

Abstract
Lesinurad is an oral inhibitor of the monocarboxylic/urate transporter URAT1 encoded by the SLC22A12 gene. Market authorization was granted in February 2016 in Europe and December 2015 in the USA. As a potentially nephrotoxic uricosuric drug acting on the kidney, nephrologists should become familiar with its indications and safety profile. The approved indication is treatment of gout in combination with a xanthine oxidase (XO) inhibitor in adult patients who have not achieved target serum uric acid levels with an XO inhibitor alone. It is not indicated for asymptomatic hyperuricaemia or for patients with estimated creatinine clearance <45 mL/min. The only authorized daily dose is 200 mg and cannot be exceeded because of the nephrotoxicity risk. Nephrotoxicity is thought to be related to uricosuria. At the 200 mg/day dose, serum creatinine more than doubled in 1.8% of lesinurad patients (versus 0% in placebo) and in 11% of these it was not reversible. In addition, it is subject to a risk management plan given the potential association with cardiovascular events. In randomized clinical trials, the association of lesinurad with either allopurinol or febuxostat achieved a greater reduction in serum uric acid (∼1 mg/dL lower) than the XO inhibitors alone, and this allowed the serum uric acid target to be met in a higher proportion of patients, which was the primary endpoint. However, no clinical differences were observed in gout flares or tophi, although these were not the primary endpoints.
Keywords: cardiovascular risk, gout, lesinurad, nephrotoxicity, probenecid, URAT1, uric acid, uricosuric
Hyperuricaemia and gout
Serum uric acid has been increasing in the last few decades in the general population of Western countries, partly because of dietary changes, such as an increased intake of red meat and sugary foods, as sucrose consists of a disaccharide of fructose and glucose and the metabolism of fructose eventually generates uric acid, as well as because of an increasing incidence of chronic kidney disease (CKD) in an ageing population and increased use of diuretics, either to treat CKD or hypertension [1, 2]. Elevated serum uric acid is universally recognized as the main cause of gout. Gout is the most common inflammatory arthritis in adults in the Western world. It is three to four times more common in males than in females and the prevalence increases with age. The global prevalence was estimated at 42 214 200 in 2015 [3]. Global disability-adjusted life-years (DALYs) and years lived with disability (YLDs) resulting from gout increased by 26% from 2006 to 2015 to 1 342 800 according to the Global Burden of Disease 2015 study [4].
Gout results from tissue deposition of monosodium urate crystals that promote a local inflammatory response [5]. The acute episode is treated with short-term anti-inflammatory drugs, but long-term prevention may be achieved by lowering serum uric acid levels. In gouty patients, urate-lowering therapies are recommended to bring uric acid levels below the threshold of 6.0 mg/dL (sometimes 5.0 mg/dL), with the aim of dissolving urate deposits and preventing new gout flares. Three categories of urate-lowering therapies are recognized, based on their mechanism of action: uricostatic agents, that is, drugs that decrease uric acid production [xanthine oxidase (XO) inhibitors: allopurinol and febuxostat]; drugs that increase urinary uric acid excretion (uricosuric agents) and drugs that enzymatically degrade uric acid (pegloticase). Newer targets for treating gout have been persistently explored since none of the currently used drugs can be called ideal. This is primarily because of ineffectiveness in achieving target serum uric acid levels and adverse effects associated with available therapies. Allopurinol, an XO inhibitor, has long been used, although it has potentially serious adverse effects, such as hypersensitivity reactions [6]. In the last decade, three new urate-lowering drugs have been approved: febuxostat, an XO inhibitor; pegloticase, a parenteral pegylated recombinant mammalian uricase; and lesinurad, a urate transporter 1 (URAT1) inhibitor uricosuric agent (Figure 1). However, marketing authorization in the European Union for pegloticase was withdrawn in 2016 following the manufacturer’s request, for commercial reasons [7]. Lesinurad has been the latest addition, approved in December 2015 by the US Food and Drug Administration (FDA) and in February 2016 by the European Medicines Agency (EMA) for the treatment of gout in combination with an XO inhibitor in patients who have not achieved target serum uric acid levels with an adequate dose of XO inhibitor alone.
Fig. 1.

Recently approved drugs for the treatment of gout and mechanism of action. About two-thirds of daily urate derives from the degradation of endogenous purines and 70% of daily uric acid disposal results from kidney excretion. About 90% of the daily burden of urate filtered by the kidneys is reabsorbed and the remaining 10% is excreted in urine: 99–100% of the filtered urate would initially be reabsorbed, a further phase of tubular secretion would return to the tubular lumen 50% of the initially filtered urate. Most would be reabsorbed back into the proximal tubule (post-secretory reabsorption). Lesinurad inhibits urate reabsorption, thus increasing urinary excretion. AMP, adenosine monophosphate; GMP, guanosine monophosphate; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; IMP, inosine monophosphate; XMP, xanthosine monophosphate; ABCG2, ATP-binding cassette subfamily G member 2, GLUT9a, glucose transporter 9a, encoded by the SLC2A9 gene; MRP4, multidrug resistance-associated protein 4, also known as multispecific organic anion transporter B (MOAT-B) or ATP-binding cassette subfamily C member 4 (ABCC4); NPT1/4, sodium-dependent phosphate transport protein 1 encoded by the SLC17A1 gene and sodium-dependent phosphate transport protein 4 encoded by the SLC17A3 gene; OAT1; OAT3; OAT4; OAT10, organic anion transporter 1, 3, 4 and 10, encoded by the SLC22A6, SLC22A8; SLC22A11 and SLC22A13 genes, respectively; URAT1, urate transporter 1 encoded by the SLC22A12 gene.
