

Abstract
Mutations in the ROMK1 potassium channel gene (KCNJ1) cause antenatal/neonatal Bartter syndrome type II (aBS II), a renal disorder that begins in utero, accounting for the polyhydramnios and premature delivery that is typical in affected infants, who develop massive renal salt wasting, hypokalaemic metabolic alkalosis, secondary hyperreninaemic hyperaldosteronism, hypercalciuria and nephrocalcinosis. This BS type is believed to represent a disorder of the infancy, but not in adulthood. We herein describe a female patient with a remarkably late-onset and mild clinical manifestation of BS II with compound heterozygous KCNJ1 missense mutations, consisting of a novel c.197T > A (p.I66N) and a previously reported c.875G > A (p.R292Q) KCNJ1 mutation. We implemented and evaluated the performance of two different bioinformatics-based approaches of targeted massively parallel sequencing [next generation sequencing (NGS)] in defining the molecular diagnosis. Our results demonstrate that aBS II may be suspected in patients with a late-onset phenotype. Our experimental approach of NGS-based mutation screening combined with Sanger sequencing proved to be a reliable molecular approach for defining the clinical diagnosis in our patient, and results in important differential diagnostic and therapeutic implications for patients with BS. Our results could have a significant impact on the diagnosis and methodological approaches of genetic testing in other patients with clinical unclassified phenotypes of nephrocalcinosis and congenital renal electrolyte abnormalities.
Keywords: Bartter syndrome, hypokalaemia, KCNJ1, nephrocalcinosis, ROMK
Introduction
Inactivating, i.e. loss-of-function, mutations in the KCNJ1 gene encoding the apical potassium inwardly-rectifying channel (ROMK) in the thick ascending limb (TAL) of the Henle’s loop cause autosomal recessive antenatal/neonatal Bartter syndrome type II (aBS II) [1].
aBS II begins in utero, accounting for polyhydramnios, which may in turn contribute to premature delivery that is typical in affected infants. aBS II presents with severe renal salt wasting resulting in life-threatening volume depletion in the neonatal period. Additional features are secondary hyperreninaemic hyperaldosteronism, metabolic alkalosis, and hypercalciuria and nephrocalcinosis. It is unique in often presenting with hyperkalaemia, rather than hypokalaemia, at birth, sometimes making the diagnosis challenging. This hyperkalaemia later evolves into hypokalaemia commonly seen in the postnatal period [1–3].
More than 40 KCNJ1 mutations have been described so far [3]. Most of these mutations are missense/nonsense mutations substituting conserved amino acids residues, predominantly within the coding exon 2, the most influential putative functional domain of ROMK [3]. Interestingly, all reported patients with KCNJ1 mutations presented a severe BS phenotype at infancy. Only one adult man has been reported with a phenotype resembling a late-onset BS due to a homozygous KCNJ1 missense mutation (c.658C > T, p.L220F). This patient initially presented with an incidental finding of nephrocalcinosis. He had modest hypokalaemic metabolic alkalosis and mild-to-moderate chronic kidney disease (CKD Grade 3A, 2012 KDIGO guidelines) [4].
Since this is the only report on BS II with clinical presentation in adulthood, it remains unclear whether aBS II can be considered also as adult-onset disease. aBS II might have evaded detection in human adulthood because Sanger sequential sequencing of multiple genes is time-consuming and costly, and subjects with incomplete or non-penetrant phenotypes might not have been investigated.
Case presentation
A 43-year-old German woman was examined after incidental findings of bilateral nephrocalcinosis by ultrasound during her second pregnancy. Figure 1 shows an ultrasound image demonstrating renal medullary nephrocalcinosis in her left kidney. Even though her recent medical history was unremarkable, the patient had claimed strong thirst and polyuria in childhood. Strong appetite for salty food was denied, even though beneficial effects after consumption of salt-rich foods were noticed. She ingested no laxatives or diuretics, nor did the patient abuse alcohol or other drugs. The patient denied nausea, vomiting, diarrhoea, weakness, fever, night sweats, weight loss and heat intolerance. The patient is mother of two healthy daughters.
Fig. 1.
