

Abstract
Genetic alterations of the transcription factor IKZF1 (“IKAROS”) are detected in around 15–30% of cases of BCR-ABL-negative B-cell precursor acute lymphoblastic leukemia. Different types of intragenic deletions have been observed, resulting in a functionally inactivated allele (“loss-of-function”) or in “dominant-negative” isoforms. The prognostic impact of these alterations especially in adult acute lymphoblastic leukemia is not well defined. We analyzed 482 well-characterized cases of adult BCR-ABL-negative B-precursor acute lymphoblastic leukemia uniformly treated in the framework of the GMALL studies and detected IKZF1 alterations in 128 cases (27%). In 20%, the IKZF1 alteration was present in a large fraction of leukemic cells (“high deletion load”) while in 7% it was detected only in small subclones (“low deletion load”). Some patients showed more than one IKZF1 alteration (8%). Patients exhibiting a loss-of-function isoform with high deletion load had a shorter overall survival (OS at 5 years 28% vs. 59%; P<0.0001), also significant in a subgroup analysis of standard risk patients according to GMALL classification (OS at 5 years 37% vs. 68%; P=0.0002). Low deletion load or dominant-negative IKZF1 alterations had no prognostic impact. The results thus suggest that there is a clear distinction between loss-of-function and dominant-negative IKZF1 deletions. Affected patients should thus be monitored for minimal residual disease carefully to detect incipient relapses at an early stage and they are potential candidates for alternative or intensified treatment regimes. (clinicaltrials.gov identifiers: 00199056 and 00198991).
Introduction
IKAROS family transcription factors have been identified as key players in lymphopoiesis.1–5 Alterations of IKZF1 in acute lymphoblastic leukemia (ALL) were first described in isolated cases in the early 1990s6,7 but it took several years to recognize the important role of IKZF1 in ALL development.8,9 The crucial role of IKZF1 in ALL development has also recently been underlined by the finding that certain non-coding single nucleotide polymorphisms in IKZF1 predispose to B lineage ALL development in later life.10–12
The first larger studies on the incidence and role of IKZF1 alterations in ALL were exclusively conducted on pediatric patients and revealed a prevalence of 15–30% of IKZF1 alterations in BCR-ABL-negative ALL3,9 compared with a particularly large fraction in BCR-ABL-positive ALL (more than 60%).8,13 IKZF1-alterated BCR-ABL-negative pediatric ALL patients were reported to have an adverse prognosis9,14–17 although this is still a subject of dispute.18 The negative prognostic effect was even found within BCR-ABL-positive pediatric19 and adult13,20 patients.
In adult BCR-ABL-negative ALL patients, studies suggested a worse outcome for IKZF1-mutated patients, albeit there have been inconsistent results concerning the prognostic impact of different IKZF1 alterations (Online Supplementary Table S1).21–24 Furthermore, to the best of our knowledge, the effect of multiple IKZF1 alterations or the impact of mutation load25,26 has not been systematically studied in this population.
The IKZF1 gene comprises eight exons, of which the first is non-coding. Its gene product is a 519 amino acid protein with six zinc finger domains.4 The two carboxyterminal zinc fingers (exon 8) are responsible for dimerization with other IKAROS family members.27 The four amino-terminal zinc fingers (exons 4–6) mediate DNA binding. Besides point mutations and the loss of the complete IKZF1 gene, various intragenic types of deletions have been experimentally observed. Loss of two or more amino-terminal zinc fingers encoded by exons 4–6 with deletion of the binding domain but retention of the dimerization domain results in dominant-negative isoforms, i.e. an isoform able to suppress the function of wild-type protein.27 Loss of exon 2 with the ATG start codon abolishes gene transcription at all and loss of exon 8 removes the dimerization domain. The latter two have historically been called “haploinsufficient”.3 Since this term implies that the other allele is still functional, which could only be proven with certainty by single cell analysis, we will use the term “loss-of-function” for these alterations.
In this study, we present an in-depth analysis of 482 BCR-ABL-negative patients with B-precursor ALL with regard to their IKZF1 status. Patients were treated uniformly in the framework of the German Multicenter ALL (GMALL) studies between 1999 and 2009. We present a detailed genetic analysis and an assessment of the prognostic impact of the various IKZF1 alterations.
Methods
Patients’ samples
Originally, 507 patients with BCR-ABL-negative B-cell precursor (BCP) ALL were studied (Figure 1). Four were excluded because of irreproducible results, and 21 for missing follow-up data (of these only breakpoint sequences are presented).
Figure 1.

Flowchart of the analysis.