Urate homeostasis
Serum uric acid levels depend on the balance between urate production/gut absorption and urate excretion [8, 9] (Figure 1). Uric acid is produced mainly in the liver, and to a lesser extent in the small intestine, from ingested or newly synthesized purines, purine recycling in cells and degradation of purines by XO. Inhibitors of XO, such as allopurinol and febuxostat, reduce uric acid synthesis, primarily in the liver and intestine. Two-thirds of daily urate production derives from the degradation of endogenous purines, with the remainder from the diet. In contrast to other mammals, humans and other primates do not have uricase, which converts uric acid (relatively insoluble) into allantoin (very soluble). The majority (70%) of uric acid excretion is renal. The remainder is removed in the gastrointestinal tract and is subsequently converted to allantoin by uricase in colon bacteria. Additionally, urate may be non-enzymatically converted to allantoin by oxidative stress [10].
The serum urate concentration is higher in men than in women due to the uricosuric effect of oestrogens. Most (90%) hyperuricaemias result from decreased renal excretion of uric acid. Renal handling of urate is complex. It consists of glomerular filtration and reabsorption in addition to tubular secretion, which occur in proximal tubules in humans (Figure 1). About 10% of urate filtered by glomeruli is excreted in urine.
The main transporters involved in proximal tubular reabsorption of uric acid are URAT1 (apical membrane), encoded by SLC22A12 (solute carrier family 22 organic anion/cation transporter member 12), and SLC2A9, encoding glucose transporter 9 (GLUT9) (basolateral membrane). More than 90% of uric acid filtered at the glomerulus is reabsorbed back into the bloodstream, mainly through URAT1 in proximal tubules. URAT1 is the main target of the classical uricosuric agents benzbromarone, probenecid, sulfinpyrazone, pyrazinamide and losartan [11, 12]. However, as discussed below for probenecid, additional transporters may also be inhibited by these agents. Interestingly, despite URAT1 being the main target of uricosuric agents, in CKD patients, serum uric acid is more closely related to single nucleotide polymorphisms in the adenosine triphosphate (ATP)-binding cassette subfamily G member 2 (ABCG2) and SLC2A9 (GLUT9) genes [13]. ABCG2 encodes an apical membrane transporter involved in urate secretion [14]. GLUT9 is a urate efflux transporter that also transports hexoses like glucose and fructose. In humans, proximal tubular reabsorption of urate also involves the apical exchange proteins organic anion transporter 4 (OAT4) and organic anion transporter 10 (OAT10). Urate uptake by URAT1 and OAT10 is accelerated by intracellular monocarboxylates such as lactate, pyrazinoate and nicotinate, while dicarboxylates accelerate uptake by OAT4 [8]. The basolateral urate/dicarboxylate OAT1 and organic anion transporter 3 (OAT3) exchangers and the apical multidrug resistance-associated protein 4 (MRP4) and ABCG2 transporters, as well as the sodium/phosphate sodium-dependent phosphate transport protein 1 (NPT1) and sodium-dependent phosphate transport protein 4 (NPT4) cotransporters participate in tubular urate secretion.
The most frequent cause of drug-induced hyperuricaemia is diuretic use. Diuretics interact with Npt4, OAT4 and MRP4. Angiotensin receptor antagonists and, above all, losartan interact with OAT3, MRP4, URAT1 and GLUT 9 and lower uric acid levels [15]. Fenofibrate increases urinary uric acid excretion through the inhibition of URAT1 by fenofibric acid, its major metabolite [16]. Glycosuria has uricosuric effects and hyperexcretory hypouricaemia has been described in type 1 and type 2 diabetics with normal renal function. SGLT2 inhibitors are used to treat diabetes by promoting glycosuria. In clinical trials, a 7% reduction in serum uric acid levels was observed [17].
Current approach to management of hyperuricaemia: what do guidelines say?
The 2012 American College of Rheumatology Guidelines for Management of Gout recommended a serum uric acid target of <6.0 mg/dL in general and <5.0 mg/dL for individual patients, as do the 2006 European League Against Rheumatism (EULAR) evidence-based recommendations for gout [18, 19]. In order to lower serum uric acid, they recommend a strategy based on achieving the target serum uric acid level by titrating an XO inhibitor to the maximal appropriate dose, followed by addition of probenecid or alternative uricosuric agents (losartan, fenofibrate) if not achieved. In the 2016 updated EULAR evidence-based recommendations for the management of gout, allopurinol is recommended as the first-line urate-lowering therapy, adjusting the dose according to renal function. If the serum uric acid target cannot be achieved with allopurinol, then febuxostat, a uricosuric or combining an XO inhibitor with a uricosuric should be considered [20]. Probenecid is the uricosuric agent of choice for monotherapy if there is intolerance to XO inhibitors. However, the recent clinical practice guideline from the American College of Physicians does not support the treat-to-target strategy and recommends that clinicians discuss benefits, harms, costs and individual preferences with patients before initiating urate-lowering therapy, including concomitant prophylaxis, in patients with recurrent gout attacks [21]. While it lists probenecid as an alternative, it does not discuss it, presumably because of insufficient evidence. In this regard, probenecid is the only widely available uricosuric in the USA and Europe. Benzbromarone was withdrawn from the US and European markets in 2004 after reports of severe hepatotoxicity [22]. However, it is still available in some European countries when there is intolerance to XO inhibitors and is the only uricosuric agent available in CKD patients with an eGFR >20 mL/min/1.73 m2 [23]. Sulfinpyrazone is only available in some European countries and not in the USA.
