

Abstract
Splenic diffuse red pulp lymphoma is an indolent small B-cell lymphoma recognized as a provisional entity in the World Health Organization 2008 classification. Its precise relationship to other related splenic B-cell lymphomas with frequent leukemic involvement or other lymphoproliferative disorders remains undetermined. We performed whole-exome sequencing to explore the genetic landscape of ten cases of splenic diffuse red pulp lymphoma using paired tumor and normal samples. A selection of 109 somatic mutations was then evaluated in a cohort including 42 samples of splenic diffuse red pulp lymphoma and compared to those identified in 46 samples of splenic marginal zone lymphoma and eight samples of hairy-cell leukemia. Recurrent mutations or losses in BCOR (the gene encoding the BCL6 corepressor) – frameshift (n=3), nonsense (n=2), splicing site (n=1), and copy number loss (n=4) – were identified in 10/42 samples of splenic diffuse red pulp lymphoma (24%), whereas only one frameshift mutation was identified in 46 cases of splenic marginal zone lymphoma (2%). Inversely, KLF2, TNFAIP3 and MYD88, common mutations in splenic marginal zone lymphoma, were rare (one KLF2 mutant in 42 samples; 2%) or absent (TNFAIP3 and MYD88) in splenic diffuse red pulp lymphoma. These findings define an original genetic profile of splenic diffuse red pulp lymphoma and suggest that the mechanisms of pathogenesis of this lymphoma are distinct from those of splenic marginal zone lymphoma and hairy-cell leukemia.
Introduction
Splenic diffuse red pulp lymphoma (SDRPL) with circulating villous lymphocytes is a rare indolent B-cell lymphoma involving the spleen, bone marrow and peripheral blood and is characterized by various clinical, morphological and immunological features.1–3 However, SDRPL was defined as a provisional entity in the World Health Organization 2008 classification and in its recent release in 2016, and assigned to the unclassifiable splenic B-cell lymphomas/leukemias.4,5 Indeed, SDRPL may present some overlapping features with other splenic B-cell lymphomas or small B-cell leukemias such as splenic marginal zone lymphoma (SMZL), hairy cell leukemia (HCL) and, especially, its variant form (HCL-v). The differential diagnosis may be difficult because of the absence of pathognomonic diagnostic markers. Recurrent mutations have been reported in HCL (BRAF V600E), HCL-v (MAP2K1) and in SMZL (KLF2, NOTCH2), indicating characteristic mutational patterns and distinctive oncogenic pathways in each of these entities.6–13 Some mutations in NOTCH1, NOTCH2, MYD88, TP53, MAP2K1, and CCND3 have recently been described in SDRPL, though the studies were non-exhaustive and lacked detailed comparisons with other B-cell malignancies.14–16 In the present study, we explored the genetic landscape of SDRPL using whole-exome sequencing of paired tumor and normal samples. We confirmed and extended our findings through the targeted sequencing of 109 mutations in a validation series of SDRPL and compared these data with those obtained for SMZL and HCL.
Methods
Case selection
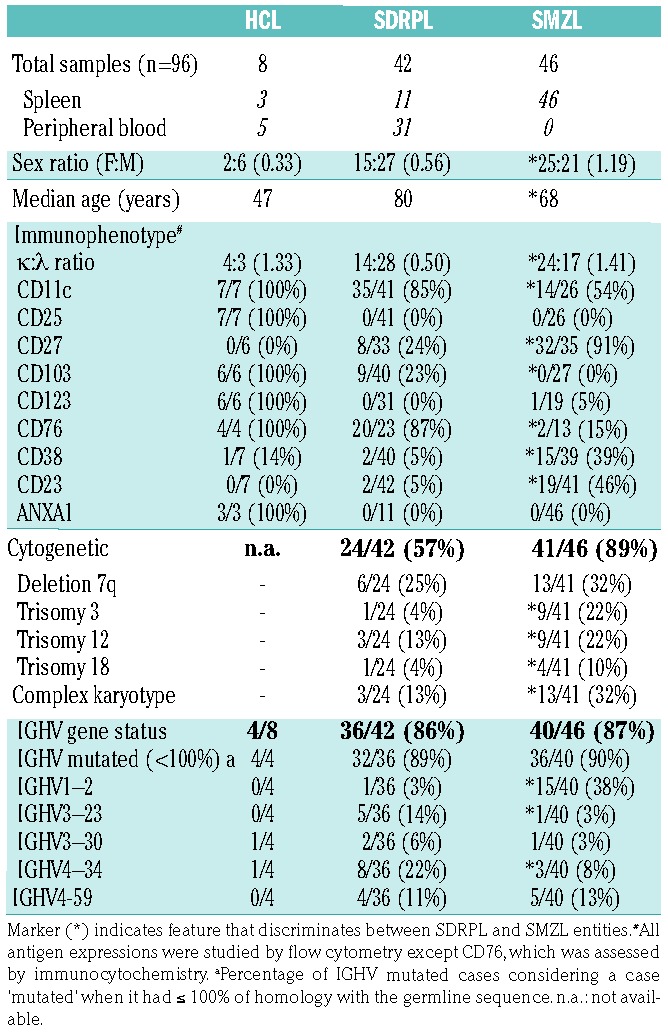