Of the remaining 482 patients who were treated within the GMALL protocols 06/99 (n=84; clinicaltrials.gov identifier: 00199056) or 07/03 (n=398; clinicaltrials.gov identifier: 00198991), we analyzed bone marrow (n=330) or peripheral blood with peripheral blasts (n=132; bone marrow or peripheral blood not specified in n=20) obtained at the time of diagnosis between 1999 and 2009 (for blast count see Online Supplementary Tables S2 and S3). Matched samples from the time of relapse were available for 16 out of 482 patients
GMALL studies
Detailed information on treatment has been published previously.28 The GMALL studies were approved by the ethics committee of the University of Frankfurt, Germany, and by local ethics committees of participating institutions, and were conducted according to the Declaration of Helsinki.
Immunophenotyping and molecular genetic analysis
At the time of diagnosis, immunophenotyping and molecular genetic analysis were performed at the GMALL central laboratory in Berlin, Germany. For all BCP-ALL patients, BCR-ABL status was determined by RT-PCR. Other molecular targets (TCF3-PBX1, ETV6-RUNX1 and MLL fusion genes) were analyzed according to our diagnostic guidelines as outlined previously.29,30
Genomic PCR for Δ4–7, Δ2–7, Δ4–8, Δ2–8
For all patients, genomic PCR was performed using HotStarTaq Polymerase Mastermix (QIAGEN) with 40–200 ng DNA and 500 nM of each primer under the following conditions: 15 minutes (min) at 95°C, followed by 35 cycles of 30 seconds (sec) at 94°C, 30 sec at 65°C and 60 sec at 72°C. Primers were located in intron 1 (F2A ACTACAGAGACTTCAGCTCTATTCCATTTC, F2B TGATTTGGATGTGTGTGTTTCATGCGTGG), intron 3 (F4 CTTAGAAGTCTGGAGTCTGTGAAGGTC), intron 7 (R7 AGGGACTCTCTAGACAAAATGGCAGGA) and 3′UTR of IKZF1 (R8 CCTCCTGCTATTGCACGTCTCGGT). For primer combinations see Online Supplementary Table S4. In all PCRs, a fragment of intron 7 or 3′UTR was amplified as internal control with primer concentration of 100 nM (F7 ACCATCAAATACAGGTCAACAGGACTGA, product 1,257 bp) or 50 nM (F8 CCCACTGCACAGATGAACAGAGCA, product 1,229 bp). Primers were manufactured by metabion (Munich, Germany) or TIB Molbiol (Berlin, Germany) and HPLC-purified.
Reverse transcriptase PCR
RT-PCR was performed with 2 μl cDNA, 500 nM of each primer and the HotStarTaq Polymerase Mastermix (QIAGEN) using the following conditions: 15 min at 95°C, followed by 35 cycles of 30 sec at 94°C, 30 sec at 64°C, and 60 sec at 72°C. Primers were located in exons 1 and 8 (RT-PCR ex1/8, primers ex1FA AAAGCGCGACGCACAAATCCA and ex8R CGTTGTTGATGGCTTGGTCCATCAC) or in exon 1 and exon 4 for detection of Δ2–3 (RT-PCR ex1/4, primers ex1FB CGAGGATCAGTCTTGGCCCCAA and ex4R GAATGCCTCCAACTCCCGACAAAG). Long IKZF1 isoforms were used as internal control. Bands of unexpected sizes were excised from the gel and sequenced.
In cases where RNA was not available for RT-PCR, we used our own and the PCR described by Meyer et al.31 as genomic screening PCR.
Quantitative PCR for Δ4–7, Δ2–7, Δ4–8
Quantitative PCR was performed in duplicates either for all patients (Δ4–7) or for patients positive in genomic PCR (Δ2–7 and Δ4–8) using a Rotorgene 6000 cycler (Corbett, Concorde, Australia), the Thermo Scientific ABsolute QPCR Mix (Life Technologies, Darmstadt, Germany) with 200–250 ng DNA per PCR and the following conditions: 15 min at 95°C, followed by 55 cycles for 15 sec at 95°C, and 60 sec at 60°C.
As DNA standard, we used the cell-line BV-173 for Δ4–7 (DSMZ, Braunschweig, Germany)32 or patient DNA (#100 for Δ2–7, #101 for Δ4–8). A PCR for the HCK gene served as internal control as described earlier.33 Oligonucleotides are given in Online Supplementary Table S4. Deletions were considered to be present in a large fraction of leukemic cells (“high deletion load”, “high-del”) when the relative PCR signal was >10−1, otherwise they were considered having a “low deletion load” (“lowdel”). The cut-off value was chosen a priori since this threshold appeared to separate samples with a high and low mutation load (Online Supplementary Figure S1). We used MLPA (SALSA MLPA P335 ALL-IKZF1 kit, MRC Holland, Amsterdam, the Netherlands) to correlelate the cut-off values of our quantitative PCRs with MLPA deletion values. We investigated a subset of patients with qPCR signals that we expected to yield a MLPA reduction of 0.3 or more (i.e. qPCR signal of 0.6 or higher). The chosen thresholds distinguishing high-del and lowdel corresponded to 5% deleted alleles in case of Δ2–7 and Δ4–7, and 10% in Δ4–8, but the latter could equally well have been placed at 5%, since there were no samples between 5% and 10%.