In contrast, no guideline recommends therapy for asymptomatic hyperuricaemia, despite evidence from open-label clinical trials that XO inhibitor may be nephroprotective in humans [24, 25]. Ongoing randomized controlled trials (RCTs) are addressing the role of treating asymptomatic hyperuricaemia in CKD and diabetic nephropathy (reviewed in [26, 27]) to slow disease progression [28] and in ischaemic heart patients to decrease cardiovascular events [29].
Lesinurad: the molecule
Lesinurad’s chemical name is 2((5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl)thio)acetic acid [30]. A total of 30–40% of the dose is excreted unchanged in urine and it additionally undergoes processing by CYP2C9. As a result, lesinurad should be used with caution in patients taking moderate inhibitors of CYP2C9 (e.g. fluconazole, amiodarone) and the therapeutic effect may be decreased in presence of inducers of CYP2C9 (e.g. rifampin). Despite the 5-h half-life, the clinical dose was adjusted for lesinurad to lower serum uric acid for at least 24 h. In this regard, doses <200 mg/day did not achieve a reduction in serum uric acid by 24 h post-administration and were not evaluated in Phase 3 RCTs. In Phase 3 RCTs, lesinurad dose-dependently increased urinary uric acid excretion and reduced serum uric acid. The short half-life and dosing schedule has two clinically relevant implications. First, it is expected to result in lower serum uric acid levels during the day than those observed 24 h later in pre-dose blood samples. Second, it results in higher urine uric acid concentration in the first few hours after administration than later in the day. In this regard, lesinurad should be taken in the morning, so the peak uric acid excretion coincides with waking hours and thus fluid intake and higher urine pH (Figure 2). This facilitates uric acid solubility, decreasing the risk of crystalluria of urolithiasis. After 6 h, a single 200 mg dose of lesinurad increased the fractional excretion of uric acid 3.6-fold and reduced serum uric acid levels by 33%. Following 400 mg/day dosing, serum uric acid reduction was 35% at 24 h post-dose, supporting once-a-day dosing [32]. At concentrations achieved in the clinic, lesinurad inhibited the activity of URAT1 and OAT4 in vitro, but not GLUT9 or ABCG2. Unlike probenecid, lesinurad did not inhibit OAT1 or OAT3 at these concentrations [33].
Fig. 2.

Serum and urine uric acid over 24 h following a single morning lesinurad dose. (A) Approximate depiction of circadian changes in urine volume and pH. The precise changes for a given individual will depend on ingestion of fluid and diet. (B) Lesinurad impact on urinary urate in the presence or absence of allopurinol. Note that a morning dosing will result in peak urate excretion coinciding with higher urinary volume and pH, thus minimizing the risk of urate precipitation [31].
Lesinurad indications and contraindications
Lesinurad is indicated in combination with an XO inhibitor for the treatment of hyperuricaemia associated with gout in patients who have not achieved target serum uric acid levels with an XO inhibitor alone [31]. It is specifically not recommended for the treatment of asymptomatic hyperuricaemia. The maximum daily dose of lesinurad is 200 mg. Contraindications include severe renal impairment, kidney transplant recipients, tumour lysis syndrome or Lesch–Nyhan syndrome. It should not be initiated if estimated creatinine clearance (ECC) is <45 mL/min and should be discontinued if ECC persistently falls below this level. The FDA-approved prescribing information includes specific warnings about the occurrence of renal dysfunction and major cardiovascular events as well as of the potential increase in the risk of renal adverse reactions if not associated with an XO inhibitor [31]. It should be emphasized that safety data were generated by estimating ECC using the Cockroft–Gault equation. In contrast, in routine clinical practice, the eGFR is calculated by the Chronic Kidney Disease Epidemiology Collaboration equation. As recently reviewed, both parameters may not be equivalent in the same individual, especially if the patient weight is well above or below expectations for age and sex [34].
Lesinurad Phase 3 randomized clinical trials
Three Phase 3 RCTs provided the backbone information for regulatory approval: CLEAR 1 (NCT01510158), CLEAR 2 (NCT01493531) and CRYSTAL (NCT01510769) [35] studied two doses of lesinurad in combination with XO inhibitors (Table 1). Overall, the clinical dose of 200 mg/day decreased serum uric acid by ∼1 mg/dL at the 24-h post-administration time point. All trials met the primary efficacy endpoint of achieving significantly more frequently a certain threshold of serum uric acid than placebo. No improvement was observed in the frequency of gout attacks or size of tophi. However, these were secondary endpoints and differences were not expected within the time frame of the trials. A fourth clinical trial testing the non-clinical dose of 400 mg/day lesinurad monotherapy in the absence of XO inhibitors is discussed under safety concerns (Table 1).