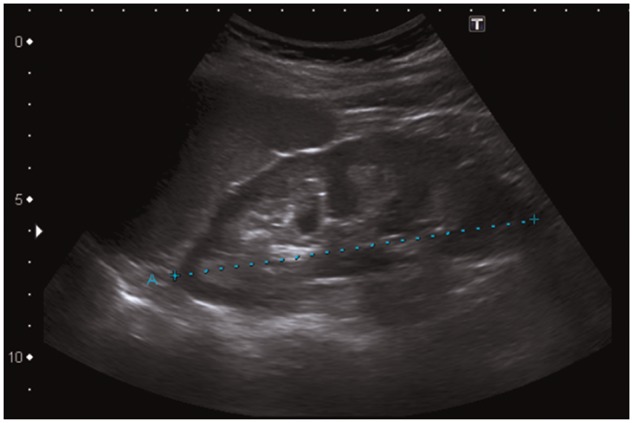
Ultrasound image demonstrating renal medullary nephrocalcinosis in the patient’s left kidney. This ultrasound image shows increased echogenicity of the renal medulla. Kidney size, 11.5 cm (blue dotted line).
Physical examination was normal. The blood pressure was normotensive (136/80 mmHg), no oedema were detected. ECG was normal. Laboratory tests showed hypokalaemia (3.0 mmol/L), low-normal total serum calcium (2.31 mmol/L), hyperaldosteronism (515 ng/L, 847 ng/L) (normal range 30–340), hyperreninaemia (43.1 ng/L, 25.6 ng/L) (normal range 2.0–24.6) and increased calcium excretion in the urine [7.5 mmol/day or 0.13 mmol/kg body weight/day (normal range <6.2 mmol/day or <0.1 mmol/kg body weight/day)]. Serum sodium (139 mmol/L), chloride (104 mmol/L), magnesium (0.87 mmol/L) and phosphate (0.9 mmol/L) levels were normal. In venous blood, pH was normal, bicarbonate levels are at the high limit of normality and there seems to be respiratory compensation (venous pH 7.37, HCO3 28.2 mmol/L, pCO2 56.9 mmHg, PO2 29.4 mmHg). In arterial blood, pH was high but bicarbonate levels were normal (pH 7.498, HCO3 25.7 mmol/L, pCO2 30.5 mmHg, PO2 118 mmHg). Fractional excretion of sodium (FENa 2.3%) was increased and tubular phosphate reabsorption (80%) was determined to be in the normal range. FE of potassium (FEK 22%, 29%) was increased, whereas FE of chloride was increased (FECl 2.52%). The trans-tubular potassium gradient (TTKG 21) was high in our patient considering the presence of hypokalaemia, indicating renal potassium wasting. Serum creatinine was 1.10 mg/dL [estimated glomerular filtration rate (eGFR) (Modification of Diet in Renal Disease, MDRD) 58 mL/min], indicating mild-to-moderate CKD (Grade 3a) (2012 KDIGO guidelines). Proteinuria and albuminuria were not detected (protein <40 mg/mL, albumin <3 mg/mL; creatinine 0.4 g/L in urine). After 14 h of water deprivation, our patient had a reduced urine osmolality [392 mOsmol/kg; normal 915 ± 91 mOsmol/kg) at a plasma osmolality of 294 mOsml/kg] [5], indicating that she cannot concentrate her urine well. Since antidiuretic hormone (AVP) in plasma is high [plasma copeptin [computed tomography (CT)-pro-AVP] levels 32.4 pmol/L; normal range 2.3–24.5 pmol/L], this decreased urine concentrating capacity does not have central cause (i.e. impaired AVP release by the pituitary gland), but has a renal origin.
Computed tomography scan densitometry was unremarkable. There was no amelogenesis imperfecta [6]. Hypothyroidism was diagnosed earlier [anti-thyroid antibodies negative (TRAK) or with non-significant titres (TPO, Tg)] and substituted with L-thyroxin. Hyperparathyroidism [serum parathyroid hormone (iPTH) levels (126 ng/L) (normal 15–65 ng/L)] was also detected earlier. Since thyroid/parathyroid ultrasound examination, computed tomography neck/thorax scan, parathyroid scintigraphy, osteodensitometry and X-rays of hands were unremarkable, the patient was initially treated with vitamin D substitution. Although this treatment resulted in a suppression of PTH levels to high-normal values, vitamin D substitution was later discontinued because of possible progression of nephrocalcinosis. The patient was treated with oral supplementation of potassium (80 mmol/day) and ramipril (10 mg/day). In the follow-up period, her serum potassium improved to a level of 3.5 mmol/L. Her serum creatinine stabilized at the level of 1.13 mg/dL (eGFR 59 mL/min) over the next 3 years of follow-up.
