

Abstract
Karenia brevis, the Florida red tide dinoflagellate produces a suite of neurotoxins known as the brevetoxins. The most abundant of the brevetoxins PbTx-2, was found to inhibit the thioredoxin-thioredoxin reductase system, whereas the PbTx-3 has no effect on this system. On the other hand, PbTx-2 activates the reduction of small disulfides such as 5, 5’-dithio-bis-(2- nitrobenzoic acid) by thioredoxin reductase. PbTx-2 has an α, β-unsaturated aldehyde moiety which functions as an efficient electrophile and selenocysteine conjugates are readily formed. PbTx-2 blocks the inhibition of TrxR by the inhibitor curcumin. Whereas curcumin blocks PbTx- 2 activation of TrxR. It is proposed that the mechanism of inhibition of thioredoxin reduction is via the formation of a Michael adduct between selenocysteine and the α, β-unsaturated aldehyde moiety of PbTx-2. PbTx-2 had no effect on the rates of reactions catalyzed by related enzymes such as glutathione reductase, glutathione peroxidase or glutaredoxin.
Keywords: Karenia brevis, brevetoxin, thioredoxin, thioredoxin reductase
Graphical abstract
INTRODUCTION
The brevetoxins (Figure 1) are a suite of structurally related polyether ladder type neurotoxins produced by the Florida red tide organism Karenia brevis. This dinoflagellate blooms almost annually in the Gulf of Mexico, resulting in massive fish kills.1 Brevetoxins have been associated with numerous marine mammal deaths, including multiple manatee epizootic events2, 3 and dolphin strandings.4 Exposure to brevetoxins as aerosols during red tide events can result in respiratory distress5 and the consumption of tainted shellfish induces a syndrome known as neurotoxic shellfish poisoning (NSP), whose symptoms include severe gastrointestinal distress, reversal of temperature sensation and paresthesia.6 Detrimental economic effects of the Florida red tide result from the closure of fisheries, loss of tourism and public health costs. Economic losses due to illness brought on by Florida Red tide have been estimated between $60,000 and $700,000 per year, but could extend up to $1,000,000 depending on the severity and length of the bloom.7 The detrimental effects of K. brevis blooms have been attributed to the interaction of the brevetoxins with site 5 of the voltage gated sodium channel. Brevetoxin binding shifts the activation potential for channel opening to more negative values (i.e. channel activation at normal resting potentials) resulting in the depolarization of excitable membranes.8
Figure 1.
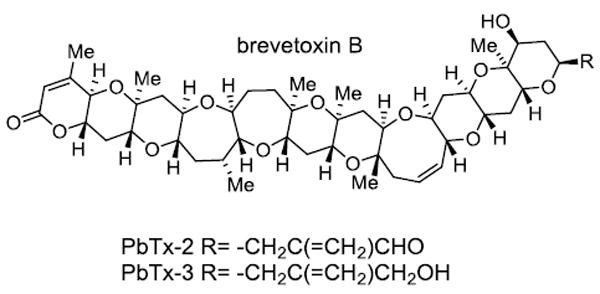
Structures of the two most abundant brevetoxins having the B-type backbone
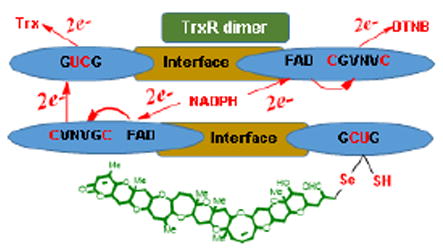
Recently, our efforts have focused on discovering the endogenous role of the brevetoxins. Using fluorescent and photoaffinity derivatives of brevetoxin, we demonstrated that exogenously applied brevetoxin localizes to the chloroplast of K. brevis where it interacts with two chloroplast proteins including thioredoxin (Trx).9 Trx is the parent of a family of enzymes which mediate the redox state within a cell via thiol disulfide exchange.10 Trx is converted from its inactive disulfide form to its active dithiol through reduction by thioredoxin reductase (TrxR). TrxR is a dimer whose two strands are organized in an antiparallel arrangement. In the enzymatic mechanism of Trx reduction by TrxR, NADPH provides the reducing equivalents and initiates a cascade of redox reactions across three redox active sites: FAD which is first reduced by NADPH, next FADH reduces an N-terminal disulfide (in human TrxR:- Cys59-Val-Asn-Val-Gly-Cys64) which undergoes thiol disulfide exchange with a C-terminal disulfide or seleno-sulfide onthe opposite chain of the dimer. The C-terminal redox center of TrxR undergoes thiol-disulfide exchange with oxidized Trx (Figure 2).
Figure 2.
Electron flow of mammalian TrxR/Trx system. Reduced sites are shown in red.
Mammalian TrxR is differentiated from those of lower organisms by replacement of the second of the two C-terminal redox-active site cysteine residues with a selenocysteine (Sec) (Cys497-Sec498-Gly- in human TrxR). Although this type of TrxR is often referred to as mammalian, the presence of a Sec residue in the C-terminal redox center has been demonstrated to be widespread in the animal kingdom.11 The rate-limiting step in the mechanism of reduction of Trx by TrxR is believed to be the transfer of electrons from the N-terminal redox center to the C-terminal redox center on the adjacent unit.7, 12 Several inhibitors of TrxR have been identified including quinones,13-16 curcumin17 and 4-hydroxy-2-nonenal.18 All of these molecules can function as electrophiles or Michael acceptors, resulting in the irreversible inhibition of TrxR.19 Curcumin was found to alkylate both the C-terminal cysteine and selenocysteine of TrxR. Whereas 4-hydroxy-2-nonenal and quinols have also been demonstrated to label the C-terminal cysteine and selenocysteine of TrxR as well as the redox active cysteines of Trx. The α,β-unsaturated aldehyde K-ring side chain of PbTx-2 may serve as an efficient electrophile, reacting with nucleophiles as a Michael acceptor. Indeed, it has long been known that the principle detoxification mechanism for PbTx-2 is the formation of cysteine and glutathione conjugates at this site20, 21 by Michael addition of the cysteine thiol. Disruption of the normal function of the TrxR/Trx system has the potential to compromise the oxidative status of the cell and redox signaling. Because Trxs are present in almost all living organisms, this suggested an alternate mechanism of toxicity in animals and humans exposed to the brevetoxins. This reasoning prompted us to examine the interaction of brevetoxins (PbTx-2 and 3) with Trx, thioredoxin reductase (TrxR) and related enzymes.