Diagnoses of HCL, SDRPL and SMZL were established by histological analyses of spleen (62 cases) or peripheral blood (38 cases) (Table 1). Given the frequent policy of watchful waiting and the low rate of splenectomy in SDRPL patients, 31 samples were included in the study after a diagnostic procedure based on thorough cytological examination of peripheral blood smears completed with bone marrow analyses, extensive flow cytometry immunophenotyping, and cytogenetic analyses. The immunophenotypic characteristics of SDRPL were previously shown to discriminate this type of lymphoma from other lymphoid malignancies.2,3 In our experience, SDRPL can be clearly distinguished from SMZL using a scoring system based on five membrane markers (CD11c, CD22, CD76, CD27 and CD38) and from HCL given that SDRPL does not co-express typical HCL markers such as CD25, CD103 and CD123.2 However, in some cases, a partial expression of CD103 may be observed in SDRPL. The criteria used to recognize each entity were in accordance with the World Health Organization 2008 classification, completed with recent published updates.2,17–23 The clinico-pathological characteristics of the HCL, SDRPL and SMZL series are detailed in Table 1. Informed consent to participation in this study was obtained from patients, and the procedures were conducted in accordance with the Helsinki Declaration and the Biological Resource Center policy of the Hospices Civils de Lyon. Furthermore, the institutional review board of the Hospices Civils de Lyon approved the research protocol (DC-2015-2566).
Table 1.
Clinico-pathological comparison between the HCL, SDRPL and SMZL cases of our series
Whole-exome sequencing
Whole-exome sequencing was performed on a discovery cohort (flowchart in Online Supplementary Figure S1), which included ten cases of SDRPL (namely, SDRPL #1, 3, 5, 7, 8, 9, 10, 12, 13, and 15). DNA from tumor cells was obtained from seven frozen spleen samples and three positively immunoselected villous lymphoma B-cell (CD19+) samples isolated from peripheral blood (to verify the origin of the samples, see Figure 1). Paired DNA from nonmalignant cells was purified from spleen fibroblasts obtained after culture of the original tissue biopsy or from the non-B-cell fraction obtained after immunoselection using a human anti-CD19 antibody-conjugated magnetic microbead kit according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of the B-cell fractions was determined by flow cytometry and systematically exceeded 90%. Non-B-cell fractions contained less than 5% CD20+ cells.
Figure 1.

Distribution of mutations in hairy-cell leukemia, splenic diffuse red pulp lymphoma and splenic marginal zone lymphoma. Each column represents a type of lymphoma and corresponding tissue (HCL: hairy cell lymphoma; SDRPL: splenic diffuse red pulp lymphoma; SMZL: splenic marginal zone lymphoma; PB: peripheral blood; SP: spleen; B+: CD19+ immunoselected sample). The orange colored boxes indicate the ten cases investigated by whole-exome sequencing. Six of them were also reanalyzed by targeted sequencing as internal controls, whereas the four remaining were not (NR: not resequenced). Each row (top) represents a gene linked to the corresponding RAS/MAPK, NF-κB or NOTCH signaling pathway. Three types of mutations (missense, nonsense, frameshift indel) are highlighted in different colors. Details of the mutations are available in Online Supplementary Table S3. Gene copy number (CN) was only reported as a gain (↑) or loss (↓) for relevant genes (❋), namely BCOR and TNFAIP3. The last row (bottom) represents the IGHV gene status, homology with germline sequence percentage, and cytogenetic findings (complex karyotype, trisomy 3, 12, 18 and deletion 7q) for each case when available.