Molecular diagnosis
Following informed consent, genomic DNA of the patient and of their healthy two daughters was extracted from peripheral blood leukocytes. Parents and other family members were not available for genetic testing. We performed DNA sequencing by a two-step approach: (i) massively parallel sequencing [next generation sequencing (NGS)]-based mutation screening combined (ii) with Sanger sequencing. High-coverage NGS-based mutation screening was performed on an Illumina HiSeq 4000 sequencing instrument followed by data analysis using two different in silico software approaches for mapping, base calling and variant detection. These approaches included data analyses by (i) Exomiser and (ii) SeqNext software modules (for details see Supplementary Methods).
(i) Both analysis approaches revealed two missense mutations in exon 2 of KCNJ1 (in compound heterozygous condition) belonging to different alleles, c.197T > A (p.I66N) and c.875G > A (p.R292Q). The heterozygous c.875G > A mutation was detected (in absence of the c.197T > A mutation) in both healthy daughters. One of the mutations, c.875G > A (p.R292Q), has already been described as pathogenic mutation in a patient with BS II, although not in this allelic combination [7]. The other mutation, causing a T-to-A transition, c.197T > A (p.I66N) (Figure 2), is novel and most likely pathogenic based on the clinical phenotype. A number of other diseases have feature that can overlap with aspects of aBS II (Supplementary Table S1). However, there were no mutations in SLC12A1 (NKCC2), CLCNKB/CLCNKA (ClC-Kb, ClC-Ka and ClC-Kb), BSND (barttin), CASR (CaSR), MAGED2 (MAGE-D2) and SLC12A3 (NCCT), which cause other forms of BS or Gitelman syndrome (Supplementary Table S1) [2, 8, 9]. Moreover, there were no mutations in CLDN16 (claudin 16) and CLDN19 (claudin 19), which cause nephrocalcinosis and renal failure, due to renal magnesium wasting with hypomagnesaemia and hypercalciuria (Supplementary Table S1) [10].
Fig. 2.

Next generation DNA sequencing pseudo-electropherograms demonstrating heterozygosity for c.197T > A (p.I66N) (A) and c.875G > A (p.R292Q) (B) in KCNJ1 in our patient. Results of forward and reverse DNA sequencing of exon 2 analysed by customized SeqNext approach. Non-synonymous mutations are shown as rectangles indicated with arrows pointing downward above the pseudo-electropherogram. In the yellow coloured boxes are shown the allelic frequencies of each substitution. In the reads sequences, substitutions are highlighted in red.
(ii) We validated the results of the NGS approaches by Sanger sequencing, which was performed on PCR amplicons from genomic DNA covering that variant position (Figure 3). We sequenced the protein coding sequence of KCNJ1, which is composed of three different splice variants of one to two exons and comprising a region of nearly 31.3 kb. The sequenced region included the neighbouring splice sites, a total of 1.905 bp. The adjustment of the found sequences data was analysed in the Ensembl transcript gene database under ENST00000392665 available sequences.
Fig. 3.

Sanger DNA sequencing electropherograms demonstrating heterozygosity for c.197T > A (p.I66N) (A) and c.875G > A (p.R292Q) (B) in KCNJ1 in our patient.
KCNJ1 mutations
The following KCNJ1 mutations were identified:
c.197T > A, heterozygote, exon 2, codon 66, protein p.I66N (ATC > AAC); Ile66Asn
c.875G > A, heterozygote, exon 2, codon 292, protein p.R292Q (CGG > CAG); Arg292Gln
Discussion
aBS II is also called the neonatal variant of BS because it presents with severe symptoms in the neonatal period. In this study, we identified a novel KCNJ1 missense mutation (p.I66N) in a compound heterozygote setting with a previously recognized pathogenic mutation (p.R292Q) [7] in an adult woman leading to an unusual mild clinical presentation of aBS II. Based on the autosomal recessive pattern of aBS II, both missense mutations are expected to disrupt KCNJ1 gene function. However, the KCNJ1 mutation p.I66N could represent a mutation causing only a mild phenotype. Our experimental approach of NGS-based mutation screening combined with Sanger sequencing proved to be a reliable approach for molecular diagnosis in our patient with a clinical unclassified phenotype resembling a late-onset BS. Two different in silico NGS software approaches were employed for mapping, base calling and variant detection. Both approaches detected the two mutations. These approaches included (i) Exomiser [11] and (ii) customized SeqNext (JSI Medical Systems) approaches for data analyses.