MATERIALS AND METHODS
General Materials and Methods
Unless otherwise noted, reagents were purchased from Sigma-Aldrich or Acros chemical companies and used without further purification. 2-Thiobarbituric acid (TBA) was purchased from MP biomedicals and 1,1,3,3-tetraethoxypropane (TEP) was purchased from Chem-Impex Int’l Inc. Brevetoxins (PbTx-2 and -3) were isolated from K. brevis cultures according to published methods.22 Enzyme assay kits (fluorescent TrxR/Trx; fluorescent glutaredoxin; colorimetric glutathione peroxidase) and rat TrxR were purchased from Cayman Chemical. Assay kits were used according to the manufacturer’s instructions with noted exceptions. Truncated human TrxR (Novus Biologicals) was purchased from Fisher Scientific. UV/Visible and fluorescence measurements were performed in 96 or 384 well microplates using a Synergy® 2 (Biotek Instrument, Inc.) or an Infinite® M1000 PRO (Tecan Group Ltd.) microplate reader.
TrxR/Trx inhibition assay
Experiments were performed in 96-well plates with 270 μL of assay buffer (50 mM Tris-HCl, 1 mM EDTA, 0.14 mg/mL BSA, pH 7.5) containing 0.13 μM rat TrxR, 0.35 mM NADPH and 0.026 μM Trx. After incubation for 30 min at 37 °C to reduce the TrxR, stock solutions of PbTx-2 or PbTx-3 (11 μL each of 1.1, 0.96, 0.64, 0.35 and 0 mM in methanol) were added to give final toxin concentrations of 35 μM, 30 μM, 20 μM, 11 μM, 0 μM. This mixture was incubated at 37 °C for 1 h. Fluorescent substrate (70 μL, 0.4 mg/mL, eosin labeled bovine insulin) was added to each aliquot (final concentration of 0.016 mg/mL). Samples were analyzed in triplicate by recording fluorescence at 545 nm (λex = 520 nm) every 10 min for 3 h.
Glutathione peroxidase (GPx) inhibition assay
The assay was carried out according to the manufacturer’s instructions except that stock solutions of PbTx-2 (15 μL of 2.2 mM, 1.3 mM and 0 mM in methanol) were each combined with the manufacturer provided assay buffer, GPx and GSH solutions to give PbTx-2 concentrations of 78 μM, 47 μM and 0 μM. This mixture was incubated for 1 h at room temperature, after which the assay was completed according to the manufacturer’s instructions. The absorbance was read once every minute at 340 nm for 10 min. Samples were analyzed in triplicate. Controls without GPx were treated as background. The final concentrations of PbTx-2 were 50 μM, 30 μM and 0 μM.
Glutaredoxin (Grx) inhibition assay
Experiments were performed in 96-well plates with 160 μL of a reaction mixture containing 0.17 M potassium phosphate, 1.7 mM EDTA, 1 mM GSH, 0.1 mg/mL BSA, 54 μM NADPH, 0.1 μM GR, pH 7.5 which was prepared according to the manufacturer’s instructions. To aliquots (160 μL) of the reaction mixture was added PbTx-2 (10μL of the following standards: 0.95, 0.63, 0.32 and 0.0 mM in methanol), 16 μL of human Grx 1 (30 nM in 0.83 M potassium phosphate and 8.3 mM EDTA, pH 7.5, final concentration 1.5 nM), 102 μL of PBS and 32 μL of eosin labeled GSH bound to BSA (0.2 mM in distilled water). Samples were analyzed in triplicate by recording fluorescence at 545 nm (λex = 520 nm) every 10 min for up to 2 h. The final concentrations of PbTx-2 were, 0 μM, 11 μM, 22 μM, 33 μM.
Activity assay for TrxR or truncated TrxR
Method A (pre-incubation with PbTx-2): Experiments were performed in 96-well plates with 160 μL of PBS containing 0.1 mM NADPH, 22 μM PbTx-2 and 0.56 μM rat TrxR or 0.68 μM truncated human TrxR. After incubation for 30 min at 37 °C to reduce the TrxR, 40 μL of a DTNB solution (Ellman’s reagent: 5,5’-dithio-bis-(2-nitrobenzoic acid) (10 mM in 0.1 M sodium phosphate and 1 mM EDTA, pH 8.0) was addedfor final concentrations of 18 μM PbTx-2 and 2 mM DTNB. The absorbance at 410 nm wasmeasured every 5 min for up to 60 min. Method B (no pre-incubation with PbTx-2): The assaywas performed as described above except that PbTx-2 and DTNB solutions were mixed and added simultaneously after NADPH reduction of TrxR.
Preparation and analysis of selenocysteine-PbTx-2 adduct
seleno-L-cystine (75 μL, 1 mg /mL) was incubated with an aqueous suspension of immobilized TCEP ((tris(2-carboxyethyl)phosphine, 30 μL, >8 μmol/mL, immobilized onto 4% crosslinked beadedagarose) for 1 h to reduce the diselenide. The reaction vial was flushed with nitrogen and centrifuged for 10 min at 14,000 x g. The supernatant was transferred via syringe to a solution of PbTx-2 (200 μL, 1 μg/μL in methanol) and stirred under nitrogen for 24 h. Accurate mass spectra was acquired using a Bruker Solarix 7.0 T using ultrahigh resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometer (FT-ICR) confirmed the formation of an oxidized (+15.999 amu) selenocysteine-PbTx-2 adduct of formula C53H77NO17Se. HRMS calc’d for [M+H+]: 1080.44355. Found: 1080.44356 (Δ = -0.56 ppm). HRMS calc’d for [M+Na+]: 1102.42545. Found: 1102.42550 (Δ = -0.12 ppm).