In cases negative for Δ4–7 by conventional PCR but positive by qPCR, qPCR measurements were repeated and were considered positive when at least 3 out of 4 measurements were positive.
Gel densitometry
When no quantification by qPCR was possible (n=41), we assessed the relative amount of cells with IKZF1 deletions (high vs. low deletion load) by gel band densitometry using the AlphaEaseFC v.4.0 software (Alpha Innotech, San Leandro, CA, USA). In deletions Δ2 (n=1) and Δ2–3 (n=17, missing values n=2), we compared deleted isoforms to full-length isoforms on RT-PCR images with a cut-off value of 0.60. In deletions Δ2–7 (n=5), Δ4–7 (n=3) and Δ5–7 (n=1) we compared deleted with long bands on RT-PCR images using a cut-off value of 1.20. In Δ2–8 (n=10) and Δ4–8 (n=2) we calculated the ratio of short PCR products to the long PCR control band with a cut-off value of 1.20.
Supplementary methods
Nucleic acid preparation, identification of rare genomic break-points (primer sequences specified in Online Supplementary Table S5),31 DNA sequencing, bioinformatic analysis,34 and statistical analysis are all described in the Online Supplementary Methods.
Results
Patients’ characteristics
All 482 patients were aged between 16 and 65 years at diagnosis (Online Supplementary Table S6). The median age was 32 years [interquartile range (IQR) 22–47]. Two hundred and eighty-five patients (59%) were male. The distribution of immunophenotypes was 111 pre-B ALL (cyIg+; 23%), 314 common ALL (cyIg−,CD10+; 65%) and 57 pro-B ALL (CD10−; 12%). Two hundred and fourteen patients (44%) were considered high risk, the remaining standard risk. All patients were BCR-ABL-negative and a MLL rearrangement was detected in 44 patients (39 MLL-AF4, 4 MLL-ENL, 1 MLL-AF9), a TCF3-PBX1 fusion in 30, and an ETV6-RUNX1 fusion in 3 cases.
Frequency of IKZF1 deletions
Two RT-PCRs were used to detect short IKZF1 isoforms (Figure 2A and Online Supplementary Figure S2A–C) and four separate PCRs to detect the Δ2–7, Δ2–8, Δ4–7 and Δ4–8 isoforms (Figure 2B–F). Deletions were then quantified using quantitative PCR or gel densitometry. Dominant-negative deletions (Δ4–7, Δ5–7) were compared to loss-of-function deletions (Δ2, Δ2–3, Δ2–7, Δ2–8, Δ4–8).
Figure 2.

Detection of IKZF1 deletions by RT-PCR and PCR screening. (A–C) RT-PCR ex1/8, PCR Δ4–7 and PCR Δ2–7 of the same 9 patients. (A) RT-PCR with primers in exon 1/8. Increased Ik6 expression in lanes 4–6 and increased Ik10 expression in lanes 6–8. Reduced full length isoform expression in lanes 1 and 7 is attributed to an additional deletion Δ2–3 in these 2 patients detected by another RT-PCR (see Online Supplementary Figure S2). (B) PCR Δ4–7. In lanes 1–3, Δ4–7 is present with a low deletion load; in lanes 4–6, the deletion is present with a high deletion load. Corresponding qPCR results are given below. Control band of 1257bp. (C) PCR Δ2–7 with low deletion load in lanes 3–4 and high deletion load in lanes 6–8. Control band of 1257bp. (D) Structure of the IKZF1 transcript isoforms Ik1 (full-length), Ik6 (loss of exons 4–7) and Ik10 (loss of exons 2–7). (E and F) PCR Δ4–8 and PCR Δ2–8 of the identical patients in lanes 10–17. Control band of 1229 bp. (E) PCR Δ4–8. See double bands in lanes 10 and 11. (F) PCR Δ2–8. See variant breakpoint in lane 17.
Overall, 128 of 482 (27%) patients carried an IKZF1 deletion (Figure 3A). Among these patients, we detected 175 different IKZF1 deletions. While 91 (19%) patients expressed only one deletion, in 37 (8%) patients more than one IKZF1 deletion was detected: 2 (n=28), 3 (n=8) or 4 (n=1) deletions (Online Supplementary Table S7; for an example, see lanes 3, 4 and 6 in Figure 2).
Figure 3.