Our findings confirm a recent observation that aBS II can present as late-onset disease in adulthood [4]. Interestingly, this case and our case presented with nephrocalcinosis as incidental presentation of BS. Although different KCNJ1 mutations were found, both patients exhibited unusual mild clinical manifestations of aBS II, i.e. hypokalaemia, modest or tendency towards metabolic alkalosis, mild hypercalciuria and modest hyperreninaemic hyperaldosteronism due to renal salt wasting. Therefore, oral potassium replacement (80 mmol/day) was sufficient for improvement of serum potassium levels to near normal values. Both patients maintained normal serum Mg2+ levels, which is consistent with previous observations demonstrating that Bartter’s patients with hypomagnesaemia rarely reach the levels seen in patients with Gitelman syndrome (Supplementary Table S1) [2]. Upregulation of NCCT via aldosterone may contribute to the increase in expression of TRPM6 Mg2+ channels in the distal convoluted tubule (DCT) and be protective against hypomagnesaemia in BS [2]. Although patients with salt-losing nephropathy have an increased salt intake (salt craving) while they consume their normal ad-lib diet [8, 12], oral salt supplementation has no or little effects on potassium homeostasis in adult BS. Klaus et al. examined a 20-year old female with BS (although not genetically tested) and found that neither low-salt diet (30 mval or 10 mval Na+) nor salt-rich diet (240 mval Na+/day) over 5 days improved hypokalaemia and potassium excretion in the urine. These manoeuvres also did not affect plasma renin levels, indicating insignificant effects on volume regulation [13]. However, it is noteworthy that the addition of potassium-sparing diuretics or an aldosterone antagonist may improve hypokalaemia in late-onset aBS II [4].
Our patient initially presented with hypercalciuric nephrocalcinosis associated with chronic renal impairment. BS patients commonly develop nephrocalcinosis. This is presumed to be due to their hypercalciuria, however the mechanisms are poorly understood. For example, nephrocalcinosis is less prevalent among Bartter’s patients with CLCNKB and BSND mutations (Supplementary Table S1), who have less hypercalciuria [2, 14]. It is possible that bone reabsorption from elevated PTH levels leads to increased urinary phosphate wasting, and that this accounts for the development of nephrocalcinosis, though this is poorly documented in patients [2]. Consistent with this possibility, Pattaragarn et al. found that suppression of PTH by a calcimimetic agent attenuated nephrocalcinosis in furosemide-treated young rats [15]. Since thyroid/parathyroid ultrasound examination, computed tomography neck/thorax scan, parathyroid scintigraphy, osteodensitometry, X-rays of hands and plasma calcium levels were unremarkable despite elevated plasma iPTH levels, there is no evidence of primary hyperparathyroidism in our patient. Instead, the slight increase of PTH can be explained by the reduction of eGFR (MDRD) (58 mL/min). Another potential mechanism contributing to the occurrence of nephrocalcinosis might involve renal luminal pH; i.e. the increased acidification of the distal nephron due to increased epithelial sodium channel (ENaC) activity might protect from calcium phosphate crystal formation in the distal parts of the nephron [2]. Kleta and Bockenhauer propose that under no circumstances should treatment with thiazides be instituted for hypercalciuria and nephrocalcinosis since the additional blockade of salt reabsorption in DCT deprives the kidney of an important compensatory mechanism with potentially disastrous consequences [16].
An additional feature of Bartter’s patients is that they produce high levels of prostaglandin E2 by cyclooxygenase 2 (COX2) in the macula densa, which can exacerbate renal salt wasting. Cyclooxygenase inhibition by indometacin is widely used for therapy of BS in children. The use of indometacin improves growth rate [14, 17] and early treatment in the neonatal period may decrease nephrocalcinosis [18]. However, data about the long-term outcome of patients with BS with indometacin treatment are not available. Instead, nephrotoxicity associated with non-steroidal anti-inflammatory drugs and COX2 inhibitors is well documented [19], which limits enthusiasm for long term treatment of Bartter’s patients with a mild phenotype in adulthood. Nevertheless, a tentative treatment with indomethacin at a low dose, with a careful monitoring of GFR, could be a therapeutic option in our patient in order to reduce (or even normalize) calcium excretion in urine, and thus to prevent progression of nephrocalcinosis.