Preparation and analysis of TrxR-PbTx-2 adduct
Rat TrxR (6 μM) and NADPH (0.16 mM) were incubated for 30 min in 44 mM Tris base, 0.9 mM EDTA. 0.4 M NaCl, 0.1 % glycerol, pH 7. PbTx-2 (5.6 mM in Me2SO) was added to give a final PbTx-2 concentration of 0.24 mM and the reaction was incubated for 12 h at room temperature. The sample was concentrated 5 fold and analyzed by SDS PAGE (12% acrylamide, 10% SDS). The band was excised and stored at -80 °C. In gel digestion and LC-Electrospray Ionization MS/MS (LC-ESI MS/MS) analysis was performed by the Proteomics and Mass Spectrometry Facility at the UMass Medical School as previously described.9
Reaction of Sel-green probe with TrxR
The selenol selective probe, Sel-green, was synthesized according to published methods.23 Rat TrxR (6 μM) and NADPH (0.1 mM) were incubated for 30 min in 45 mM Tris base, 0.9 mM EDTA. 0.5 M NaCl, 0.1 % glycerol, pH 7. PbTx-2 (1.1 mM in methanol) was added for a final toxin concentration of 48 μM. Samples were prepared in triplicate. A control sample was prepared by the addition of an equal volume of methanol without PbTx-2. A blank sample was prepared without TrxR. The solutions were incubated for 1 h. Sel-green probe (9 μL, 481 μM in 20% acetone in PBS) was added to each reaction mixture (82 μL). After 55 min, fluorescence at 502 (λex = 370 nm) was measured. Calibration standards were prepared as follows: A stock solution of seleno-L-cystine (1.0 mL, 27 μM, in PBS) was reduced using immobilized TCEP (1.0 mL, > 8 μmol/mL) for 1 h. After centrifugation (14000 x g, 2 min), the supernatant was transferred to a clean tube to provide a stock solution of L-selenocysteine (54 μM). Aliquots of the stock solution of L-selenocysteine was diluted with Sel-green probe in PBS for final concentrations of L-selenocysteine of 0 μM, 3 μM, 6 μM, 12 μM, 25 μM and 43 μM Sel-green probe, incubated for 55 min, and fluorescence was measured at 502 nm (λex = 370 nm). Calibration standards were prepared in triplicate. For this experiment, the concentration of FAD (flavin adenine dinucleotide) was calculated to be 5.3 μM by measuring the absorbance of at 450 nm and using an extinction coefficient ε450 = 11,300 M–1cm–1 for FAD.24
Cell culture
The human lymphoblast cell line (GM02152) was purchased from Coriell Institute for medical research. Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 15% fetal bovine serum (FBS) and 2 μM sodium selenite. The cells were maintained in a humidified incubator at 37°C and 5% CO2 until they reached log phase.
Cytotoxicity assay
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which is based on cleavage of tetrazolium salt by metabolically active cells to form formazan dye.25 The MTT-formazan crystals are solubilized in DMSO after the specified incubation period. GM02152 cells were seeded in 96-well plates at 8×104 cells/well with fresh medium containing different doses of PbTx-2 or trolox. After a 24 h incubation period, 200 μL fresh medium containing 0.5 mg/mL MTT (Amresco, Solon, OH) were added in each well and incubated for an additional 4 h. Culture medium containing MTT was subsequently removed and replaced with 200 μL DMSO to dissolve the resulting formazan crystals. The absorbance of each well at 570 nm (A570) was measured by a microplate reader (BioTek, Winooski, VT). Cells were treated with PbTx-2 from 0-4μg/mL or 0-500 μM trolox (6- hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) for 24 h and then subject to MTT assay. Untreated cells served as the control. Cell viability was calculated by the equation: cell viability (%) = (A570 of treated group/A570 of control group)×100%. The MTT assay was repeated three times. The dose response curve was analyzed using the Graphpad Prism 7 software (La Jolla, CA).
Preparation of cell lysates for biochemical assays
GM02152 cells (2.5 ×107) were seeded in 75 cm2 flasks containing growth medium. After the cells reached log phase, growth medium was removed by centrifugation and replaced with fresh medium containing: 1 μg/mL PbTx-2 or 1μg/mL PbTx-2 +100 μM trolox or DMSO alone. The cells were exposed for 24 hours after which they were resuspended in growth medium and centrifuged (720 × g, 4 minutes). The supernatant was discarded and the cells were lysed by the addition of 100 μL lysis buffer (10 mM Tris-HCl, 200 mM KCl, 2 mM EDTA, 40% Glycerol, 0.2% TritonX-100, pH 7.5) by rotating at 4 °C for 2 h as previously described.26 Cellular debris was removed by centrifugation at 17000 × g at 4°C for 30 min. Protein concentration of the lysate was determined by the Bradford method 27 using the Coomassie protein assay reagent in duplicate. Typical concentrations of the lysate ranged from 10 – 15 mg/mL.
DTNB assay on cell lysates
DTNB (100 μL, 5 mM in 0.1 M sodium phosphate, 1mM EDTA pH 8) solution was added to the cell lysate (90 μg of total protein/well) and absorbance was measured immediately at 410 nm. The assay was performed in triplicate.
Thiobarbituric acid-reactive substances (TBARS) assay on cell lysates
Lipid peroxidation was determined by measuring the level of thiobarbituric acid-reactive substances (TBARS) in cells.28 Proteins were precipitated from the cell lysate (160 μg of total protein) by the addition of trichloroacetic acid (500 μL, 20%) followed by centrifugation at 14,000 × g for 15 min. To the supernatant thiobarbituric acid solution (500 μL of 0.67% in 10% DMSO) was added. The mixture was then heated for 15 minute at 90°C. Absorbance was measured in triplicate at 532 nm.
Sel green assay on cell lysates
In a 96 well plate, Sel green probe (10 μL, 48 μM final concentration in 20 % acetone in PBS) was added to cell lysate (75 μg of total protein/well) to quantify the amount of selenocysteine. The mixture was incubated for 5 minutes and the fluorescence was measured at λex = 502 and λem = 370 nm). Each treatment was analyzed in triplicate.
Statistical analysis of cytotoxicity and cell lysate assays
All data values are given as mean ± standard error. ANOVA was performed by a Tukey Honestly Significant Difference test using the JMP 13.0.0 Software.
RESULTS
Inhibition of mammalian TrxR/Trx by brevetoxins
We first examined the effect of PbTx-2 and PbTx-3 on the mammalian TrxR/Trx system using a commercially available kit which is based on Trx reduction of eosin modified insulin.29 Trx is reduced by TrxR with reducing equivalents ultimately provided by the NADPH dependent reduction of TrxR. PbTx-2 and PbTx-3 at concentrations ranging from 10 to 35 μM were incubated with a mixture of pre-reduced TrxR and Trx for one hour, after which eosin-modified insulin was added and the fluorescence was monitored. Interestingly, PbTx-2 exhibited a dose dependent inhibition of Trx/TrxR activity with an IC50 of 25 μM, whereas PbTx-3 had no effect on the rate of insulin reduction (Figures 3A and 3B).
