).
Specific lncRNA signature in CN-AML with NPM1 mutation
To identify the differentially-expressed lncRNAs that were associated with NPM1 mutations in the 40 CN-AML patients, we compared lncRNA expression in NPM1-mutated AML patients (n=14) with NPM1-wild type AML samples (n=26), using the EdgeR package. We focused our study on the highly-expressed lncRNAs by selecting those that had at least 10 cpm (counts per million) in at least 14 of the 40 patients. Among the 5333 lncRNAs selected, 107 were significantly (>4-fold change) differentially-expressed (FDR<10−3) between the two groups, with 26 of these up-regulated in NPM1-mutated patients and 81 down-regulated.
To validate our strategy we then applied the same approach to mRNAs and compared mRNA expression in NPM1-mutated and NPM1-wild-type AML patients. The 71 most differentially-expressed mRNAs included the HOX genes, which were up-regulated, and the CD34, BAALC and MN1 genes, which were down-regulated (Online Supplementary Table S3). These have already been described in several previous studies and validate our lncRNA approach.19
Among the 107 lncRNAs that were found to be differentially-expressed in NPM1-mutated patients, 74 are located within the introns or exons of protein-coding genes and 33 are intergenic or anti-sense lncRNAs. To avoid the quantification of pre-mRNA by RT-qPCR, we focused our attention on the 33 intergenic and anti-sense lncRNAs (Online Supplementary Table S4). Hierarchical clustering of these clearly separated NPM1-mutated from NPM1-wild-type AML patients, as shown in Online Supplementary Figure S1A (P=1.16×10−9). In order to validate the RNA-seq data, we used RT-qPCR (Fluidigm), but were not able to amplify lncRNA XLOC_051554. A Pearson’s correlation between the two techniques showed a large positive correlation for all the lncRNAs except XLOC_085385 (Online Supplementary Table S5). Hierarchical clustering using the 31 lncRNAs validated by Fluidigm confirmed the separation of the NPM1-mutated and NPM1-wild-type groups (P=1.16×10−9) (Figure 2B). In order to validate this lncRNA signature, a validation set (n=134) composed of 80 new NPM1-mutated AML patients and 54 NPM1-wild-type patients was used (Figure 1, Cohort 2, and Online Supplementary Table S1). Hierarchical clustering again allowed us to discriminate between NPM1-mutated and NPM1-wild-type patients (P=2.60×10−17) (Figure 2C).
Identification of a minimal signature composed of 12 lncRNAs able to discriminate NPM1-mutated patients from NPM1-wild-type patients
In order to reduce the number of discriminating lncRNAs we then used the Sparse partial least squares discriminant analysis (Sparse PLS-DA) approach on the 31 lncRNAs identified from the RNA-seq data. This allowed us to identify a minimal set of 12 lncRNAs that were able to discriminate NPM1-mutated patients from NPM1-wild-type patients in both Cohort 1 (P=5.17×10−9) and Cohort 2 (P=9.07×10−20) (Figure 3 and Online Supplementary Figures S1B and S2).
Figure 3.

A minimal set of 12 lncRNAs is able to discriminate between NPM1-mutated and NPM1-wild-type acute myeloid leukemia (AML) patients. (A) Sparse partial least squares discriminant analysis (sPLS-DA) plot of NPM1-mutated versus NPM1-wild-type patients based on 12 discriminating lncRNAs. (B) Variable plot of the 12 discriminative lncRNAs. (C) Hierarchical clustering of 40 cytogenetically normal acute myeloid leukemia (CN-AML) patients (Cohort 1) and associated heatmap of the 12 lncRNA signature identified by Sparse PLS-DA to compare NPM1-mutated (NPM1+) patients with NPM1-wild-type (NPM1wt) patients (Fluidigm data). (D) Hierarchical clustering of 134 CN-AML patients (Cohort 2: NPM1+, n=80; NPM1wt, n=54) and associated heatmap of the 12 lncRNA signature identified by Sparse-PLS-DA (Fluidigm data).