Table 1.
Phase 3 RCTs of lesinurad for gout
| RCT | Indication | XO inhibitor | Lesinurad dose (mg/day) | n, total | Duration (months) | Age (years) | Uric acid inclusion criterion (mg/dL) | Baseline uric acid (mg/dL) | ECC <60 mL/ min (%) | Met primary endpoint (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Study 303 [30] | Gout, XO inhibitor intolerant | None | 400 | 214 | 6 | 54 ± 12 | ≥6.5 | 9.33 ± 1.51 | 27 | 2-30a |
| CLEAR 1 [36] | Goutb | Allopurinolb | 200–400 | 603 | 12 | 52 ± 11 | ≥6.5b | 6.94 ± 1.27 | 29 | 28-54-59c |
| CLEAR 2 [37] | Gouta | Allopurinola | 200–400 | 610 | 12 | 51 ± 11 | ≥6.5a | 6.90 ± 1.19 | 20 | 23-55-66c |
| CRYSTAL [30] | Goutd | Febuxostatd | 200–400 | 324 | 12 | 54 ± 11 | ≥6.0d | 5.27 ± 1.63 | 29 | 47-57-76e |
Percentage of patients with serum uric acid <6.0 mg/dL at 6 months, results provided as placebo, 400 mg lesinurad.
On allopurinol ≥300 mg/day (≥200 mg/day for moderate renal impairment ECC <60 mL/min), two or more gout flares in the prior 12 months required.
Percentage of patients with serum uric acid <6.0 mg/dL at 6 months, results provided as placebo, 200 mg lesinurad, 400 mg lesinurad.
On Febuxostat 80 mg/day, presence of gouty tophi.
Percentage of patients with serum uric acid <5.0 mg/dL at 6 months, results provided as placebo, 200 mg lesinurad, 400 mg lesinurad.
What are the benefits? Are there advantages of lesinurad over other uricosuric agents?
The only widely available uricosuric in the USA and Europe is probenecid. A recent Cochrane systematic review found only two studies comparing benzbromarone with probenecid. It found moderate-quality evidence based on one study (62 participants) that participants taking benzbromarone were more likely to achieve serum uric acid normalization and also that benzbromarone was associated with fewer adverse effects and withdrawals due to adverse effects [38]. However, as discussed above, benzbromarone is not available in the USA and many European countries because of concerns over hepatotoxicity.
While lesinurad has not been formally compared with probenecid in an RCT, potential advantages may include a higher efficacy in lowering serum uric acid and better tolerance. In addition, probenecid has a number of drug–drug interactions, and, indeed, it was initially developed with the goal of reducing the renal excretion of antibiotics [39]. It is also used clinically to prevent nephrotoxicity induced by drugs that are toxic to proximal tubular cells and accesses these cells via probenecid-sensitive transporters, such as cidofovir [40]. Unfortunately, probenecid was approved for clinical use many years ago and detailed studies, such as those available for lesinurad, regarding the potential to induce kidney injury are not available. A PubMed search for probenecid and acute kidney injury (AKI) did not return any publication on humans on 5 March 2017. Thus we found no data on probenecid-induced kidney injury.
What are the dangers?
Two safety signals were observed at the higher (400 mg/day) lesinurad dose tested in Phase 3 placebo-controlled RCTs: renal and cardiovascular, emphasizing the potential dangers of this dose.
The main adverse effect of lesinurad is nephrotoxicity, which is dose dependent. Thus only the 200-mg dose got regulatory approval and only in association with XO inhibitors, as the 400 mg/day dose posed a nephrotoxicity risk. To correctly assess the nephrotoxicity results, we should emphasize that an increase of serum creatinine (sCr) ≥1.5-fold over baseline may be considered AKI if certain time-course criteria are met [41].
Of the four Phase 3 RCTs with available results, only CLEAR 1 [36] and CLEAR 2 [37] have been published in a peer-reviewed journal to date. In both, the XO inhibitor used was allopurinol. However, information on the other two Phase 3 RCTs (lesinurad monotherapy and lesinurad/febuxostat) is available to the public from presentations to the EMA and FDA (Table 1) [30].
In CLEAR 1, a trend towards a dose-dependent decrease in ECC was observed from baseline to the last value on treatment (Figure 3A). However, this was no longer apparent at the last value off treatment at follow-up [36], which is reassuring. Similar results were observed in CLEAR 2, in which resolution of sCr elevations occurred while patients continued on the study medication in approximately two-thirds of sCr elevations [37]. A trend towards a dose-dependent decrease in ECC was observed from baseline to the last value on treatment (Figure 3B). However, in the subset of 133 patients who did not enter the extension study, there was no global decrease in ECC from baseline to the last value off treatment at follow-up (ECC change 1.8 ± 11.7, 2.7 ± 10.0 and 1.1 ± 24.2 ‘mg/dL’; ‘mg/dL’ is assumed to represent mL/min) [37]. However, the high standard deviation for patients in the highest lesinurad dose suggests the presence of some patients with persistently decreased ECC.
Fig. 3.