Figure 3.
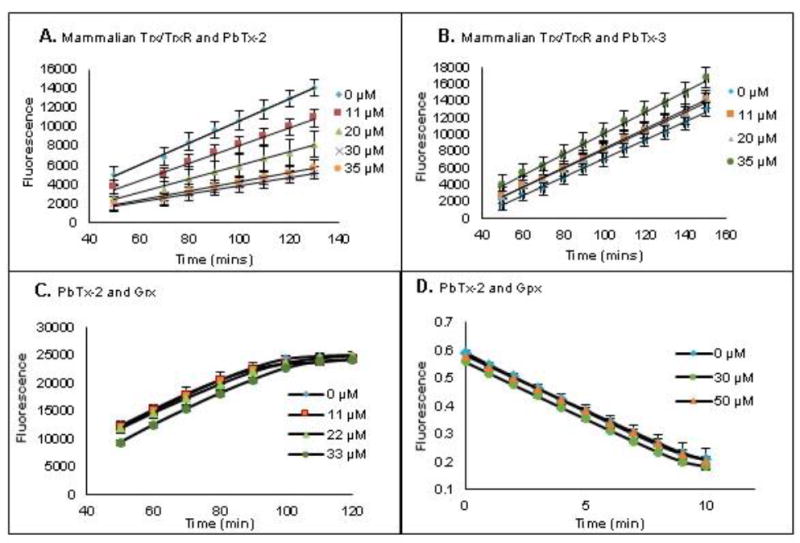
The effect of brevetoxin on TrxR/Trx system, TrxR, Grx and GPx. (A/B) The effect of PbTx-2 (A) or PbTx-3 (B) on the TrxR/Trx system in the fluorescent insulin reduction assay. Rat TrxR and human Trx pre-reduced with NADPH (0.35 mM) for 30 min followed by incubation with brevetoxins for 1 h prior to addition of insulin substrate. (C) The effect of PbTx-2 on Grx. (D) The effect of PbTx-2 on GPx.
The effect of PbTx-2 on glutaredoxin (Grx), glutathione peroxidase (GPx) and glutathione reductase (GR)
The activity of Grx and GPx in the presence and absence of PbTx-2 was examined using commercially available assay kits (Figures 3C and 3D). No inhibition was observed for either enzyme in the presence of PbTx-2 (up to 56 μM for Grx and 30 μM for GPx). Because these kits contain GR to provide a supply of reduced glutathione, we can conclude that PbTx-2 also does not inhibit GR.
The effect of brevetoxin (PbTx-2) on mammalian TrxR
Because we observed inhibition of the two enzyme TrxR/Trx system by PbTx-2, our next step was to examine one of these enzymes independently. Mammalian TrxR has a broad substrate specificity and reduces low molecular weight disulfides in addition to Trx. We therefore examined the activity of rat TrxR towards the reduction of the disulfide of 5,5’-dithio-bis-(2-nitrobenzoic acid) (DTNB or Ellman’s reagent) in the presence and absence of PbTx-2. When pre-reduced TrxR was incubated with PbTx-2 (22 μM) prior to addition of substrate, we found that the initial rate of reduction of DTNB by TrxR was actually increased by 2.5-fold when compared to the control sample which lacked PbTx-2. The same result was obtained when PbTx-2 was added simultaneously with DTNB. In other words, without pre-incubation of TrxR and PbTx-2. (Figures 4A and 4B).
Figure 4.
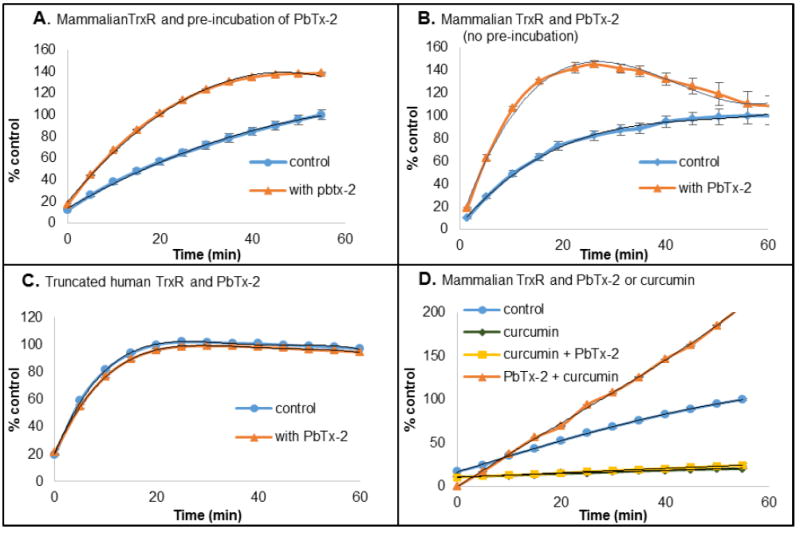
(A/B) Rat TrxR was pre-reduced with NADPH (0.10 mM) for 30 min either with (A) or without (B) PbTx-2 (18 μM), followed by addition of DTNB (2 mM). (C) Truncated human TrxR was pre-reduced with NADPH (0.10 mM) for 30 min in the presence of PbTx-2 (18 μM), followed by DTNB (2 mM). D. Rat TrxR was pre-reduced with NADPH (0.10 mM) for 30 min followed by incubation with curcumin (22 μM, 30 min) or curcumin (22 μM, 30 min) then PbTx-2 (22 μM, 30 min) or PbTx-2 (22 μM, 30 min) then curcumin (22 μM, 30 min), prior to addition of DTNB (2 mM). Data are expressed as % of control at 60 min.
Mammalian TrxR is a selenocysteine containing enzyme having C-terminal redox residues (Cys497-Sec498 for human TrxR) which alternate between the selenosulfide and thiol + selenol. It is this redox center that is responsible for the reduction of the active site disulfide of Trx (Figure 2). However, truncated TrxR, which lacks the C-terminal Sec residue retains its ability to reduce low molecular weight disulfides via its N-terminal dithiol redox center. In order to identify the redox center which is affected by brevetoxin, the activity of truncated human TrxR was examined in the presence of PbTx-2. At 22 μM, PbTx-2 had no effect on the activity of truncated hTrxR towards the reduction of DTNB when compared to the control sample which lacked PbTx-2 (Figure 4C). This suggests that PbTx-2 interacts with the C-terminal redox center of TrxR.