Walsh et al. reported that the degree of hypokalaemia (as an index of chronic potassium depletion) does not correlate with GFR, and that on-going sodium and water losses, consequent secondary hyperaldosteronism, may play a more important role in the aetiology of chronic renal impairment in BS and possibly other hypokalaemic nephropathies [20]. This view is supported by renal histopathological findings in BS, which are initially normal except for hyperplasia of the juxtaglomerular apparatus and glomerular atrophy [21]. Importantly, this contrasts with chronic kaliopenic nephropathy by laxative abuse, which is characterized by vacuolar changes in renal tubular epithelium accompanied by inflammatory interstitial changes in patients with potassium losses [22]. Therefore, Walsh et al. suggested that long-term blockade of the mineralocorticoid receptor in BS with spironolactone or eplerenone (or perhaps even be direct renin inhibition) may be justified more for renal protection than for trying to correct hypokalaemia per se. Similarly, angiotensin-converting-enzyme inhibitors may be justified for renal protection in Bartter’s patients, unless they produce symptomatic orthostasis, hypotension or lethargy.
Our study suggests a high degree of variability of aBS II regarding severity of disease as shown in another report in adolescence [23]. Mutations in KCNJ1 should thus be considered even beyond the neonatal period in patients who present with nephrocalcinosis, hypokalaemia, metabolic alkalosis or symptoms of renal salt wasting (Supplementary Table S1).
Conclusions
We report an unusual, in severity mild, variant of BS II associated in a female patient with a remarkably late onset of the disease. This phenotype is associated with compound heterozygous KCNJ1 mutations, consisting of a novel c.197T > A (p.I66N) and a previously reported c.875G > A (p.R292Q) KCNJ1 mutation. Our results demonstrate that aBS II may be suspected in patients with a late-onset phenotype resembling BS. Our experimental approach of NGS-based mutation screening combined with Sanger sequencing proved to be a reliable approach for molecular diagnosis in our patient and can be implemented in genetic testing approaches in various clinical conditions. Our methodological approach and results may help to diagnose and classify other subjects with clinical unclassified renal phenotypes of nephrocalcinosis and electrolyte disorders.
Supplementary data
Supplementary data [24-27] are available online at http://ckj.oxfordjournals.org.
Supplementary Material
Acknowledgements
We would like to thank the patient for participating in this study. We also thank Claudia Langnick (Max Delbrück Center for Molecular Medicine, Berlin, Germany) for expert technical assistance and Manuel Holtgrewe (Berlin Institute of Health, Bioinformatics Core Unit) for data file processing. This work is supported by the Deutsche Forschungsgemeinschaft (DFG).
Conflict of interest statement
None declared.