XLOC_109948 lncRNA expression serves as a prognostic biomarker in CN-AML
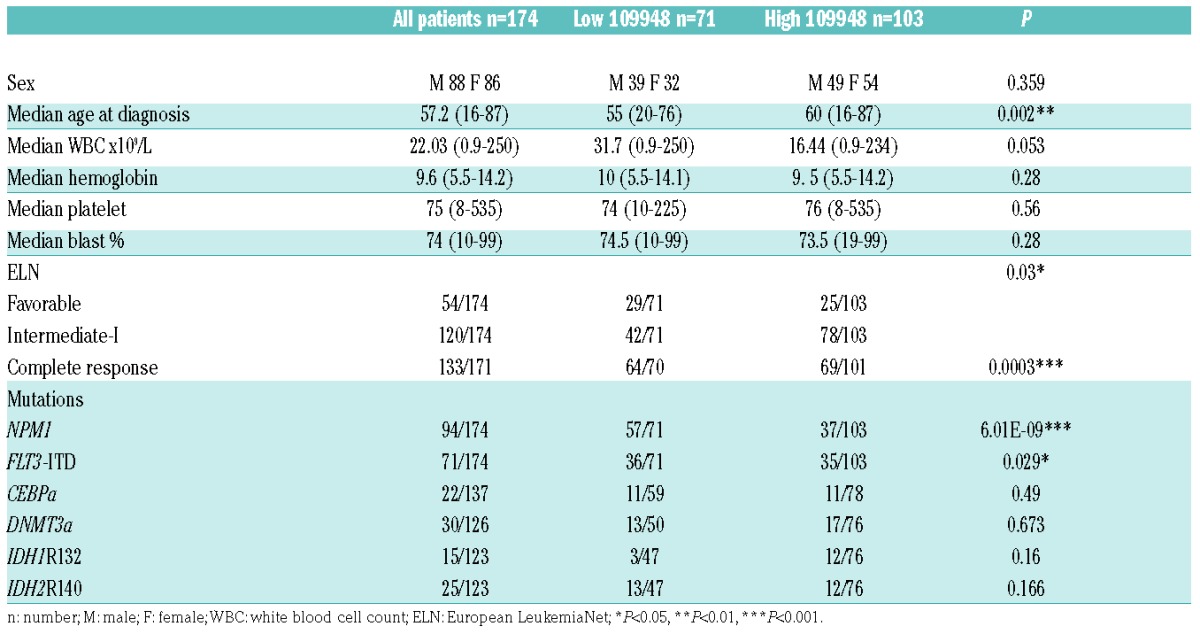
Although the lncRNAs identified in the minimal signature were differentially expressed between NPM1-mutated and NPM1-wild-type patients, we noticed a heterogeneous expression among patients. We asked whether one of them could be used as a prognostic biomarker. To this end, we selected patients who matched the following criteria: 1) those who had received intensive chemotherapy; and 2) those for whom we had clinical data available. This accounted for 25 of the 40 patients in Cohort 1 and all 134 patients in Cohort 2 (Figure 1 and Online Supplementary Table S6). We used ROC curves to predict the best cut off (−ΔCt value) for each lncRNA for event-free survival (EFS) in Cohort 1. This cut off was then used to define subgroups with a high or low expression of each lncRNA and the log-rank test was used to compare the survival distribution in the two groups. Only XLOC_005798 and XLOC_109948 were found to be good putative biomarkers for EFS in Cohort 1 (Figure 4A). Using the previously identified cut off, both lncRNAs could also separate patients according to overall survival (OS) and disease-free survival (DFS) in Cohort 1 (Figure 4A). In order to validate these lncRNAs as good biomarkers, we tested EFS, OS and DFS in the validation cohort (Cohort 2, n=134) using the cut off identified for Cohort 1 (Figure 4A). These analyses allowed us to validate XLOC_109948 as a good biomarker in CN-AML. Indeed, patients in Cohort 2 with high and low XLOC_109948 transcript levels had median EFS times of 474 and 1128 days, respectively, and estimated 5-year EFS rates of 22% and 45%, respectively (P=0.003) (Figure 4B). Patients with high XLOC_109948 expression levels also had significantly worse OS (estimated 5-year OS values of 36% and 55%, respectively; P=0.021) and DFS (estimated 5-year DFS values of 28% and 48%, respectively; P=0.046) than those with low XLOC_109948 levels (Figure 4B). Interestingly, we also observed that XLOC_109948 is almost unexpressed in CD33+ bone marrow cells from healthy samples (Online Supplementary Figure S3). Table 1 lists the clinical characteristics of the entire study population (Cohort 1 and Cohort 2) and of the patient subgroups distinguished by high versus low expression of XLOC_109948 lncRNA. We found that high XLOC_109948 expression was associated with older patients (P=0.002) and that the patients with low XLOC_109948 expression were enriched in the favorable ELN group (P=0.03) and had higher complete remission (CR) rates (P=0.0003). Low expression of XLOC_109948 also strongly correlated with NPM1+ (P=6.01×10–9) and FLT3-ITD (P=0.029) mutations.