Curcumin is an inhibitor of TrxR, which acts by alkylation of the C-terminal Cys497 and Sec498.17 At pH 8.5 both the Cys497 and Sec498 are alkylated. However, at pH 6.5 only the Sec498 is alkylated by curcumin. The activity of TrxR towards DTNB reduction in the presence of curcumin and in the presence of both PbTx-2 and curcumin was examined at pH 7.0. At this pH, one would expect alkylation by curcumin principally at the Sec residue. As anticipated, the reduction of DTNB by TrxR is completely inhibited after incubation of TrxR with 22 μM curcumin for 30 min (Figure 4D). Furthermore, DTNB reduction by TrxR is inhibited completely when TrxR is first incubated with curcumin for 30 min followed by incubation with PbTx-2 for 30 min (Figure 4D). This observation was also not unexpected as the alkylation by curcumin is irreversible at this pH. However, most noteworthy is the effect on DTNB reduction by TrxR when first incubated with 22 μM PbTx-2 for 30 min followed by 22 μM curcumin for 30 min. Under these conditions, activation of DTNB reduction by TrxR was observed. These results demonstrate that PbTx-2 exerts it effect on TrxR by alkylating the C-terminal redox center at the Sec residue. The alkylation by PbTx-2 blocks alkylation by curcumin and the end result is activation rather than inhibition. To the best of our knowledge, no other TrxR inhibitor behaves in this way.
Adduct formation between PbTx-2 and selenocysteine and TrxR
Overnight incubation of a 2:1 mixture of seleno-L-cysteine and PbTx-2 revealed two prominent peaks in the mass spectrum (m/z = 1080.44355) corresponding to [M + H] and (m/z = 1102.42563) corresponding to [M + Na] for C53H77NO17Se, an oxidized adduct of PbTx-2 and selenocysteine Methionine is readily oxidized to methionine sulfoxide during electrospray MS analysis.30 One would anticipate that oxidation of the PbTx-2/Sec conjugate to the selenoxide would be even more facile as the oxidation potential of selenium is significantly lower than sulfur.31 An m/z for PbTx-2 (894. 4766) was not observed, indicating the reaction was quantitative for PbTx-2. Following overnight incubation of reduced TrxR with either with a 20-fold excess of PbTx-2 in Me2SO or Me2SO alone, samples were analyzed by SDS PAGE. The bands of anticipated size were excised from the gel and subjected to in-gel trypsin digestion, treatment of the tryptic digest with iodoacetamide (IAC) followed by MS analysis. MS analysis of the peptides from the control sample (no PbTx-2) revealed the presence of numerous peptides of predicted size according to the amino acid sequence and the anticipated IAC modified fragments. These included an observed m/z of 629.7179 which corresponds to the predicted C-terminal peptide SGGDILQSGC497U498G, doubly charged and dialkylated with IAC at the cysteine and selenocysteine residues, for an actual mass of 1257.4212 (Δ = 0.9 ppm) for the most abundant isotopes. Selenium has five stable naturally occurring isotopes and the isotopic distribution for this peptide was consistent with that predicted based on the isotope ratios confirming the presence of selenium. From the PbTx-2 incubated TrxR sample, we had anticipated observing a PbTx-2 modified C-terminal peptide SGGDILQSGC497U498(PbTx-2)G. However, this PbTx-2- peptide adduct was not observed. Nonetheless, in the PbTx-2 treated TrxR, the peptide sequence SGGDILQSGC497U498G either modified with IAC or unmodified was conspicuously absent.
Reaction of TrxR with Sel-green probe in the presence and absence of PbTx-2
The selenol selective probe, Sel-green was prepared as previously reported23 to monitor for Sec in the presence and absence of PbTx-2. The Sel-green probe undergoes a nucleophilic aromatic substitution (SNAr) reaction selectively with selenols to release a fluorescent coumarin reporter (Scheme 1). After incubation of reduced TrxR with an 8 fold excess of PbTx-2, Sel-green was added and the fluorescence increase was measured at 502 nm (λex = 370 nm). In the absence of PbTx-2 an increase in fluorescence over the control sample (which contained no TrxR) was observed. We anticipated a 1:1 stoichiometry for the reaction between TrxR and the Sel-green probe and while the concentration of TrxR (5.3 μM based on FAD absorbance) is detectable, but below the limit for accurate quantitation for the reporter, the fluorescence in the absence of PbTx-2 is comparable to the 6 μM control. Fluorescence is clearly reduced in the presence of PbTx-2 to a level which is only slightly higher than the control sample (Figure 5).
Scheme 1.
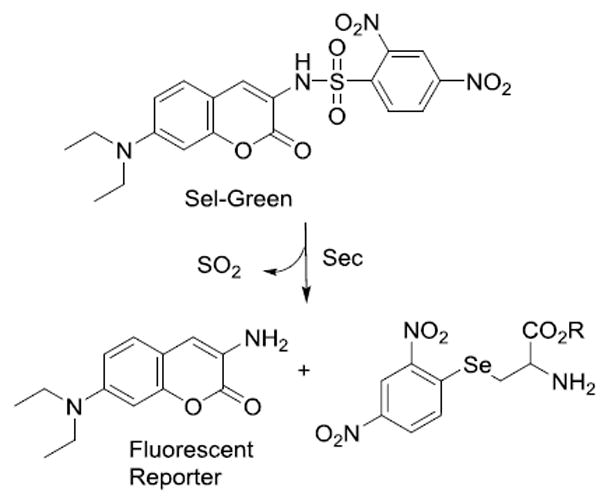
Reaction of Sel-Green probe with selenocysteine.
Figure 5.
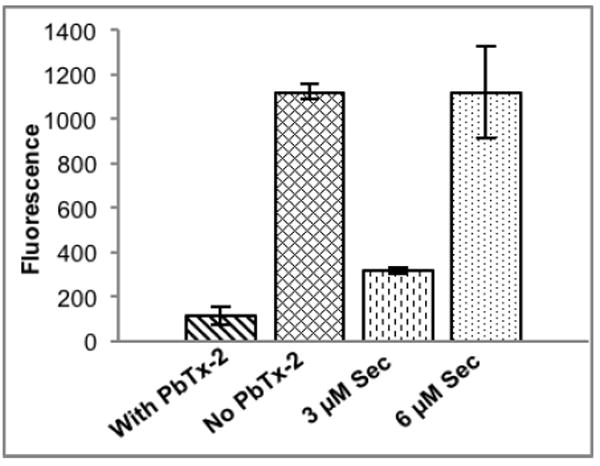
Release of fluorescent reporter from Sel-green probe upon incubation with TrxR in the presence and absence of PbTx-2 compared to standards of L-selenocysteine (3 μM and 6 μM).