References
- 1. Simon DB, Karet FE, Rodriguez-Soriano J. et al. Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nature Genet 1996; 14: 152–156 [DOI] [PubMed] [Google Scholar]
- 2. Scholl UI, Lifton RP.. Molecular genetics of Gitelman's and Bartter's syndromes and their implications for blood pressure variation In: Lifton RP, Somlo S, Giebisch GH. et al. (eds) Genetic Diseases of the Kidney. London, Oxford, Boston, New York and San Diego: Elsevier, 2009, 229–247 [Google Scholar]
- 3. Fretzayas A, Gole E, Attilakos A. et al. Expanding the spectrum of genetic mutations in antenatal Bartter syndrome type II. Pediatr Int 2013; 55: 371–373 [DOI] [PubMed] [Google Scholar]
- 4. Huang L, Luiken GP, van Riemsdijk IC. et al. Nephrocalcinosis as adult presentation of Bartter syndrome type II. Neth J Med 2014; 72: 91–93 [PubMed] [Google Scholar]
- 5. Zittema D, Boertien WE, van Beek AP. et al. Vasopressin, copeptin, and renal concentrating capacity in patients with autosomal dominant polycystic kidney disease without renal impairment. Clin J Am Soc Nephrol 2012; 7: 906–913 [DOI] [PubMed] [Google Scholar]
- 6. Martelli-Junior H, Ferreira SP, Pereira PC. et al. Typical features of amelogenesis imperfecta in two patients with Bartter’s syndrome. Nephron Extra 2012; 2: 319–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schulte U, Hahn H, Konrad M. et al. pH gating of ROMK (K(ir)1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc Natl Acad Sci USA 1999; 96: 15298–15303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roser M, Eibl N, Eisenhaber B. et al. Gitelman syndrome. Hypertension 2009; 53: 893–897 [DOI] [PubMed] [Google Scholar]
- 9. Laghmani K, Beck BB, Yang SS. et al. Polyhydramnios, transient antenatal Bartter's syndrome, and MAGED2 mutations. N Engl J Med 2016; 374: 1853–1863 [DOI] [PubMed] [Google Scholar]
- 10. Viering DH, de Baaij JH, Walsh SB. et al. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smedley D, Jacobsen JO, Jager M. et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc 2015; 10: 2004–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cruz DN, Simon DB, Nelson-Williams C. et al. Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension 2001; 37: 1458–1464 [DOI] [PubMed] [Google Scholar]
- 13. Klaus D, Bocskor A, Seif F.. Regulation der Reninsekretion beim Bartter-Syndrom. Klin Wschr 1968; 46: 1201–1206 [Google Scholar]
- 14. Brochard K, Boyer O, Blanchard A. et al. Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol Dial Transplant 2009; 24: 1455–1464 [DOI] [PubMed] [Google Scholar]
- 15. Pattaragarn A, Fox J, Alon US.. Effect of the calcimimetic NPS R-467 on furosemide-induced nephrocalcinosis in the young rat. Kidney Int 2004; 65: 1684–1689 [DOI] [PubMed] [Google Scholar]
- 16. Kleta R, Bockenhauer D.. Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol 2006; 104: p73–p80 [DOI] [PubMed] [Google Scholar]
- 17. Vaisbich MH, Fujimura MD, Koch VH.. Bartter syndrome: benefits and side effects of long-term treatment. Pediatr Nephrol 2004; 19: 858–863 [DOI] [PubMed] [Google Scholar]
- 18. Matsumoto J, Han BK, Restrepo de Rovetto C. et al. Hypercalciuric Bartter syndrome: resolution of nephrocalcinosis with indomethacin. AJR Am J Roentgenol 1989; 152: 1251–1253 [DOI] [PubMed] [Google Scholar]
- 19. Fletcher JT, Graf N, Scarman A. et al. Nephrotoxicity with cyclooxygenase 2 inhibitor use in children. Pediatr Nephrol 2006; 21: 1893–1897 [DOI] [PubMed] [Google Scholar]
- 20. Walsh SB, Unwin E, Vargas-Poussou R. et al. Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome. QJM 2011; 104: 939–944 [DOI] [PubMed] [Google Scholar]
- 21. Bartter FC, Pronove P, Gill JR Jr. et al. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med 1962; 33: 811–828 [DOI] [PubMed] [Google Scholar]
- 22. Elitok S, Bieringer M, Schneider W. et al. Kaliopenic nephropathy revisited. Clin Kidney J 2016; 9: 543–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharma A, Linshaw MA.. A novel compound heterozygous ROMK mutation presenting as late onset Bartter syndrome associated with nephrocalcinosis and elevated 1,25(OH)(2) vitamin D levels. Clin Exp Nephrol 2011; 15: 572–576 [DOI] [PubMed] [Google Scholar]
- 24. Hou J, Goodenough DA.. Claudin-16 and claudin-19 function in the thick ascending limb. Curr Opin Nephrol Hypertens 2010; 19: 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Claverie-Martin F. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: clinical and molecular characteristics. Clin Kidney J 2015; 8: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsvetkov D, Hohmann M, Anistan YM. et al. A CD2AP mutation associated with focal segmental glomerulosclerosis in young adulthood. Clin Med Insights Case Rep 2016; 9: 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gollasch B, Basmanav FB, Nanda A. et al. Identification of a novel mutation in RIPK4 in a kindred with phenotypic features of Bartsocas-Papas and CHAND syndromes. Am J Med Genet Part A 2015; 167A: 2555–2562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.