Genomic DNA was enriched in protein-coding sequences using the in-solution exome capture SureSelect Human All Exon 50-Mb kit (Agilent Technologies) according to the manufacturer’s protocol. The captured targets were subjected to sequencing using the Illumina HiSEQ2000 analyzer (Illumina) with the paired-end 2 × 75 bp read option. Exome capture, massively parallel sequencing and quality controls were performed at IntegraGen (Evry, France). Paired-end reads obtained by high-throughput sequencing were aligned with the human genome reference hg19/NCBI GRCh37, and differences from the reference sequence were identified separately for tumor and normal samples using the CASAVA pipeline (Illumina) (IntegraGen), as well as another pipeline based on BWA-MEM, SAMBAMBA, and GATK (INSERM UMR S910, Marseille, France). Merge analysis, data mining and manual review were performed using VarAFT (http://varaft.eu) and ALAMUT software (Interactive Biosoftware, Rouen, France). To investigate genomic copy number aberrations (e.g., copy number gains and copy number losses), we used the Bioconductor DNACopy package (DNAcopy 1.32.0), comparing the DNA exome data with the paired reference sample data and used the circular binary segmentation algorithm to segment DNA copy number data. All changes were compared to the catalogue of the Database of Genomic Variants to provide a comprehensive summary of structural variations in the human genome.
Targeted gene sequencing
A panel of 109 genes was selected from the exploratory exome data for subsequent analyses; these genes were selected according to their previous description in mutational landscape studies of other B-cell malignancies, to their well-established role in B-cell physiology or to the type of mutation they produce (predicted to be deleterious or damaging according to SIFT and PolyPhen2-score). The genes, exon positions and coverage are listed in Online Supplementary Table S2. High-throughput sequencing was performed on a validation series of 38 SDRPL samples, consisting of six samples from the discovery cohort used as positive controls (namely, SDRPL #1, 3, 5, 8, 9 and 12) and 32 new SDRPL samples, as well as 46 SMZL and eight HCL samples. The origin of the samples is shown in Figure 1. DNA was extracted from frozen spleen samples, from CD19+ immunoselected cells, or from nonimmunoselected peripheral blood mononuclear cells after Ficoll-isolation using patients’ samples with a total lymphocyte count exceeding 5×109/L (mean 14×109/L; minimum: 5.8×109/L; maximum: 92×109/L). All of the spleen tissue samples were infiltrated with over 70% B-cell lymphoma tumor cells.
Library preparation, capture, sequencing, variant detection, and annotation were performed by IntegraGen. Exons of genomic DNA samples were captured using the Agilent in-solution enrichment methodology with the biotinylated oligonucleotide probe library, followed by paired-end 75-bp massively parallel sequencing on an Illumina HiSEQ2000. Image analysis and base calling were performed using the Illumina Real Time Analysis Pipeline version 1.14 and default parameters. Bioinformatics analysis of the sequencing data was based on the Illumina pipeline (CASAVA1.8.2). Only positions included in the bait coordinates were conserved. Genetic variation annotation was performed using the IntegraGen in-house pipeline, which consists of gene annotation (RefSeq) and detection of known polymorphisms (dbSNP 132, 1000Genome) followed by characterization of the mutations (exonic, intronic, silent, nonsense, etc.). For each position, exomic frequencies were determined using the IntegraGen Exome database, and exome results were provided by HapMap.
The procedure for detecting copy number variation was adapted from previously published methods; read depths of each exon from all of the target genes were obtained for each sample using DeCovA.24,25 Exon read depths (ERD) were then normalized against the sum of all ERD from the same sample. A copy number ratio (CNR) was obtained by dividing the ERD of the sample by the median ERD of all of the samples as a control: CNRx=(ERDx/∑ERD1>n)sample/(ERDx/∑ERD1>n)control. For the two genes located on chromosome Xp (BCOR and KDM6A), a 2-fold extrapolation was applied to the normalized ERD for males. Copy number ratios <0.7 and >1.25 were considered to represent a loss and gain, respectively. Chromosomal abnormalities (chromosome X loss or gain) were confirmed by copy number variation detection by whole-exome sequencing using a circular binary segmentation algorithm and compared to that of the paired reference genome or karyotype analysis when available.
Microarray-based comparative genomic hybridization
A 60K oligonucleotide microarray (Agilent Technologies) was used according to the manufacturer’s instructions to confirm the microdeletion of the BCOR locus, identified by the detection, by whole-exome sequencing, of copy number variation (SDRPL case: VL_218).