Prevalence of IKZF1 deletions at the time of diagnosis. (A) Frequency of all deletions as detected by PCR (Δ2–7, Δ2–8, Δ4–7, Δ4–8) and RT-PCR (exon 1/4, exon 1/8). (B) Only deletions classified as high deletion load by quantitative PCR and densitometry.
Among the 175 IKZF1 deletions, Δ4–7 was the most frequent (n=71). Δ2–7 was found in 47, Δ4–8 in 26, Δ2–3 in 19 and Δ2–8 in 10 patients. Rare deletions were Δ5–7 (n=1) and Δ2 (n=1). In summary, 56 patients (12%) carried only loss-of-function deletions, 50 (10%) had only dominant-negative deletions while 22 patients exhibited both types of deletions (5%).
We then quantified the amount of cells with IKZF1 deletions, as a variable deletion load was apparent from gel images (Figure 2B and C). We avoided the simple terminology “clonal” and “subclonal” since we did not prove clonality in a strict sense and did not investigate clonal relationships. Instead, we adopted the terms “high deletion load” (highdel) and “low deletion load” (lowdel) for IKZF1 aberrations present either in the vast majority of leukemic cells or only in a small fraction.
Out of 173 quantifiable deletions (n=2 not quantified), 106 (61%) were considered to have a high deletion load. At least one highdel IKZF1 deletion could be found in 98 of 482 (20%) patients (Figure 3B). Among these, 50 had a highdel loss-of-function deletion only, 44 patients had a highdel dominant-negative deletion only, and there was a group of 4 patients expressing both deletions with a high deletion load level.
qPCR screening revealed 50 additional cases positive for Δ4–7 with a low deletion load not detectable by our conventional PCR. In 41 of these cases, the lowdel Δ4–7 was the only IKZF1 deletion, while in 9 cases a loss-of-function deletion had been detected by conventional PCR. Patients with a lowdel Δ4–7 detected by qPCR only were considered IKZF1 wild-type.
Prognostic impact of IKZF1 deletions
Four hundred and twenty-eight (89%) patients reached a complete remission, 31 patients (6%) died during induction, and 23 patients (5%) had a treatment failure after induction. The overall survival was 55% at five years.
We first calculated the effect of any IKZF1 deletion (n=128 vs. wild-type n=354) and then analyzed loss-of-function (n=78 vs. negative n=404) and dominant-negative deletions (n=72 vs. negative n=410) separately. We compared the effect of high to low deletion load and no deletion in the group of loss-of-function (n=54/23/404, missing value n=1) and dominant-negative deletions (n=48/24/410).
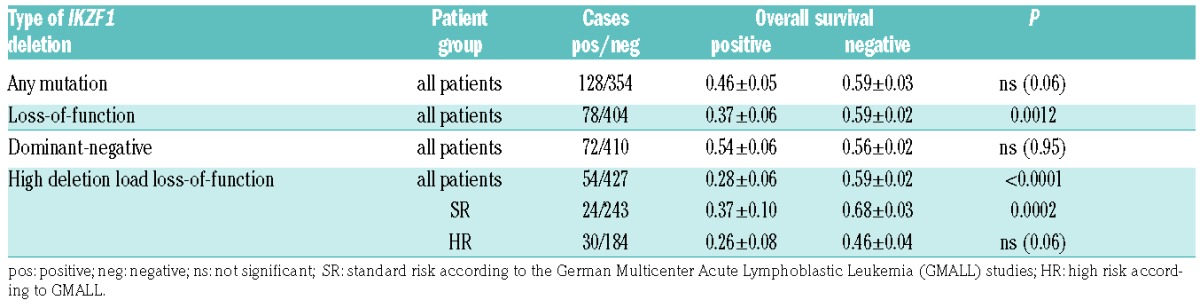
There was a non-significant trend towards inferior overall survival (OS) for patients with any IKZF1 deletion (0.46 vs. 0.59; P=0.06) (Online Supplementary Figure S3A). Patients carrying a loss-of-function IKZF1 deletion had a reduced OS (0.37 vs. 0.59; P=0.0012) (Figure 4A) while dominant-negative deletions had no effect on OS (0.54 vs. 0.56; P=0.95) (Figure 4B). Patients with both dominant-negative and loss-of-function deletions showed a clinical course comparable to loss-of-function deletions only (Online Supplementary Figure S3B). Analysis of the amount of IKZF1-deleted cells showed that the inferior survival in loss-of-function deletions was an effect of highdel loss-of-function deletions only (Figure 4C). Lowdel loss-of-function deletions did not influence the clinical course. In dominant-negative deletions, OS was not associated with the relative amount of IKZF1-deleted cells (Figure 4D).
Figure 4.