Nephrotoxicity of lesinurad in placebo-controlled Phase 3 RCTs with 6 (monotherapy: M) or 12 months of follow-up (associated with XO inhibitors: CLEAR1, CLEAR 2, CRYSTAL). The 400-mg dose (red) is not indicated in routine clinical practice because of nephrotoxicity risk. (A, B) Change in ECC according to lesinurad dose in Phase 3 RCTs, CLEAR1 and CLEAR2. In both RCTs lesinurad was used together with allopurinol. Change in ECC (mL/min) in CLEAR 2 from baseline to last follow-up on drug. (C–E) Incidence of nephrotoxicity as defined by different thresholds of fold change in sCr for patients with any baseline eGFR. (F–H) Incidence of nephrotoxicity defined as an increase in sCr for patients at different baseline eGFR categories in patients treated with XO inhibitors. Data obtained from [30, 33, 40]. M stands for lesinurad monotherapy.
In this regard, further analysis available in the documentation presented to regulatory agencies and their response that integrates several Phase 3 RCTs [30] show clear-cut, dose-dependent increases in the percentage of patients who developed diverse degrees of renal dysfunction, as assessed by increases of sCr ≥1.5-fold, ≥2.0-fold and ≥3.0-fold over baseline values (Figure 3C–E). For every criterion, the risk was higher for patients in any lesinurad dose than in placebo controls and higher in 400 mg/day lesinurad monotherapy than for lesinurad associated with XO inhibitors. In this regard, 25% of patients on 400 mg/day lesinurad monotherapy developed a ≥1.5-fold increase in sCr within 6 months. Lesinurad was also associated with a higher incidence of renal dysfunction at any baseline eGFR assessed (Figure 3F–H). While the incidence of nephrotoxicity increased as baseline eGFR decreased, at the lowest eGFR tested, the difference between placebo and lesinurad was the lowest.
No patients in placebo groups had doubling of sCr. Among patients on lesinurad, time to resolution of doubling of sCr increased with the dose of lesinurad (Figure 4), suggesting intrinsic kidney injury rather than pre-renal or post-renal causes. Of these patients, 11% of those treated with 200 mg/day, 20% of those on 400 mg/day plus XO inhibitors and 46% of those on 400 mg/day monotherapy had not recovered baseline renal function at the last follow-up, >3 months after the nephrotoxicity episode (Figure 4) [30].
Fig. 4.
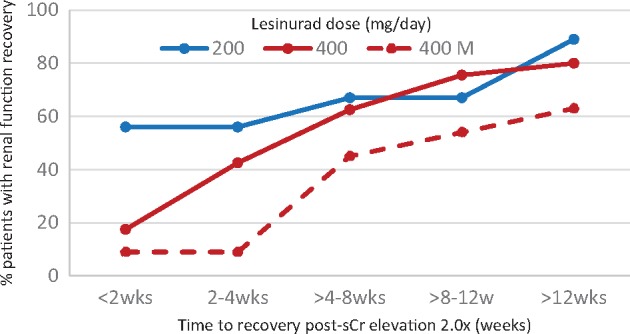
Nephrotoxicity of lesinurad in placebo-controlled Phase 3 RCTs. Time to resolution in patients with sCr elevations ≥2-fold of baseline. Data from [30].
More worrisome is the safety signal regarding death from cardiovascular events (Figure 5). A clear dose response was observed; death increased from placebo to 200 mg/day lesinurad, 400 mg/day and 400 mg/day monotherapy (Figure 5A). For major cardiovascular events, the difference between the 200 mg/day dose and placebo was minimal or non-existent, while 400 mg/day more than doubled the risk, without a further increase with 400 mg/day monotherapy, contrary to observations for death and nephrotoxicity (Figure 5B and C). Since these were not large trials, the 95% confidence interval is wide. Nevertheless, the percentage of patients with major cardiovascular events within the first year was 34% higher with lesinurad 200 mg/day than with placebo (Figure 5C). This is well within the range considered potentially clinically significant. As an example, a 30% difference is within the range of protection offered by interventions such as statins. In this regard, lesinurad is subject to a risk management plan given the potential association with cardiovascular events [42]. This consists of a post-marketing observational cohort safety study through 2019 for cardiovascular safety and an RCT to assess effectiveness and safety for patients with an ECC of 30–46 mL/min.
Fig. 5.

Death or major cardiovascular events (MACE) in placebo-controlled Phase 3 RCTs with 6–12 months of follow-up. (A) Deaths expressed per 100 patient-years. (B) MACE expressed per 100 patient-years. (C) MACE expressed as percentage of patients with MACE. One 6-month RCT did not use XO inhibitor (lesinurad monotherapy, indicated by M). Placebo groups were grouped together in (A) and (B) since data are expressed per 100 patient-years, but not in (C) since monotherapy (M) placebo follow-up was 6 months. The number of patients on placebo was 623 (107 to monotherapy placebo), on 200 mg lesinurad 511, on 400 mg lesinurad 510 and on lesinurad monotherapy (400 M) 107. Data from [30].
In the lesinurad 400 mg/day monotherapy trial, 19% of patients on lesinurad discontinued the drug within 6 months due to adverse effects, as opposed to 6% in the placebo arm [30]. Again, we should emphasize that lesinurad is not licensed to be used at daily doses >200 mg or in monotherapy.