Results
Whole-exome sequencing of the discovery cohort of SDRPL resulted in the identification of more than 300 unique somatic mutations among the ten different tumor samples (Online Supplementary Table S1). These included exon missense (83%), frameshift (5%), nonsense (4%), untranslated regions (5%), and intron splicing site (3%) alterations. Some of the mutated genes were previously associated with various other malignancies. These mutated genes encode proteins in various pathways, including cell cycle regulation (CCND3, MGA, MYC), epigenetic regulation (BCOR, EZH1, HIST1H1D, HIST1H2AD, HIST4H4, KAT6A, KDM6A, NCOA6), the RAS-MAPK pathway (HRAS, KIF26A, NRAS, RAF1, WNK1), NF-κB signaling (IKBKB, TBK1, TRAF3), NOTCH signaling (NOTCH1, NOTCH2, DTX3L, DTX1, SPEN), and cytoskeleton and cell-matrix interactions (ARHGAP20, ARHGEF15, ARHGEF17, ROCK1, MYLK, DOCK6, DNAH5, DNAH7, DNAI1, RAPGEF2, RFTN1, DSP, DTNB). In the SDRPL discovery cohort, we detected only three recurrently mutated genes [CCND3 (cyclin D3), HIST4H4 (histone cluster 4, H4), and RFTN1 (raftlin, lipid raft linker 1)] in two of the ten SDRPL samples (Online Supplementary Table S1).
Given the characteristic mutational profiles previously identified in other B-cell malignancies and after extensive manual review and data mining of the SDRPL discovery cohort, we selected a panel of 109 target genes the functions of which may be relevant during lymphomagenesis and the types of mutations of which were predicted by the SIFT and PolyPhen2-score to be deleterious or damaging (Online Supplementary Table S2). This panel was used to further explore the validation cohort, including SDRPL, SMZL and HCL samples, by high-throughput sequencing and to assess the frequency of mutations in each of these lymphoma/leukemia entities. All of the mutations detected by whole-exome sequencing were confirmed by targeted gene sequencing for SDRPL cases, sequenced using both methodologies. All of the samples of the validation cohort, except one, displayed at least one of the 109 target gene mutations, with the number of mutations ranging from 1–13 in some samples. Only a few recurrent and discriminative mutations were identified and are described hereafter, with a promising candidate being mutations or losses in BCOR since these affected 10/42 SDRPL patients compared to 1/46 patients with SMZL and 0/8 of those with HCL (Figure 1).
Recurrent BCOR mutations and deletions in splenic diffuse red pulp lymphoma
A single mutation in a splicing site of BCOR was initially detected in the discovery cohort and was confirmed to be somatic (Figure 1 and Online Supplementary Table S3). This mutation was identified in the same splicing site position as another mutation reported in the COSMIC database as being COSM521431. Additional BCOR mutations were identified in five SDRPL cases of the validation set. These mutations were distributed along the gene (Figure 2) within exons 4, 5 and 11 (BCOR gene coverage: 100%; mean depth analysis: 345 reads). These mutations were characterized by splicing site (1/6), nonsense (2/6) and frameshift (3/6) alterations. The mutations exhibited a high variant allele frequency, with the exception of two frameshift mutations that exhibited a lower variant allele frequency. The tumor cell content was elevated in these two cases, which also harbored some mutations in other genes with a higher variant allele frequency, possibly indicating a subclonal change (Online Supplementary Table S3). Finally, the pattern of these mutations strongly suggests that they would result in loss of function of BCOR.
Figure 2.

BCOR mutations in splenic diffuse red pulp lymphoma, lymphoid and hematologic malignancies. The distribution of mutations along the BCOR sequence was drawn using cBioPortal (http://www.cbioportal.org). Mutation diagram circles are colored with respect to the corresponding mutation types. Six distinct mutations were detected in our SDRPL series (top), compared to mutations reported in lymphoid (middle) or hematologic (bottom) malignancies.43,44 BCOR: BCL-6 corepressor, non-ankyrin-repeat region (1202 - 1414); Ank_2: Ankyrin repeats (3 copies) (1467 - 1560); PUFD: BCORL-PCGF1-binding domain (1634 - 1747).
Since such loss of function may arise from the deletion of BCOR on chromosome Xp11.4, we further analyzed our data to look for copy number variations. A single BCOR allele is supposed to be functional in both males (one copy on the single chromosome X) and females (because of X-inactivation). Copy number losses of the BCOR locus were identified by analysis of exon read depths in four SDRPL female patients displaying no BCOR mutation within the single remaining allele (Figure 3A). Three of these losses involved the whole chromosome X, whereas the last deletion was a microdeletion of approximately 669 kb assessed by microarray-comparative genomic hybridization [arr[GRCh37] Xp11.4(39576746_40245183)×1], only targeting an uncharacterized long non-coding RNA (LOC101927476) and the BCOR gene (Figure 3B). Two copy number gains of the BCOR locus were also observed in two SDRPL cases. In one male patient, who also had a BCOR frameshift mutation, the copy number gain explained the observed variant allele frequency of 40% in that individual (VL#208) (Figures 1 and 3A, Online Supplementary Table S3). Some of these abnormalities were confirmed by copy number variation detection from whole-exome sequencing or by karyotype analysis when available (Figure 3). Taken together, these findings demonstrated an alteration in BCOR in 11/42 SDRPL cases, most of them (10/11) being potentially pathogenic (mutations or loss). Furthermore, the percentage of BCOR alterations in cases with spleen histology (2/11 cases, 18%) was similar to that of the cases without spleen histology (8/31, 26%).