Figure 4.

LncRNA XLOC_109948 expression levels can predict clinical outcome. (A) Clinical outcomes in patient subgroups defined according to cut offs (−ΔCt value) and identified with ROC curves for each lncRNA. (B) Prognostic value of XLOC_109948 lncRNA expression in a validation cohort (Cohort 2, n=134) of cytogenetically normal acute myeloid leukemia (CN-AML) patients. Kaplan-Meier plots show the event-free survival (EFS), overall survival (OS) and disease-free survival (DFS) of patient subgroups with high versus low transcript levels of XLOC_109948 lncRNA. (C) Multivariate analyses of clinical outcome in 134 patients (Cohort 2). (D) Risk stratification of patients with CN-AML according to NPM1 mutational status and XLOC_109948 expression level (Cohort 2). EFS, OS and DFS are shown for the four subgroups defined by NPM1 and XLOC_109948 status. P values are given for the overall comparison across all four groups. optcut: optimal cut off; HR: hazard ratio.
Table 1.
Disease and patients’ characteristics according to XLOC_109948 expression level.
Given the correlations we had observed between the various molecular risk markers, we then performed multivariate analyses to identify the factors that independently predicted prognosis in CN-AML. Multivariate analysis was conducted for Cohort 2 (n=134). In a multivariate model, XLOC_109948 expression was a significant prognostic factor for EFS (P=0.008) and DFS (P=0.026). NPM1 mutational status, age and WBC count were also prognostic factors for EFS, and NPM1 mutational status and age for DFS (Figure 4C). Patients with high XLOC_109948 expression had shorter OS rates (P=0.042) than those with low expression after analysis adjustment for WBC count (Figure 4C).
Finally, we investigated patient stratification by using a combination of XLOC_109948 expression and NPM1 mutational status. Patients with low XLOC_109948 expression and mutated NPM1 constituted a favorable subset of patients in terms of EFS (P=0.0002), OS (P=0.007) and DFS (P=0.003) (Figure 4D). These results suggest that XLOC_109948 lncRNA may be a useful marker in the risk stratification of patients with CN-AML, especially among patients with the NPM1 mutation.
XLOC_109948 lncRNA downregulation induces apoptosis in OCI-AML3 cells
As XLOC_109948 low expression was found to be of good prognosis for patients with NPM1 mutation, we decided to down-regulate it in the only available NPM1-mutated AML cell line, OCI-AML3.20
In order to adopt the best strategy to inactivate the XLOC_109948 lncRNA, we first evaluated its subcellular localization by OCI-AML3 cell fractionation. XLOC_109948 is mainly located into the nucleus (Figure 5A). Since the efficiency of siRNAs in the nucleus remained controversial,21 we chose to use a GapmeR strategy. GapmeR are antisense LNA oligonucleotides which are able to induce the cleavage of the target RNA by endogenous RNase H, a ubiquitous enzyme cleaving the RNA part of RNA/DNA hybrids.22
Figure 5.

Downregulation of XLOC_109948 lncRNA enhances drug sensitivity in OCI-AML3 cell line. (A) Subcellular localization of XLOC_109948 lncRNA. The RNA level of XLOC_109948 in nuclear and cytoplasmic fraction was evaluated by RT-qPCR after OCI-AML3 cell fractionation. GAPDH was a positive control for cytoplasmic fraction and Snord44 was a positive control for nuclear fraction. (B) Quantification of XLOC_109948 expression in OCI-AML3 cells transiently transfected with two different GapmeRs against XLOC_109948 (a and b), GapmeR Negative Control or water. The RNA expression levels were evaluated by quantitative real-time PCR, normalized to the expressions of TBP and ABL1, and presented as fold change [2−ΔΔCt]±Standard Deviation (SD) (n≥3) relative to cells transfected with the GapmeR Negative Control; ***P<0.0005. (C and D) Apoptosis assay. One day post transfection, cells were treated with (C) Ara-C (10 μM) or (D) ATRA (1 μM), and annexinV/PI staining was performed respectively 24 hours (h) or 48 h later. One representative flow cytometry plot is shown. The histogram represents the average of apoptotic cells (Annexin V+) from four independent experiments. *P<0.05, **P<0.01.