Mechanisms of nephrotoxicity
The most obvious candidate for the nephrotoxicity of lesinurad is uric acid crystalluria. In this regard, hereditary tubular urate transport defects resulting in hypouricaemia may be complicated by repeated AKI episodes [43]. Furthermore, evidence of nephrotoxicity was more frequent in subjects with higher baseline serum uric acid levels and in the absence of XO inhibitors (Table 1) [30], and no further increase in incidence of nephrotoxicity was observed in individuals with the lowest eGFR studied (Figure 3H). These low eGFR individuals would be expected to be more sensitive to nephrotoxic drugs, unless the drug effect depended on a preserved eGFR. However, kidney biopsies are not available and direct kidney toxicity cannot be completely excluded. In rat chronic toxicity studies, kidney toxicity was lethal at the highest tested doses (43-fold for the 400 mg dose tested in human Phase 3 trials) and was characterized by tubular cell death [30]. At a lower dose (20-fold for the 400 mg dose tested and 43-fold for the clinical dose of 200 mg/day), tubular dilation and biochemical changes were observed. Nephrotoxicity in rats is unlikely to be related to uric acid crystalluria, given the very low uric acid levels in rats since, unlike humans, they have functional uricase. Interference with sCr tubular secretion is unlikely, given that in Phase 2 trials the difference in mean change from baseline sCr at 4 weeks was ≤0.1 mg/dL for doses of 200–600 mg/day [44], increased serum urea levels were also observed and the long time until functional recovery [30].
This is not the first uricosuric drug with significant nephrotoxicity. Clinical development of PF-06743649, a dual URAT1/XO inhibitor, was terminated because of development of AKI in the first 3 days after the first dose in healthy volunteers [45]. Tienilic acid (ticrynafen) was a diuretic with uricosuric properties that is no longer in clinical use. Several cases of AKI were reported from uric acid urolithiasis, biopsied acute interstitial nephritis, tubular cell injury or suspected urate nephropathy [46–48]. However, the URAT1 inhibitor benzbromarone has a good renal safety profile [49] and probenecid may protect from AKI induced by nephrotoxins that enter proximal tubular cells through probenecid-sensitive transporters [40, 50]. Renal hypouricaemia due to SLC22A12 or SLC2A9 deficiency is associated with exercise-induced AKI and acute tubular necrosis [51–53]. This has been linked to excess uric acid excretion, since allopurinol prevented exercise-induced AKI both in humans and in an animal model [54, 55].
Lesinurad in CKD
In Phase I studies, mild (ECC 60–89 mL/min), moderate (ECC 30–59 mL/min) and severe (ECC <30 mL/min) renal impairment increased the area under the plasma concentration–time curve for lesinurad by 34, 54–65 and 102%, respectively. The serum uric acid–lowering effect of a single dose of lesinurad was reduced in moderate impairment and greatly decreased in severe impairment, despite higher lesinurad exposures [56]. These findings were confirmed in Phase 3 RCTs, where there was inconclusive evidence of benefit to patients with ECC <45 mL/min) [30].
Conclusions
Lesinurad is a novel addition to the therapeutic armamentarium against gout. As a uricosuric agent, a number of precautions should be taken to prevent pathological hyperuricosuria. In this regard, lesinurad should be prescribed at a maximal dose of 200 mg/day and always in association with XO inhibitors to decrease the risk of nephrotoxicity. While nephrotoxicity is usually reversible, this is not always the case. A safety concern regarding cardiovascular risk should be clarified in the first years of widespread clinical use. It is likely that nephrologists will be consulted if patients develop increased sCr while on lesinurad and they should become familiar with its kidney side effects.
Funding
FIS ISCIII FEDER funds PI15/00298, PI16/02057, PI16/01900, ISCIII-RETIC REDinREN RD12/0021 RD16/0009, CYTED IBERERC, Programa Intensificación Actividad Investigadora (ISCIII) to A.O.
Conflict of interest statement
A.O., J. L. and M.G. have received speaker fees from Menarini. J.L. has received fees from a Grunenthal Pharma Advisory Board.