Figure 3.

Copy number evaluation at the BCOR Xp11.4 locus in hairy-cell leukemia, splenic diffuse red pulp lymphoma and splenic marginal zone lymphoma. (A) Copy number (CN) variation was detected from exon sequencing of the target genes after normalization using DeCovA.25,24 Each row represents a lymphoma case (HCL: hairy-cell lymphoma; SDRPL: splenic diffuse red pulp lymphoma; SMZL: splenic marginal zone lymphoma). Each column represents an exon of two different genes (BCOR and KDM6A) located at locus Xp11. A progressive gradation of CN variation distinguishes loss (red) and gain (green). Right: mean copy number ratio of the BCOR locus, with some references in brackets corresponding to the next inserts (B–G). (B) Microarray-based comparative genomic hybridization confirmed a monoallelic microdeletion of approximately 669 kb (arr[GRCh37] Xp11.4(39576746_40245183)×1), encompassing the BCOR locus (SDRPL female patient VL_218), whereas the KDM6A gene was unaffected. (C–D) Copy number variation of chromosome X detected by whole-exome sequencing using a circular binary segmentation algorithm and compared to a paired reference genome (log2 ratio). These two female patients with SDRPL acquired complete monosomy X. (E–G) Detailed karyotype results confirmed monosomy X (E), tetrasomy X (F) or trisomy X (G).
A single BCOR frameshift mutation (distinct from the previous ones identified in SDRPL samples) was detected in the 46 SMZL samples (2%) (Figure 1 and Online Supplementary Table S3). Three copy number variations of the BCOR locus were also observed in three SMZL patients, and in all three cases entailed a gain. No BCOR alteration (mutation, gain or deletion) was observed in the eight HCL samples.
MAP2K1 and BRAF mutations
While no MAP2K1 (MEK1) mutation was detected in the SMZL samples (Figure 1), three mutated cases were observed in SDRPL samples (3/42; 7%). Two mutations (MAP2K1 p.I103N and p.C121S) were previously reported in HCL-v.7 Of the three patients with MAP2K1 mutations in our series, two also harbored a BCOR mutation (VL#201, VL#208). In contrast to HCL-v cases with MAP2K1 mutations, these two SDRPL cases did not use the IGHV4-34 gene, as indicated in Online Supplementary Table S3 (comments column).
Whereas BRAF p.V600E is the hallmark of HCL, we identified one distinct BRAF mutation (p.G469A) in 1/42 SDRPL samples (2%). This case was characterized by an unmutated immunoglobulin IGHV4-34 sequence and a non-HCL immunological profile (CD25−/CD103+/CD123−) and harbored both BRAF p.G469A and MAP2K1 p.I103N mutations but no BCOR mutation.
A single sample of the 46 SMZL (2%) cases harbored both a BRAF p.V600E mutation and a frameshift mutation of KLF2 (MZ#104). The diagnosis of SMZL was confirmed after further review of the spleen histology in this patient, who also had a typical IGHV1-2*04 rearrangement and trisomy 3q.
KLF2, TNFAIP3 and MYD88 mutations
Mutations in KLF2, TNFAIP3 and MYD88, which are known to activate the NF-κB pathway, were observed in 14/46 (30%), 9/46 (20%) and 4/46 (9%) of SMZL patients, respectively (Figure 1 and Online Supplementary Table S3).4,8,11,26 TNFAIP3 and MYD88 mutations were not detected in either SDRPL or HCL samples.
Only one KLF2 mutant (1/42; 2%) was observed in an SDRPL patient. The diagnosis of SDRPL in this case was based on cytological features of a peripheral blood smear with more than 60% villous lymphoma cells among the lymphoid cells, an immunological SDRPL score of 0/5 (dimCD11c+/dimCD22+/CD38−/CD27−/CD76+) and the fact that IGHV3-23 usage is more frequently observed in SDRPL than SMZL.2 One other KLF2 mutation was detected in a HCL sample, which also displayed a BRAF V600E mutation.