Transient transfections of OCI-AML3 cells with two different GapmeR against XLOC_109948, a GapmeR Control or H2O were performed and the efficiency of XLOC_109948 downregulation was evaluated by RT-qPCR 48 h post transfection. Both GapmeRs against XLOC_109948 were able to down-regulate it with respectively 30% and 70% of efficiency (Figure 5B).
In order to evaluate the role of XLOC_109948 in drug sensitivity, we treated OCI-AML3 transfected cells with Ara-C (10μM) or ATRA (1μM), two drugs used in the clinic for AML treatment, and apoptosis was measured by flow cytometry 24 h post treatment for Ara-C and 48 h post treatment for ATRA. Both GapmeRs against XLOC_109948 enhanced apoptosis in cells treated with Ara-C or ATRA compared to cells transfected with the GapmeR control or water (Figure 5C and D).
Discussion
The analysis of 200 AML genomes as part of the Atlas Genome Project has highlighted the genetic and epigenetic architecture of CN-AML.3 It has also provided important new information on the genetic alterations involved in CN-AML development and has identified potential tools for risk stratification of CN-AML, as well as proposing new diagnostic and therapeutic targets. However, the studies based on this data have so far mainly focused on the genetic alterations that impact either on protein coding genes or on microRNAs. The discovery of long non-coding RNAs (lncRNAs) has provided a unique opportunity to identify new biomarkers and key players in leukemogenesis.
In this study, we sought to determine lncRNA expression profiles in CN-AML and to correlate them with the most common mutations underlying CN-AML and outcome. Using RNA sequencing approach, we detected a broad spectrum of lncRNA molecules (11036 lncRNAs). Compared with the previous study of Garzon et al., who quantified lncRNA expression by microarray,11 our results improve the overall knowledge of lncRNAs, as our RNA-seq approach has led to the identification of more than 8000 new lncRNAs.
A major finding of this study is the discovery that NPM1-mutated AML patients show a distinct global lncRNA expression profile. NPM1 is a nucleo-cytoplasmic shuttling protein mainly localized in the nucleolus that plays a key role in many biological processes, including ribosome biogenesis,23 histone assembly,24 and the maintenance of genomic stability.25,26 In addition, by regulating the activity and stability of crucial tumor suppressors such as ARF27 and p53,28 NPM1 can also affect cell proliferation and apoptosis. With rare exceptions, NPM1 mutations in AML occur in exon 12 and result in the loss of two key tryptophans, creating a new nuclear export signal leading to aberrant cytoplasmic NPM1 localization.14 NPM1 mutations have been reported to occur in up to 60% of CN-AML adult cases14 and are related to distinct expression profiles of both mRNA (including the downregulation of CD34 and the upregulation of HOX genes) and microRNA (including the upregulation of miR-10a and miR-10b).19 When taken together with other features of NPM1 mutations, such as their mutual exclusivity with other AML recurrent cytogenetic abnormalities, their high specificity for AML, their stability at relapse, and their association with unique gene expression and microRNA profiles,29 our findings that NPM1-mutated AML displays a distinct lncRNA signature further supports the view that this leukemia represents a distinct disease entity, in accordance with the recent update of the WHO classification.30 By using differential and statistical analysis, we have identified a minimal signature of 12 lncRNAs that are able to discriminate between NPM1-mutated and NPM1-wild-type patients.
NPM1 mutations are associated with a favorable prognosis in the absence of an accompanying internal tandem duplication mutation in FLT3 (FLT3-ITD), and with an intermediate prognosis if FLT3-ITD co-exists.29 In our study, we showed that low expression of XLOC_109948 lncRNA is significantly associated with better prognosis, especially among NPM1-mutated patients, thus constituting a potentially useful new biomarker to better stratify the risk for NPM1-mutated CN-AML patients. In accordance with this observation, we demonstrated that inactivation of XLOC_109948 sensitizes NPM1-mutated OCI-AML3 cell line to drug treatment, suggesting that XLOC_109948 quantification could be used to predict the response to chemotherapy.