The effect of brevetoxin (PbTx-2) on of human lymphoblast cells (GMO2125)
The dose response curve for 24-hour exposure of human lymphoblast (GMO2125) cells to PbTx-2 determined by the MTT assay25 yielded an EC50 of 2.3 μM (Figure 6A). Treatment with 1 μg/mL PbTx-2 for 24 hr reduces GMO2152 cell viability to 79 % of the control (Figure 6B). However, simultaneous treatment of GMO2152 cells with 1 μg/mL PbTx-2 and 100 μM trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) a water soluble vitamin E analog, increases cell viability to 96 % of the control. There is a statistically significant difference in cell viability of the PbTx-2 and PbTx-2/trolox treatments (p = 0.05) while the difference between the control and the PbTx-2/trolox treatment is not statistically significant. Cellular glutathione (GSH) content is reduced to 94 % of the control for both the PbTx-2 and PbTx-2/trolox treatments. Fluorescence from the Sel-green probe is reduced to 89 and 87 % of the control for the PbTx-2 and PbTx-2/trolox treatments respectively. On the other hand, lipid peroxidation, as determined by the TBARS assay, is increased slightly, to 105 % of the control for the PbTx-2 treatment whereas for the PbTx-2/trolox treatment lipid peroxidation is not statistically different from the control sample.
Figure 6.
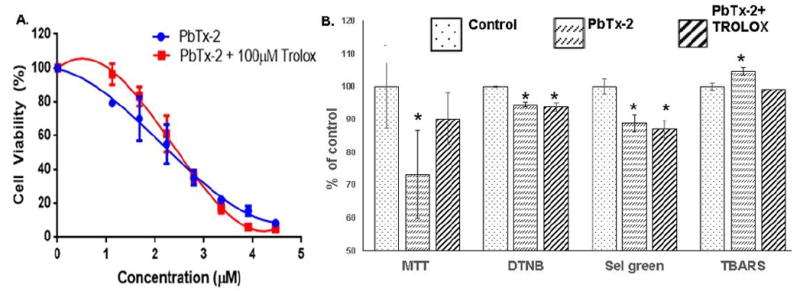
A. 24 hr dose response curve for human lymphoblast cells (GMO2125) in the presence of PbTx-2. EC50 for PbTx-2 is 2.4 μM and 2.4 μM in the presence of 100 μM trolox. B. Cell viability (MTT), cellular glutathione (DTNB), selenol content (Sel green) and lipid peroxidation (TBARS) for PbTx-2 (1 μg/mL) treated and PbTx-2 (1 μg/mL) treatment simultaneous with trolox (100 μg/mL). *Indicates a statistically significant difference from the control (p ≤ 0.05).
DISCUSSION
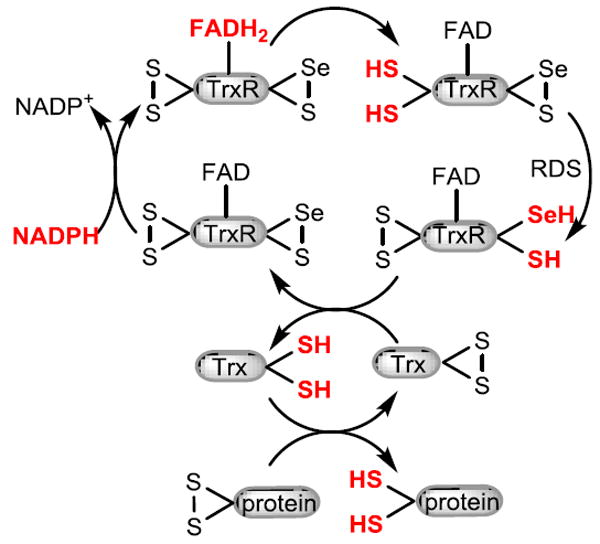
Our studies demonstrate that PbTx-2 but not PbTx-3 is an inhibitor of the mammalian TrxR/Trx system with an IC50 of 25 μM. The reactivity of PbTx-2 towards Michael donors, coupled with the observation that PbTx-3 failed to inhibit the TrxR/Trx system suggests that PbTx-2 alkylates one or more of the redox residues of TrxR or Trx resulting in the inhibition of insulin reduction. It has been previously demonstrated that small molecules such as DTNB can be reduced by the N-terminal reaction center of TrxR when the C-terminal center is either alkylated with electrophiles or absent as in the case of truncated TrxR (Figure 7).32-34 The observation that DTNB reduction by TrxR is not only retained but actually enhanced in the presence of PbTx-2 was unexpected. However, a review of the relevant literature reveals that this behavior was previously predicted. Lothrop and co-workers hypothesized that modification of the Sec residue in TrxR could, under some circumstances, result in a conformational change that would allow access of small molecule substrates to the N-terminal redox center.32 This enhancement of activity can be blocked by pre-incubation of TrxR with the TrxR inhibitor curcumin at pH 7.0. Under these conditions, curcumin alkylates the C-terminal redox center at the Sec residue. Pre-incubation of TrxR with PbTx-2 followed by curcumin also results in enhancement of activity or, in other words, prevents curcumin inhibition. These observations suggest that modification by PbTx-2 occurs at the C-terminal redox center. Indeed, a 2.3-fold increase in kcat and 1.3 fold increase in kcat/Km a for truncated TrxR (missing eight C terminal amino acids) when compared to the wild type has been observed.35 This conclusion is supported by the observation that PbTx-2 has no effect on the relative rates of reduction of DTNB by the truncated TrxR, which lacks the C-terminal redox center, when compared to the control reaction without PbTx-2. The observation that the simultaneous addition of PbTx-2 and DTNB to prereduced TrxR showed the same rate enhancement as pre-incubation of reduced TrxR with PbTx-2 indicates that the alkylation of the C-terminal redox residue(s) of TrxR by PbTx-2 occurs rapidly relative to the reduction of DTNB. Crystal structures of TrxR with and without Trx have shown that the C-terminal (Cys497/Sec498) residues of TrxR are located at the end of a mobile arm. Prior to docking with Trx, the mobile arm is in what is known as the “reduced waiting position” and the C-terminal residues are buried in the interior of the enzyme. Upon docking with Trx, the mobile arm swings out of this position to the surface of the enzyme.36 It is possible that alkylation with the large brevetoxin molecule also causes the mobile are to move away from the N-terminal disulfide making it more accessible by DTNB, bypassing the slow reduction of the C-terminal selenosulfide resulting in a rate enhancement. It seems most likely that modification of TrxR takes place at the Sec residue as selenols are better nucleophiles than thiols: selenols have a lower pKa than thiols and would be deprotonated to the selenide under the experimental conditions. We have demonstrated the formation of an adduct between PbTx-2 and selenocysteine. While our MS experiments