Overall survival (OS) depending on IKZF1 deletions. (A) OS of patients with loss-of-function IKZF1 deletions. (B) OS of patients with dominant-negative deletions. (C) OS of patients with high or low deletion load loss-of-function IKZF1 deletions. (D) OS of patients with high or low deletion load dominant-negative IKZF1 deletions.
Patients with highdel loss-of-function deletions showed a reduced OS (0.28 vs. 0.59; P<0.0001) (Table 1). In subgroups according to risk stratification, highdel loss-of-function IKZF1 deletions conferred a negative prognostic effect on standard-risk patients (0.37 vs. 0.68; P=0.0002), while in high-risk patients, the trend towards inferior OS narrowly missed statistical significance (0.26 vs. 0.46; P=0.06).
Table 1.
Effect of IKZF1 deletions on overall survival.
Clinico-biological characteristics of patients with IKZF1 deletions
Patients with IKZF1 deletion showed a common immunophenotype significantly more often than patients without IKZF1 deletions (98 in 128, 77%, vs. 216 in 354, 61%; P=0.0064). The former were also significantly more likely to be CD34-positive (112 in 127, 88%, vs. 209 of 353, 59%; P<0.0001; n=2 CD34 N/A). The occurence of IKZF1 deletions was not associated with patients’ age, gender, WBC or GMALL risk group, neither for all deletions (Online Supplementary Table S8) nor for different types of deletion (Online Supplementary Table S9).
TCF3-PBX1 and IKZF1 deletions were mutually exclusive (0 of 30 TCF3-PBX1+ vs. 64 of 250 TCF3-PBX1−; P=0.0004). One in 3 ETV6-RUNX1-positive patients showed an IKZF1 deletion. There was a trend towards a lower frequency of IKZF1 deletions in MLL-positive patients (7 of 44 MLL+, 16% vs. 7 of 26 MLL−, 26%; P=0.3556).
Oligoclonality is more common in loss-of-function deletions
Some patients showed more than one IKZF1 deletion (e.g. Δ2–7 and Δ4–7). Forty out of 175 deletions (23%) showed more than one chromosomal breakpoint resulting in the same type of RNA transcript. This oligoclonality may arise from multiple alterations in a single hyperdipoid clone or from alterations in different clones. This was evident either by gel electrophoresis (9 patients; see lanes 9–10 in Figure 2E and F) or by multiple sequences in chromatograms (2 breakpoints in 5 patients, Figure 5A; more than two breakpoints in 26 patients, Figure 5B). This kind of oligoclonal pattern occurred more often in loss-of-function deletions (31 of 103 deletions, 30%) compared with dominant-negative (9 of 72, 13%; P=0.0064).
Figure 5.

Distribution of IKZF1 breakpoints and clonality of deletions. (A) Chromatogram of patient #189 showing two distinguishable clones (sequenced sense and antisense reverse complement). (B) Chromatogram of patient #395 showing oligoclonality at the breakpoint junction in both sequencing directions. (C) Distribution of breakpoints in the IKZF1 gene locus. Proximal breakpoints are shown in black, distal breakpoints in blue. There are four major breakpoint clusters within intron 1, 3, 7 and 3′UTR of IKZF1.
Breakpoint sequences
Sequencing of 193 breakpoints revealed four clusters (Figure 5C; for all breakpoints see Online Supplementary Table S10). In intron 1, 66 of 83 were located within 30bp. In intron 3, 106 of 108 proximal breakpoints were located within 40bp. All 132 distal breakpoints in intron 7 clustered within 43bp. Thirty-six of 42 breakpoints in the 3′UTR region were located in a 27bp region, and an additional 5 breakpoints clustered around 500bp proximally.
The remaining 17 breakpoints in intron 1 were more diverse, covering a region of 7kb. Distal (3′) breakpoints in intron 3 (Δ2–3) were scattered all over the 40kb intron. In 183 of 193 (95%) molecularly characterized breakpoints, putative cryptic recombination signal sequences, either with 23bp or 12bp spacer, were identified at both breakpoint sites (5′ and 3′). This was the case for the four major breakpoint clusters (Figure 5 and Online Supplementary Table S11) but also true for the majority of the atypical breakpoints in intron 1 and 3. In 10 of 25 atypical break-points, only one cRSS could be identified (8 only on the 3′ site, 2 only on the 5′ site) (Online Supplementary Table S11). There was no evidence of somatic hypermutation near the break sites.
Detection of deletions by RT-PCR
In 13 of 17 patients positive for Δ2–3 in RT-PCR ex1/4, a genomic breakpoint could be identified by eyer et al.’s PCR (Online Supplementary Figure S2A).31 In the remaining 4 patients, breakpoints were identified by a newly developed PCR (Online Supplementary Figure S2B). We also identified Δ2 once by RT-PCR ex1/4 and confirmed the genomic deletion. One patient expressed isoform Δ2–4 in RT-PCR ex1/8 but we could only find a deletion Δ2–3 on the genomic level and no deletion Δ2–4 or Δ4.