Other mutations found in both splenic diffuse red pulp lymphoma and splenic marginal zone lymphoma
Finally, different mutations (already reported in other B-cell malignancies) likely implicated in the activation of the Notch pathway (i.e., mutations in NOTCH2, NOTCH1, and SPEN) were identified in 7/42 SDRPL (17%) and 14/46 SMZL (30%) samples (Figure 1 and Online Supplementary Table S3).6,8,9
Other mutations were recurrently observed but were distributed across both SDRPL and SMZL samples, in particular CCND3 [in 9 (21%) of 42 SDRPL and 6 (13%) of 46 SMZL samples], BIRC3, TP53, MYC and CXCR4. No additional recurrent mutations were observed in HIST4H4 and RFTN1, previously identified in two cases of the SDRPL discovery cohort. These findings show that, in addition to the original somatic mutation pattern described above, SDRPL can also share some mutations with its closest B-cell malignancy.
Discussion
We explored the mutational landscape of SDRPL with circulating villous lymphocytes in an attempt to identify diagnostic pathognomonic markers associated with this rare unclassifiable (World Health Organization 2016 classification) splenic B-cell lymphoma. One of the limitations of our study was the restricted number of patients having undergone histological spleen analyses, and we thus partially relied on stringent immunophenotypic and cytological criteria to select SDRPL cases from peripheral blood samples. This methodology is consistent with the current medical management of patients suffering from splenic lymphoma, who are preferentially administered rituximab rather than being submitted to splenectomy. However, irrespective of the origin of the SDRPL tissues analyzed, their genetic landscape was clearly distinct from the SMZL and HCL samples used herein as comparative B-cell malignancy cases. Indeed, we identified recurrent BCOR mutations or losses in 10/42 SDRPL cases (24%), while these remained rare in SMZL (1/46) and absent in HCL (0/8). Most BCOR mutations (identified in 6 cases), as well as BCOR deletions (in 4 cases including a microdeletion), were speculated to result in inactivation of gene function. These mutations were not reported in the COSMIC database, except for one case with a BCOR p.S340Vfs*41 mutation similar to the reported pS340* mutation (COSM5945498). There were no particular associations between BCOR alterations and cytogenetic features, IGHV gene usage, or the mutational pattern of SDRPL cases.
Germline BCOR mutations have been detected in patients with inherited oculofaciocardiodental and Lenz microphthalmia syndromes.27 Recently, massively parallel sequencing has identified inactivating somatic BCOR mutations at a very low frequency in patients with various types of solid neoplasia but also in patients with hematologic malignancies, such as acute myeloid leukemia, myelodysplastic syndrome, T-cell prolymphocytic leukemia, and extranodal NK/T-cell lymphoma, nasal type.28–36 Altogether, these data underline the critical role of BCOR in cell differentiation and oncogenesis.
BCOR was first identified as a corepressor whose product interacts specifically with BCL6.37,38 Through epigenetic modifications, the enzymatic activity of the BCOR complex provides a mechanism for silencing BCL6 targets. Although BCOR and BCL6 play key roles in germinal center formation, and BCL6 alterations are involved in the transformation of germinal center B cells, their functions in other B cells remain elusive.39,40 The predicted inactivating mutations of BCOR or acquired hemizygosity observed in SDRPL may lead to the loss of BCL6 repression, but BCL6 expression is not usually detected by immunohistochemistry in SDRPL. Recent data also indicate that BCL6/BCOR inhibits Notch-activated target genes during embryonic development.40 Given the frequency of mutations of genes involved in the Notch pathway in SDRPL (17%), it would be interesting to investigate whether BCOR loss-of-function might represent an alternative mechanism for activating this pathway in B cells. Overall, how BCOR mutations or deletions might participate in SDRPL oncogenesis remains unknown.
Recently, recurrent CCND3 mutations have been described in six of 25 (24%) of another series of SDRPL cases.16 In our series, we also observed recurrent CCND3 mutations in nine of 42 SDRPL cases (21%), but CCND3 mutations were also detected in six of 46 SMZL cases (13%). Case selection based on diagnostics, obtained either following spleen histology or through morphological and immunological examination of peripheral blood, may potentially account for these distinct findings. Further studies are, therefore, required to identify the spectrum of CCND3 mutations among splenic lymphomas more precisely. A nonsense mutation in CDKN1B, the gene that encodes the cyclin-dependent kinase inhibitor and that interacts with cyclin D3, was found in another case of SDRPL.16 The immunohistochemical expression of CCND3 showed a selective expression of cyclin D3 by most neoplastic cells in SDRPL spleen tissues. Considering that cyclin D3 works downstream of BCL6 in germinal center development, it could be hypothesized that dysregulation of the BCL6/BCOR complex on the one hand or CCND3 dysregulation on the other hand may represent two aspects of the same pathophysiology in SDRPL; this remains to be explored more thoroughly.41
Some MAP2K1 mutations were also reported in the present series of SDRPL cases but at a low frequency (7%). In contrast, a high prevalence (48%) of MAP2K1 mutations was previously reported in HCL-v and IGHV4-34-expressing HCL, suggesting a possible degree of overlap between HCL-v and SDRPL.7 Among the 12 lymphoma samples in the present series sharing the IGHV4-34 pattern (8 SDRPL, 3 SMZL and 1 HCL that was BRAF V600E-positive), we identified only one case of IGHV4-34 in a SDRPL patient with a MAP2K1 mutation, also harboring a BRAF G469A mutation (VL#203), but lacking the BCOR mutation. These previously unknown molecular findings may result in tools for the diagnosis of an atypical form of HCL rather than a true case of SDRPL. The two other MAP2K1 mutants were identified in samples with distinct IGHV rearrangements that also harbored a BCOR mutation, which was the most frequent abnormality in our SDRPL series. Further screening of BCOR mutations in the HCL-v series may, therefore, refine our understanding of the relationship between HCL-v and SDRPL.