References
References
- 1. Johnson RJ, Titte S, Cade JR. et al. Uric acid, evolution and primitive cultures. Sem in Nephrol 2005; 25: 3–8 [DOI] [PubMed] [Google Scholar]
- 2. Johnson RJ, Nakagawa T, Sanchez-Lozada LG. et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013; 62: 3307–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators Allen C, Arora M. et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016; 388: 1545–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. GBD 2015 DALYs and HALE Collaborators, Arora M, Barber RM. et al. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016; 388: 1603–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rock KL, Kataoka H, Lai J-J.. Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol 2013; 9: 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stamp LK, Day RO, Yun J.. Allopurinol hypersensitivity: investigating the cause and minimizing the risk. Nat Rev Rheumatol 2016; 12: 235–242 [DOI] [PubMed] [Google Scholar]
- 7.Krystexxa. Withdrawal of the marketing authorisation in the European Union. http://www.ema.europa.eu/docs/en_GB/document_library/Public_statement/2016/07/WC500210911.pdf (21 February 2017, date last accessed)
- 8. So A, Thorens B.. Uric acid transport and disease. J Clin Invest 2010; 120: 1791–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richette P, Bardin T.. Gout. Lancet 2010; 375: 318–328 [DOI] [PubMed] [Google Scholar]
- 10. Kaur H, Halliwell B.. Action of biologically-relevant oxidizing species upon uric acid. Identification of uric acid oxidation products. Chem Biol Interact 1990; 73: 235–247 [DOI] [PubMed] [Google Scholar]
- 11. Enomoto A, Kimura H, Chairoungdua A. et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002; 417: 447–452 [DOI] [PubMed] [Google Scholar]
- 12. Ichida K, Hosoyamada M, Hisatome I. et al. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J Am Soc Nephrol 2004; 15: 164–173 [DOI] [PubMed] [Google Scholar]
- 13. Bhatnagar V, Richard EL, Wu W. et al. Analysis of ABCG2 and other urate transporters in uric acid homeostasis in chronic kidney disease: potential role of remote sensing and signaling. Clin Kidney J 2016; 9: 444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woodward OM. ABCG2: the molecular mechanisms of urate secretion and gout. Am J Physiol Renal Physiol 2015; 309: F485–F488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smink PA, Bakker SJL, Laverman GD. et al. An initial reduction in serum uric acid during angiotensin receptor blocker treatment is associated with cardiovascular protection: a post-hoc analysis of the RENAAL and IDNT trials. J Hypertens 2012; 30: 1022–1028 [DOI] [PubMed] [Google Scholar]
- 16. Derosa G, Maffioli P, Sahebkar A.. Plasma uric acid concentrations are reduced by fenofibrate: a systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol Res 2015; 102: 63–70 [DOI] [PubMed] [Google Scholar]
- 17. Zinman B, Wanner C, Lachin JM. et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373: 2117–2128 [DOI] [PubMed] [Google Scholar]
- 18. Khanna D, Fitzgerald JD, Khanna PP. et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res 2012; 64: 1431–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang W, Doherty M, Bardin T. et al. EULAR evidence-based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2006; 65: 1312–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richette P, Doherty M, Pascual E. et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis 2017; 76: 29–42 [DOI] [PubMed] [Google Scholar]
- 21. Qaseem A, Harris RP, Forciea MA, Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the american college of physicians. Ann Intern Med 2017; 166: 58–68 [DOI] [PubMed] [Google Scholar]
- 22. Jansen TLTA, Reinders MK, van Roon EN. et al. Benzbromarone withdrawn from the European market: another case of "absence of evidence is evidence of absence"? Clin Exp Rheumatol 2004; 22: 651. [PubMed] [Google Scholar]
- 23. Fujimori S, Ooyama K, Ooyama H, Moromizato H.. Efficacy of benzbromarone in hyperuricemic patients associated with chronic kidney disease. Nucleosides, Nucleotides & Nucleic Acids 2011; 30: 1035–1038. [DOI] [PubMed] [Google Scholar]
- 24. Goicoechea M, Garcia de Vinuesa S, Verdalles U. et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis 2015; 65: 543–549 [DOI] [PubMed] [Google Scholar]
- 25. Goicoechea M, de Vinuesa SG, Verdalles U. et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol 2010; 5: 1388–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fernandez-Fernandez B, Ortiz A, Gomez-Guerrero C. et al. Therapeutic approaches to diabetic nephropathy—beyond the RAS. Nat Rev Nephrol 2014; 10: 325–346 [DOI] [PubMed] [Google Scholar]
- 27. Perez-Gomez MV, Sanchez-Niño MD, Sanz AB. et al. Horizon 2020 in Diabetic Kidney Disease: the clinical trial pipeline for add-on therapies on top of renin angiotensin system blockade. J Clin Med 2015; 4: 1325–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hosoya T, Kimura K, Itoh S. et al. The effect of febuxostat to prevent a further reduction in renal function of patients with hyperuricemia who have never had gout and are complicated by chronic kidney disease stage 3: study protocol for a multicenter randomized controlled study. Trials 2014; 15: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackenzie IS, Ford I, Walker A. et al. Multicentre, prospective, randomised, open-label, blinded end point trial of the efficacy of allopurinol therapy in improving cardiovascular outcomes in patients with ischaemic heart disease: protocol of the ALL-HEART study. BMJ Open 2016; 6: e013774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Center for Drug Evaluation and Research. Application Number: 207988Orig1s000. Cross Discipline Team Leader Review. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207988Orig1s000CrossR.pdf (12 February 2017, date last accessed)