RT-PCR revealed 3 patients positive for Ik10 (lacking exons 2–7) but negative for Δ2–7 by genomic PCR due to a more proximal 5′ breakpoint (Online Supplementary Figure S4A). In all 70 cases of RT-PCR positive for Ik6 (lacking exons 4–7) and negative for Ik6Δ (lacking exons 4–7 but with an additional 60 bp cryptic exon 3b),7,35 genomic PCR was positive for deletion Δ4–7. In one patient with Ik6 and Ik6Δ we found two deletions Δ4–7, one with common breakpoints, one with a 5′ breakpoint distal to the 60bp insert (Online Supplementary Figure S4B). The second patient with Ik6/Ik6Δ showed only a deletion Δ5–7 that was supposedly the reason for overexpression of Ik6 and Ik6Δ (Online Supplementary Figure S4C).
Comparison between diagnosis and relapse
DNA at the time of relapse was available from 16 patients carrying 20 IKZF1 deletions. Four in 7 (57%) Δ4–7 and 9 in 13 (69%) loss-of-function deletions were conserved (P=0.65) (Online Supplementary Table S12). Eleven in 15 (73%) highdel and 1 in 4 lowdel deletions were conserved (P=0.12; 1 deletion not quantified). All genomic breakpoints were identical at the time of diagnosis and relapse. No newly acquired deletion Δ2–7, Δ2–7, Δ4–7 or Δ4–8 could be detected in relapse samples. We also investigated 5 relapse samples from patients who had shown a lowdel Δ4–7 IKZF1 deletion at diagnosis, detectable only by quantitative PCR. None of these cases evolved into a major clone, i.e. with high deletion load at relapse.
Discussion
IKZF1 alterations have been recognized as recurrent aberrations in B-precursor ALL but their prognostic impact in adult ALL is still not well defined. Two major studies involving more than 200 patients have focused on the prognostic impact in BCR-ABL-negative adult BCP ALL.
Moorman et al.21 investigated 304 patients and found IKZF1 deleted patients (29%) to have a lower OS, but this was only seen in a univariate analysis. The authors stated cautiously that “there was evidence to suggest that the poor outcome was not linked to the expression of the IK6 isoform but rather to other types of IKZF1 deletions”.21 Beldjord et al.22 investigated 216 younger adults and observed a significantly higher cumulative incidence of relapse in patients with focal IKZF1 alterations (25%) but not with whole gene deletion. No statistically significant difference between patients with different focal alterations was observed.
Our present study included 482 homogenously treated patients and revealed IKZF1 alterations in 128 cases. The incidence of focal deletions (27%) was comparable to both studies mentioned above. Our study is the first to systematically address the issue of IKZF1 mutation load and its implications for prognosis on a larger scale. This is of diagnostic interest if IKZF1 alterations are to be used as molecular markers for risk stratification and/or for detecting minimal residual disease.15,26 Ninety-eight patients revealed a high deletion load IKZF1 aberration while 29 patients showed low deletion load IKZF1 alterations only (n=1 not quantified). Regarding clinical implications, only high deletion load loss-of-function IKZF1 alterations were of prognostic relevance and conferred an adverse prognosis while low deletion load IKZF1 alterations or dominant-negative IKZF1 alterations did not have a prognostic effect.
In animal studies, double IKZF1 knock-out mice show a total absence of B cells.36 Mice with only IKZF1 deletions did not develop BCP ALL, but haploinsufficiency of IKZF1 in BCR-ABL-transgenic mice significantly accelerated the development of BCP ALL.37 Current evidence suggests that IKZF1 alterations alone are not sufficient to cause leukemia in humans but are an important co-factor or secondary event in the development and acceleration of ALL disease.
It may seem unexpected that the loss of one IKZF1 allele without apparent functional alteration of the other allele should have such a significant prognostic effect. However, this is supported by the above mentioned mouse model of Virely et al.37 The observation that loss-of-function IKZF1 deletions frequently occur in a small fraction of cells, but only seem to have an impact on prognosis if they are found in a large fraction, requires some explanation. A hypothetical explanation is the assumption that RAG-mediated IKZF1 deletions occur sporadically during all stages of B-cell maturation because of the ongoing process of VDJ recombination.38,39 However, only those IKZF1 aberrations occurring at a very early maturation stage are thought to result in a cell phenotype with the full capacity of self-renewal, i.e. a “leukemia stem cell phenotype”.40 IKZF1 alterations occurring at later stages of B-cell maturation should result in low deletion load aberrations.