Finally, the frequent BCOR mutations and the absence of alterations in genes regulating the NF-κB pathway (triple-negative for KLF2, TNFAIP3 and MYD88 mutations) or the absence of a BRAF mutation appear to delineate a specific genetic pattern of SDRPL, which is distinct from that already identified in SMZL, HCL or HCL-v. Exploration of these mutational patterns should improve the differential diagnosis of splenic B-cell lymphoma/leukemia and other lymphoproliferative disorders, in combination with morphological and immunological criteria currently used for their diagnosis. This will be essential for pursuing the biological and clinical characterization of this range of B-cell lymphoproliferative disorders. Our findings also pave the way for additional functional studies to characterize the oncogenic mechanism by which BCOR may promote lymphomagenesis and how this may potentially lead to the development of novel therapeutics.
Supplementary Material
Acknowledgments
We acknowledge Prof Damien Sanlaville, Dr Nicolas Chatron, Audrey Labalme and Jessica Michel for assistance with the microarray-CGH experiment and the Centre de Ressources Biologiques Sudbiothèque of the Hospices Civils de Lyon. We thank Dr Brigitte Manship for improving the use of English in the manuscript and for her critical reading.
Footnotes
Funding
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/10/1758
References
- 1.Traverse-Glehen A, Baseggio L, Bauchu EC, et al. Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: a distinct clinicopathologic and molecular entity? Blood. 2008;111(4):2253–2260. [DOI] [PubMed] [Google Scholar]
- 2.Baseggio L, Traverse-Glehen A, Callet-Bauchu E, et al. Relevance of a scoring system including CD11c expression in the identification of splenic diffuse red pulp small B-cell lymphoma (SRPL). Hematol Oncol. 2011;29(1):47–51. [DOI] [PubMed] [Google Scholar]
- 3.Miguet L, Lennon S, Baseggio L, et al. Cell-surface expression of the TLR homolog CD180 in circulating cells from splenic and nodal marginal zone lymphomas. Leukemia. 2013;27(8):1748–1750. [DOI] [PubMed] [Google Scholar]
- 4.Piris MA, Foucar K, Mollejo M, Campo E, Falini B. Splenic B-cell lymphoma/leukaemia unclassifiable. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. IARC Press Lyon; 2008;191–193. [Google Scholar]
- 5.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364(24):2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34–expressing hairy-cell leukemias. Nat Genet. 2013;46(1):8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossi D, Trifonov V, Fangazio M, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209(9):1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiel MJ, Velusamy T, Betz BL, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209(9):1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martínez N, Almaraz C, Vaqué JP, et al. Whole-exome sequencing in splenic marginal zone lymphoma reveals mutations in genes involved in marginal zone differentiation. Leukemia. 2013;28(6):1334–1340. [DOI] [PubMed] [Google Scholar]
- 11.Clipson A, Wang M, de Leval L, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia. 2014;29(5):1177–1185. [DOI] [PubMed] [Google Scholar]
- 12.Piva R, Deaglio S, Fama R, et al. The Krüppel-like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia. 2014;29(2):503–507. [DOI] [PubMed] [Google Scholar]
- 13.Parry M, Rose-Zerilli MJ, Ljungstrom V, et al. Genetics and prognostication in splenic marginal zone lymphoma: revelations from deep sequencing. Clin Cancer Res. 2015;21(18):4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez D, Navarro A, Martinez-Trillos A, et al. NOTCH1, TP53, and MAP2K1 mutations in splenic diffuse red pulp small B-cell lymphoma are associated with progressive disease. Am J Surg Pathol 2015;40(2):192–201. [DOI] [PubMed] [Google Scholar]
- 15.Traverse-Glehen A, Verney A, Gazzo S, et al. Splenic diffuse red pulp lymphoma has a distinct pattern of somatic mutations amongst B-cell malignancies. Leuk Lymphoma. 2016;58(3):666–675. [DOI] [PubMed] [Google Scholar]
- 16.Curiel-Olmo S, Mondejar R, Almaraz C, et al. Splenic diffuse red pulp small B-cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood. 2017;129(8):1042–1045. [DOI] [PubMed] [Google Scholar]
- 17.Traverse-Glehen A, Baseggio L, Callet-Bauchu E, et al. Hairy cell leukaemia-variant and splenic red pulp lymphoma: a single entity? Br J Haematol. 2010;150(1):113–116. [DOI] [PubMed] [Google Scholar]