- 31. FDA Prescribing Information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207988lbl.pdf (14 January 2017, date of accessed); http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM467951.pdf (14 January 2017, date last accessed)
- 32. Shen Z, Rowlings C, Kerr B. et al. Pharmacokinetics, pharmacodynamics, and safety of lesinurad, a selective uric acid reabsorption inhibitor, in healthy adult males. Drug Des Devel Ther 2015; 9: 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miner J, Tan PK, Hyndman D. et al. Lesinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibition of urate transporters in the kidney. Arthritis Res Ther 2016; 18: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fernandez-Prado R, Castillo-Rodriguez E, Velez-Arribas FJ. et al. Creatinine clearance is not equal to glomerular filtration rate and cockcroft-gault equation is not equal to CKD-EPI collaboration equation. Am J Med 2016; 129: 1259–1263 [DOI] [PubMed] [Google Scholar]
- 35. Hoy SM. Lesinurad: first global approval. Drugs 2016; 76: 509–516 [DOI] [PubMed] [Google Scholar]
- 36. Saag KG, Fitz-Patrick D, Kopicko J. et al. Lesinurad combined with allopurinol: a randomized, double-blind, placebo-controlled study in gout patients with an inadequate response to standard-of-care allopurinol (a US-based study). Arthritis Rheum 2017; 69: 203–212 [DOI] [PubMed] [Google Scholar]
- 37. Bardin T, Keenan RT, Khanna PP. et al. Lesinurad in combination with allopurinol: a randomised, double-blind, placebo-controlled study in patients with gout with inadequate response to standard of care (the multinational CLEAR 2 study). Ann Rheum Dis 2017; 76: 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kydd ASR,, Seth R,, Buchbinder R. et al. Uricosuric medications for chronic gout. Cochrane Database Syst Rev 2014; 11: CD010457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robbins N, Koch SE, Tranter M, Rubinstein J.. The history and future of probenecid. Cardiovasc Toxicol 2012; 12: 1–9 [DOI] [PubMed] [Google Scholar]
- 40. Ortiz A, Justo P, Sanz A. et al. Tubular cell apoptosis and cidofovir-induced acute renal failure. Antivir Ther 2005; 10: 185–190 [PubMed] [Google Scholar]
- 41. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2012; 2: 1–138 [Google Scholar]
- 42. Summary of the Risk Management Plan (RMP) for Zurampic (Lesinurad). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Risk-management-plan_summary/human/003932/WC500198831.pdf (21 February 2017, date last accessed)
- 43. Mou L, Jiang L, Hu Y.. A novel homozygous GLUT9 mutation cause recurrent exercise-induced acute renal failure and posterior reversible encephalopathy syndrome. J Nephrol 2015; 28: 387–392 [DOI] [PubMed] [Google Scholar]
- 44. Perez-Ruiz F, Sundy JS, Miner JN. et al. Lesinurad in combination with allopurinol: results of a phase 2, randomised, double-blind study in patients with gout with an inadequate response to allopurinol. Ann Rheum Dis 2016; 75: 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dua P, Gurrell R, Kirby S. et al. Acute kidney injury observed during phase 1 clinical trials of a novel xanthine oxidase/URAT1 dual inhibitor PF-06743649. Clin Rheum 2016; 35: 2045–2051 [DOI] [PubMed] [Google Scholar]
- 46. Powers D, Vaziri ND, Muhalwas K. et al. Ticrynafen-induced acute renal failure. Clin Toxicol 1981; 18: 425–430. [DOI] [PubMed] [Google Scholar]
- 47. Sanchez MA,, Frier S,, Hallac R. et al. Renal tubular degeneration after administration of ticrynafen. Arch Pathol Lab Med 1980; 104: 656. [PubMed] [Google Scholar]
- 48. Walker RG, Whitworth JA, Kincaid-Smith P.. Acute interstitial nephritis in a patient taking tienilic acid. Br Med J 1980; 280: 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee M-HH, Graham GG, Williams KM. et al. A benefit-risk assessment of benzbromarone in the treatment of gout. Was its withdrawal from the market in the best interest of patients? Drug Saf 2008; 31: 643–665 [DOI] [PubMed] [Google Scholar]
- 50. Baudoux TER, Pozdzik AA, Arlt VM. et al. Probenecid prevents acute tubular necrosis in a mouse model of aristolochic acid nephropathy. Kidney Int 2012; 82: 1105–1113 [DOI] [PubMed] [Google Scholar]
- 51. Ito O, Hasegawa Y, Sato K. et al. A case of exercise-induced acute renal failure in a patient with idiopathic renal hypouricemia developed during antihypertensive therapy with losartan and trichlormethiazide. Hypertens Res 2003; 26: 509–513 [DOI] [PubMed] [Google Scholar]
- 52. Kikuchi Y, Koga H, Yasutomo Y. et al. Patients with renal hypouricemia with exercise-induced acute renal failure and chronic renal dysfunction. Clin Nephrol 2000; 53: 467–472. [PubMed] [Google Scholar]
- 53. Ninomiya M, Ito Y, Nishi A. et al. Recurrent exercise-induced acute renal failure in renal hypouricemia. Acta Paediatr 1996; 85: 1009–1011 [DOI] [PubMed] [Google Scholar]
- 54. Yeun JY, Hasbargen JA.. Renal hypouricemia: prevention of exercise-induced acute renal failure and a review of the literature. Am J Kidney Dis 1995; 25: 937–946. [DOI] [PubMed] [Google Scholar]
- 55. Hosoyamada M, Tsurumi Y, Hirano H. et al. Urat1-Uox double knockout mice are experimental animal models of renal hypouricemia and exercise-induced acute kidney injury. Nucleosides Nucleotides Nucleic Acids 2016; 35: 543–549 [DOI] [PubMed] [Google Scholar]
- 56. Gillen M, Valdez S, Zhou D. et al. Effects of renal function on pharmacokinetics and pharmacodynamics of lesinurad in adult volunteers. Drug Des Devel Ther 2016; 10: 3555–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]