The extremely narrow clustering of breakpoints in regions comprising only a few nucleotides strongly argues in favor of a specific mechanism. The analysis of the breakpoint junctions revealed four breakpoint clusters in the vicinity of recombination signal sequences suggestive of a break mechanism involving the immunoglobulin VDJ recombination enzyme complex. RAG1 and RAG2 and other genes involved in VDJ rearrangement are not expressed at a very early stage of differentiation but only after lymphoid committment,41 which would be in line with the assumption that IKZF1 deletions are a later event in the path towards the malignant phenotype. The fact that cRSS could not be identified in 10 out of 193 breakpoints may be explained by limitations of the RSSsite software, since some of these breaks occurred in near vicinity, suggesting a specific mechanism.
The PCR method used in this study has the advantage that it can also detect IKZF1 alterations in a small fraction of leukemic cells, which is not possible when using MLPA.26 Since we analyzed the final IKZF1 cDNA transcript, we were in principle also able to detect deletions or aberrant splice isoforms arising from alterations involving only a few nucleotides that would escape detection by MLPA. However, MLPA has the advantage of also detecting whole gene deletions that are not detectable with our PCR-based approach. As long as there are no reliable PCR-based detection methods for the former, and given the fact that low deletion load alterations are prognostically irrelevant, we consider MLPA to be a suitable detection method.
To summarize, we detected partial IKZF1 gene deletions in approximately 27% of cases of adult BCR-ABL-negative adult ALL. Only high deletion load loss-of-function IKZF1 alterations, but not dominant-negative IKZF1 alterations, had negative prognostic implications and should thus be monitored closely, while those that were found in a small fraction of cells did not influence prognosis. We report extensive molecular data on these alterations which should help to establish suitable diagnostic methods for their detection and which shed additional light on the molecular pathogenesis.
Supplementary Material
Acknowledgments
The authors are grateful for the excellent technical work of D. Gröger, R. Lippoldt and colleagues and the members of the MPI sequencing team in Cologne. They thank all involved patients and physicians for participating in the GMALL studies. TB was supported by DFG grant BU 2453/1-1.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/10/1739
References
- 1.Georgopoulos K, Bigby M, Wang JH, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79(1):143–156. [DOI] [PubMed] [Google Scholar]
- 2.Georgopoulos K. Haematopoietic cell-fate decisions, chromatin regulation and ikaros. Nat Rev Immunol. 2002;2(3):162–174. [DOI] [PubMed] [Google Scholar]
- 3.Kastner P, Dupuis A, Gaub MP, Herbrecht R, Lutz P, Chan S. Function of Ikaros as a tumor suppressor in B cell acute lymphoblastic leukemia. Am J Blood Res. 2013;3(1):1–13. [PMC free article] [PubMed] [Google Scholar]
- 4.Olsson L, Johansson B. Ikaros and leukaemia. Br J Haematol. 2015;169(4):479–491. [DOI] [PubMed] [Google Scholar]
- 5.John LB, Ward AC. The Ikaros gene family: Transcriptional regulators of hematopoiesis and immunity. Mol Immunol. 2011;48(9–10):1272–1278. [DOI] [PubMed] [Google Scholar]
- 6.Sun L, Heerema N, Crotty L, et al. Expression of dominant-negative and mutant isoforms of the antileukemic transcription factor Ikaros in infant acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1999;96(2):680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L, Crotty ML, Sensel M, et al. Expression of dominant-negative Ikaros isoforms in T-cell acute lymphoblastic leukemia. Clin Cancer Res. 1999;5(8):2112–2120. [PubMed] [Google Scholar]
- 8.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–114. [DOI] [PubMed] [Google Scholar]
- 9.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treviño LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burmeister T, Bartels G, Gröger D, et al. Germline variants in IKZF1, ARID5B, and CEBPE as risk factors for adult-onset acute lymphoblastic leukemia: an analysis from the GMALL study group. Haematologica. 2014;99(2):e23–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009;27(31):5202–5207. [DOI] [PubMed] [Google Scholar]
- 14.Kuiper RP, Waanders E, van der Velden VH, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24(7):1258–1264. [DOI] [PubMed] [Google Scholar]
- 15.Waanders E, van der Velden VH, van der Schoot CE, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. 2011;25(2):254–258. [DOI] [PubMed] [Google Scholar]
- 16.Dörge P, Meissner B, Zimmermann M, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. 2013;98(3):428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clappier E, Grardel N, Bakkus M, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children’s Leukemia Group study 58951. Leukemia. 2015;29(11):2154–2161. [DOI] [PubMed] [Google Scholar]