using TrxR and PbTx-2 did not provide direct evidence for Sec alkylation, the conspicuous absence of the IAC modified C-terminal peptide, SGGDILQSGC497U498G, which was identified in the control sample, suggests modification at this site. Alkylation of this peptide by PbTx-2 would prevent IAC alkylation, and suggests that the PbTx-2 modified peptide may not ionize efficiently. Interestingly, the peptide fragment (Gln463-Lys486) which is immediately N-terminal to the Sec498 containing peptide fragment, as identified in this sample, indicating that the trypsin digestion was not inhibited by alkylation with PbTx-2. Because the MS analysis of PbTx-2 treated TrxR failed to provide direct evidence of alkylation of Sec, we prepared the selenol selective Sel-green probe to monitor for the presence of the free reduced selenol of TrxR in the presence and absence of PbTx-2. As anticipated, in the absence of PbTx-2 the release of an equivalent of fluorescent coumarin reporter was observed upon the addition of Sel-green to reduced TrxR. However, in the presence of PbTx-2 the observed fluorescence was comparable to the control sample indicating that Sec had been alkylated with PbTx-2.
Figure 7.
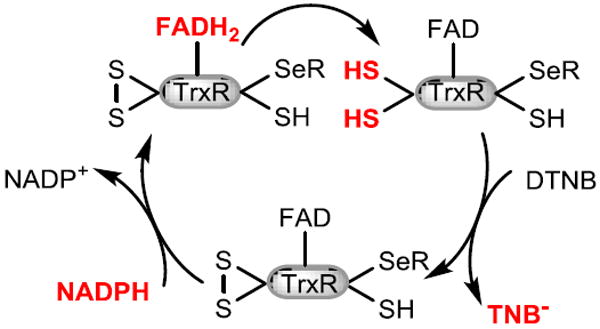
Electron flow in alkylated TrxR.
Trx is the parent of a family of oxidoreductases which function through thiol-disulfide exchange including Grx. While Trx and Grx show low sequence homology, they exhibit similar threedimensional topology including the so-called thioredoxin fold, consisting of an N-terminal βαβ motif and a C-terminal ββα motif with the central β-sheets surrounded by the α-helices and an active site CXXC motif which protrudes from the hydrophobic surface of the protein.37, 38 On the other hand, both GR and TrxR belong to the pyridine nucleotide-disulfide oxidoreductase family of dimeric flavoenzymes.39 Each monomer contains an FAD prosthetic group, an NADPH binding domain, and an active site containing a redox-active disulfide bond. The N-terminal domains of TrxR resemble those of GR, including the N-terminal redox-active CVNVGC motif. However, GR lacks the C-terminal redox center (GCUG) of TrxR.40 Finally, GPx catalyzes the reduction of hydroperoxides with glutathione. Most eukaryotic GPx have an active site Sec residue in which the catalytic cycle includes the formation of a mixed selenosulfide with glutathione. None of these oxidoreductases (Gx, GR or GPx) were inhibited by PbTx-2 demonstrating the selectivity of PbTx-2 for TrxR.
The principle substrate for TrxR is Trx. There are, however a few alternate substrates including other protein disulfides, and small molecules such as dehydroascorbate, lipoic acid and quinones. TrxR is generally considered to have antioxidant activity via Trx and inhibition of TrxR severely compromises redox homeostasis within a cell. The roles of Trx include regulating the redox status of numerous disulfide containing proteins via thiol disulfide exchange, facilitation of cell growth through the reduction of ribonucleotide reductase and redox regulation via reduction of peroxiredoxin (Prx) which scavenge reactive oxygen and nitrogen species.10 Furthermore, the N-terminal (Cys59/Cys64) redox center of TrxR may have multiple roles that are not yet well understood32 including pro-oxidant activity which occurs when the Cys497/Sec498 redox center is compromised.41 Indeed, SecTRAPS (selenium compromised thioredoxin reductase apoptotic proteins) which are derived both from the chemical modification of the selenocysteine residue or truncation of the C-terminal selenocysteine, are believed to promote both apoptosis and necrosis via oxidative stress and increased intracellular reactive oxygen species (ROS) production.42 This has been proposed to be the mechanism of action for anticancer drugs which target TrxR, such as auranofin,43 cisplatin44 and arsenic trioxide.45 Both curcumin and juglone modified TrxR have demonstrated strongly induced NADPH oxidase activity and produced ROS in the presence of oxygen via the N-terminal (Cys59/Cys64) redox center.13, 17, 41 Anestal determined that SecTRAP induced cell death could be reduced to background levels in human A549 cells (human lung carcinoma) by pre-treatment with 100 μM ascorbic acid (vitamin C) or α-tocopherol (vitamin E).46 Treatment of human lymphoblast (GMO2125) cells with PbTx-2 (1μg/mL) for 24 h reduced cell viability to 73% of the untreated cells (Figure 6B). We demonstrated that cell viability could be increased to control levels when cells were treated simultaneously with PbTx-2 and trolox, a water soluble analog of vitamin E. The inhibition of TrxR will lead to an oxidized intracellular environment. Cellular glutathione and free selenol content were reduced by PbTx-2 treatment whereas lipid peroxidation increased. These observations are consistent with PbTx-2 induced oxidative stress. While the simultaneous treatment with PbTx-2 and trolox had no effect on the reduced glutathione content or free selenol present in the cells when compared to the PbTx-2 treated cells, trolox did reduce lipid peroxidation to levels comparable to the control samples.