- 18.Traverse-Glehen A, Baseggio L, Salles G, Felman P, Berger F. Splenic marginal zone B-cell lymphoma. Curr Opin Oncol. 2011;23(5):441–448. [DOI] [PubMed] [Google Scholar]
- 19.Traverse-Glehen A, Baseggio L, Salles G, Coiffier B, Felman P, Berger F. Splenic diffuse red pulp small-B cell lymphoma: toward the emergence of a new lymphoma entity. Discov Med. 2012;13(71):253–265. [PubMed] [Google Scholar]
- 20.Wotherspoon AC. Extranodal and splenic small B-cell lymphoma. Mod Pathol. 2013;26:S29–S41. [DOI] [PubMed] [Google Scholar]
- 21.Kanellis G, Mollejo M, Montes-Moreno S, et al. Splenic diffuse red pulp small B-cell lymphoma: revision of a series of cases reveals characteristic clinico-pathological features. Haematologica. 2010;95(7):1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baseggio L, Traverse-Glehen A, Petinataud F, et al. CD5 expression identifies a subset of splenic marginal zone lymphomas with higher lymphocytosis: a clinico-pathological, cytogenetic and molecular study of 24 cases. Haematologica. 2010;95(4):604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponzoni M, Kanellis G, Pouliou E, et al. Bone marrow histopathology in the diagnostic evaluation of splenic marginal-zone and splenic diffuse red pulp small B-cell lymphoma. Am J Surg Pathol. 2012;36(11):1609–1618. [DOI] [PubMed] [Google Scholar]
- 24.Pasmant E, Parfait B, Luscan A, et al. Neurofibromatosis type 1 molecular diagnosis: what can NGS do for you when you have a large gene with loss of function mutations? Eur J Hum Genet. 2014;23(5):596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimassi S, Simonet T, Labalme A, et al. Comparison of two next-generation sequencing kits for diagnosis of epileptic disorders with a user-friendly tool for displaying gene coverage, DeCovA. Appl Transl Genom. 2015;7:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi D, Deaglio S, Dominguez-Sola D, et al. Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118(18):4930–4934. [DOI] [PubMed] [Google Scholar]
- 27.Ng D, Thakker N, Corcoran CM, et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36(4):411–416. [DOI] [PubMed] [Google Scholar]
- 28.Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488(7409):106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto Y, Abe A, Emi N. Clarifying the impact of polycomb complex component disruption in human cancers. Mol Cancer Res. 2014;12(4):479–484. [DOI] [PubMed] [Google Scholar]
- 30.Moreira AL, Won HH, McMillan R, et al. Massively parallel sequencing identifies recurrent mutations in TP53 in thymic carcinoma associated with poor prognosis. J Thorac Oncol. 2015;10(2):373–380. [DOI] [PubMed] [Google Scholar]
- 31.Abe H, Kaneda A, Fukayama M. Epstein-Barr virus-associated gastric carcinoma: use of host cell machineries and somatic gene mutations. Pathobiology. 2015;82(5):212–223. [DOI] [PubMed] [Google Scholar]
- 32.Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153–6163. [DOI] [PubMed] [Google Scholar]
- 33.Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169–3177. [DOI] [PubMed] [Google Scholar]
- 34.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Eng J Med. 2015;373(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82–94. [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Park HY, Kang SY, et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget. 2015;6(19): 17764–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Gen Dev. 2000;14(14): 1810–1823. [PMC free article] [PubMed] [Google Scholar]
- 38.Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med. 2014;20(6):343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polo JM, Dell’Oso T, Ranuncolo SM, et al. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat Med. 2004;10(12):1329–1335. [DOI] [PubMed] [Google Scholar]
- 40.Sakano D, Kato A, Parikh N, et al. BCL6 canalizes notch-dependent transcription, excluding mastermind-like1 from selected target genes during left-right patterning. Dev Cell. 2010;18(3):450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cato MH, Chintalapati SK, Yau IW, Omori SA, Rickert RC. Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol Cell Biol. 2010;31(1):127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5): 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.