- 18.Palmi C, Valsecchi MG, Longinotti G, et al. What is the relevance of Ikaros gene deletions as a prognostic marker in pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia. Haematologica. 2013;98(8):1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. 2014;123(11):1691–1698. [DOI] [PubMed] [Google Scholar]
- 20.DeBoer R, Koval G, Mulkey F, et al. Clinical impact of ABL1 kinase domain mutations and IKZF1 deletion in adults under age 60 with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL): molecular analysis of CALGB (Alliance) 10001 and 9665. Leuk Lymphoma. 2016;57(10):2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moorman AV, Schwab C, Ensor HM, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. 2012;30(25):3100–3108. [DOI] [PubMed] [Google Scholar]
- 22.Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123(24):3739–3749. [DOI] [PubMed] [Google Scholar]
- 23.Mi JQ, Wang X, Yao Y, et al. Newly diagnosed acute lymphoblastic leukemia in China (II): prognosis related to genetic abnormalities in a series of 1091 cases. Leukemia. 2012;26(7):1507–1516. [DOI] [PubMed] [Google Scholar]
- 24.Dhédin N, Huynh A, Maury S, et al. Role of allogeneic stem cell transplantation in adult patients with Ph-negative acute lymphoblastic leukemia. Blood. 2015; 125(16):2486–2496. [DOI] [PubMed] [Google Scholar]
- 25.Dupuis A, Gaub MP, Legrain M, et al. Biclonal and biallelic deletions occur in 20% of B-ALL cases with IKZF1 mutations. Leukemia. 2013;27(2):503–507. [DOI] [PubMed] [Google Scholar]
- 26.Caye A, Beldjord K, Mass-Malo K, et al. Breakpoint-specific multiplex polymerase chain reaction allows the detection of IKZF1 intragenic deletions and minimal residual disease monitoring in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2013;98(4):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun L, Liu A, Georgopoulos K. Zinc finger-mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. EMBO J. 1996; 15(19):5358–5369. [PMC free article] [PubMed] [Google Scholar]
- 28.Brüggemann M, Raff T, Flohr T, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107(3):1116–1123. [DOI] [PubMed] [Google Scholar]
- 29.Burmeister T, Meyer C, Schwartz S, et al. The MLL recombinome of adult CD10-negative B-cell precursor acute lymphoblastic leukemia: results from the GMALL study group. Blood. 2009;113(17):4011–4015. [DOI] [PubMed] [Google Scholar]
- 30.Burmeister T, Gökbuget N, Schwartz S, et al. Clinical features and prognostic implications of TCF3-PBX1 and ETV6-RUNX1 in adult acute lymphoblastic leukemia. Haematologica. 2010;95(2):241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer C, zur Stadt U, Escherich G, et al. Refinement of IKZF1 recombination hotspots in pediatric BCP-ALL patients. Am J Blood Res. 2013;3(2):165–173. [PMC free article] [PubMed] [Google Scholar]
- 32.Nakayama M, Suzuki H, Yamamoto-Nagamatsu N, et al. HDAC2 controls IgM H- and L-chain gene expressions via EBF1, Pax5, Ikaros, Aiolos and E2A gene expressions. Genes Cells. 2007;12(3):359–373. [DOI] [PubMed] [Google Scholar]
- 33.Burmeister T, Marschalek R, Schneider B, et al. Monitoring minimal residual disease by quantification of genomic chromosomal breakpoint sequences in acute leukemias with MLL aberrations. Leukemia. 2006;20(3):451–457. [DOI] [PubMed] [Google Scholar]
- 34.Merelli I, Guffanti A, Fabbri M, et al. RSSsite: a reference database and prediction tool for the identification of cryptic Recombination Signal Sequences in human and murine genomes. Nucleic Acids Res. 2010;38 (Web Server Issue):W262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payne KJ, Dovat S. Ikaros and tumor suppression in acute lymphoblastic leukemia. Crit Rev Oncog. 2011;16(1–2):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JH, Nichogiannopoulou A, Wu L, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5(6):537–549. [DOI] [PubMed] [Google Scholar]
- 37.Virely C, Moulin S, Cobaleda C, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia.[letter]. Leukemia. 2010;24(6):1200–1204. [DOI] [PubMed] [Google Scholar]
- 38.Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood. 2009;114(10):2159–2167. [DOI] [PubMed] [Google Scholar]
- 39.Yu W, Nagaoka H, Jankovic M, et al. Continued RAG expression in late stages of B cell development and no apparent reinduction after immunization. Nature. 1999;400(6745):682–687. [DOI] [PubMed] [Google Scholar]
- 40.Warner JK, Wang JC, Hope KJ, Jin L, Dick JE. Concepts of human leukemic development. Oncogene. 2004;23(43):7164–7177. [DOI] [PubMed] [Google Scholar]
- 41.Nagaoka H, Yu W, Nussenzweig MC. Regulation of RAG expression in developing lymphocytes. Curr Opin Immunol. 2000;12(2):187–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.