Altogether, our data suggest that lncRNAs should be considered for use as biomarkers and could be used as therapeutic targets in the pathogenesis of AML. Determining the molecular mechanisms of these lncRNAs will be of great interest in the future.
Supplementary Material
Acknowledgments
The authors would like to thank all participating investigators from the GOELAMS. We also thank V. De Mas and S. Bertoli for the update of the clinical information for patients collected at the HIMIP. We thank C. Daugrois for her help with multivariate analysis. English proofreading was performed by Scientific Scripts (http://scientificscripts.com).
Footnotes
Funding
MB was supported by a fellowship from ARC (Association pour la Recherche sur le Cancer). EDC was supported by a fellowship from LABEX. This work was also supported by the Fondation ARC pour la Recherche sur le Cancer, the Association Laurette Fugain, the Ligue Régionale Contre le Cancer (Comités du Gers et de la Haute-Garonne), the Institut Universitaire de France (PB) and by the Associazione Italiana per la Ricerca sul Cancro (AIRC, IG 2013 n.14595).
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/10/1718
References
- 1.Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23(26):6285–6295. [DOI] [PubMed] [Google Scholar]
- 2.Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109(2):431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long non-coding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vance KW, Ponting CP. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014;30(8):348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrieri C, Cimatti L, Biagioli M, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491(7424):454–457. [DOI] [PubMed] [Google Scholar]
- 7.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14(11):699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu W, Yuan B, Flygare J, Lodish HF. Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev. 2011;25(24):2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitagawa M, Kitagawa K, Kotake Y, Niida H, Ohhata T. Cell cycle regulation by long non-coding RNAs. Cell Mol Life Sci. 2013;70(24):4785–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang G, Lu X, Yuan L. LncRNA: A link between RNA and cancer. Biochim Biophys Acta. 2014;1839(11):1097–1109. [DOI] [PubMed] [Google Scholar]
- 11.Garzon R, Volinia S, Papaioannou D, et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc Natl Acad Sci USA. 2014;111(52):18679–18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morlando M, Ballarino M, Fatica A. Long Non-Coding RNAs: New Players in Hematopoiesis and Leukemia. Front Med. 2015;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans JR, Feng FY, Chinnaiyan AM, et al. The bright side of dark matter: lncRNAs in cancer. J Clin Invest. 2016;126(8):2775–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–266. [DOI] [PubMed] [Google Scholar]
- 15.Fröhling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002; 100(13):4372–4380. [DOI] [PubMed] [Google Scholar]
- 16.Pabst T, Mueller BU, Zhang P, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27(3):263–270. [DOI] [PubMed] [Google Scholar]
- 17.Ley TJ, Ding L, Walter MJ, et al. DNMT3A Mutations in Acute Myeloid Leukemia. N Engl J Med. 2010;363(25):2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcucci G, Maharry K, Wu Y-Z, et al. IDH1 and IDH2 Gene Mutations Identify Novel Molecular Subsets Within De Novo Cytogenetically Normal Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J Clin Oncol. 2010;28(14):2348–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker H, Marcucci G, Maharry K, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28(4):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quentmeier H, Martelli MP, Dirks WG, et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005; 19(10):1760–1767. [DOI] [PubMed] [Google Scholar]
- 21.Lennox KA, Behlke MA. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016; 44(2):863–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurreck J, Wyszko E, Gillen C, Erdmann VA. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002;30(9):1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y, Maggi LB, Brady SN, et al. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol. 2006;26(10):3798–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okuwaki M, Matsumoto K, Tsujimoto M, Nagata K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001;506(3):272–276. [DOI] [PubMed] [Google Scholar]
- 25.Okuda M, Horn HF, Tarapore P, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103(1):127–140. [DOI] [PubMed] [Google Scholar]
- 26.Ziv O, Zeisel A, Mirlas N, et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nat Comm. 2014;25;5:5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colombo E, Bonetti P, Lazzerini Denchi E, et al. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol. 2005;25(20):8874–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurki S, Peltonen K, Latonen L, et al. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5(5):465–475. [DOI] [PubMed] [Google Scholar]
- 29.Falini B, Martelli MP, Bolli N, et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011;117(4):1109–1120. [DOI] [PubMed] [Google Scholar]
- 30.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.