The occurrence of NSP among humans is rare due to effective monitoring for brevetoxins in shellfish, in regions where the Florida red tide is common. The last documented case involved a family of three who harvested and consumed whelks during an active red tide in 1996.47 While plasma brevetoxin levels were not reported, the highest concentration of brevetoxin reported in urine was 117 ng/mL (0.13 μM) four hours post ingestion. In rescued manatees48 and turtles49, plasma brevetoxin levels have reached 20 nM and 60 nM respectively. Only a few studies have examined the relationship between brevetoxin exposure and oxidative stress, with the majority of those studies focused on marine organisms. A significant correlation was found between plasma brevetoxin levels and oxidative stress markers such as superoxide dismutase (SOD), reactive oxygen and reactive nitrogen species in rescued Florida manatees after exposure to a red tide bloom.48, 50 Rescued loggerhead turtles exhibited an up to two-fold increase in SOD, when compared to healthy captive turtles, which correlated to plasma brevetoxin levels.49 Brevetoxin exposure in coral larvae induced an increase in lipid hydroperoxide content and catalase activity in coral larvae51 and induced lipid peroxidation in fish gills.52 PbTx-2 induced apoptosis in human Jurkat cells.53 Furthermore, the brevetoxins have been shown using the comet assay, to induce DNA damage in rat liver, 54 human lymphocytes55 and Jurkat cells.56 A mechanism for brevetoxin induced oxidative stress has not been identified. We propose that the mechanism is We propose that the mechanism is the alkylation of Sec 422 of TrxR.
Supplementary Material
Highlights.
Brevetoxin (PbTx-2) inhibits thioredoxin reduction by thioredoxin reductase.
Brevetoxin (PbTx-2) activates DTNB reduction by thioredoxin reductase.
Brevetoxin (PbTx-2) reacts with the active site selenocysteine of thioredoxin reductase.
Brevetoxin (PbTx-2) competes with curcumin for the active site of thioredoxin reductase.
Acknowledgments
Funding Sources
Research described in this publication was supported, in part by the National Institute of General Medical Sciences of the National Institutes of Health under award number R25 GM061347 and National Institutes of Health grant R01ES023569 to Y. Liu. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- BSA
bovine serum albumin
- DTNB
Ellman’s reagent: 5,5’-dithio-bis-(2-nitrobenzoic acid)
- ESI
electrospray ionization
- FAD
flavin adenine dinucleotide
- FT-ICR
Fourier transform ion cyclotron resonance
- GPx
glutathione peroxidase
- GR
glutathione reductase
- Grx
glutaredoxin
- HRMS
high resolution mass spectrometry
- IAC
iodoacetamide
- PbTx
brevetoxin
- ROS
reactive oxygen species
- Sec or U
selenocysteine
- SNAr
nucleophilic aromatic substitution
- SOD
superoxide dismutase
- TCEP
tris(2-carboxyethyl)phosphine
- Trx
thioredoxin
- TrxR
thioredoxin reductase
Footnotes
Conflict of Interest Statement
The authors declare there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kirkpatrick B, Fleming LE, Squicciarini D, Backer LC, Clark R, Abraham W, Benson J, Cheng YS, Johnson D, Pierce R, Zaias J, Bossart GD, Baden DG. Literature Review of Florida Red Tide: Implications for Human Health Effects. Harmful algae. 2004;3:99–115. doi: 10.1016/j.hal.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flewelling LJ, Naar JP, Abbott JP, Baden DG, Barros NB, Bossart GD, Bottein M-YD, Hammond DG, Haubold EM, Heil CA, Henry MS, Jacocks HM, Leighfield TA, Pierce RH, Pitchford TD, Rommel SA, Scott PS, Steidinger KA, Truby EW, Van Dolah FM, Landsberg JH. Brevetoxicosis: Red tides and marine mammal mortalities. Nature. 2005;435:755–756. doi: 10.1038/nature435755a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bossart GD, Baden DG, Ewing RY, Roberts B, Wright SD. Brevetoxicosis in manatees (Trichechus manatus latirostris) from the 1996 epizootic: gross, histologic, and immunohistochemical features. Toxicol Pathol. 1998;26:276–282. doi: 10.1177/019262339802600214. [DOI] [PubMed] [Google Scholar]
- 4.Fire SE, Flewelling LJ, Stolen M, Noke Durden W, de Wit M, Spellman AC, Wang Z. Brevetoxin-associated mass mortality event of bottlenose dolphins and manatees along the east coast of Florida, USA. Mar Ecol: Prog Ser. 2015;526:241–251. [Google Scholar]
- 5.Fleming LE, Kirkpatrick B, Backer LC, Bean JA, Wanner A, Dalpra D, Tamer R, Zaias J, Cheng YS, Pierce R, Naar J, Abraham W, Clark R, Zhou Y, Henry MS, Johnson D, Van De Bogart G, Bossart GD, Harrington M, Baden DG. Initial evaluation of the effects of aerosolized Florida red tide toxins (brevetoxins) in persons with asthma. Environ Health Perspect. 2005;113:650–657. doi: 10.1289/ehp.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.James KJ, Carey B, O’Halloran J, van Pelt FNAM, Škrabakova Z. Shellfish toxicity: human health implications of marine algal toxins. Epidemiology & Infection. 2010;138:927–940. doi: 10.1017/S0950268810000853. [DOI] [PubMed] [Google Scholar]
- 7.Hoagland P, Jin D, Beet A, Kirkpatrick B, Reich A, Ullmann S, Fleming LE, Kirkpatrick G. The human health effects of Florida red tide (FRT) blooms: an expanded analysis. Environ Int. 2014;68:144–153. doi: 10.1016/j.envint.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Jeglitsch G, Rein K, Baden DG, Adams DJ. Brevetoxin-3 (PbTx-3) and its derivatives modulate single tetrodotoxin-sensitive sodium channels in rat sensory neurons. J Pharmacol Exp Ther. 1998;284:516–525. [PubMed] [Google Scholar]
- 9.Cassell RT, Chen W, Thomas S, Liu L, Rein KS. Brevetoxin, the dinoflagellate neurotoxin, localizes to thylakoid membranes and interacts with the lightharvesting complex II (LHCII) of photosystem II. ChemBioChem. 2015;16:1060–1067. doi: 10.1002/cbic.201402669. [DOI] [PubMed] [Google Scholar]
- 10.Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: from Cofactors to Antioxidants to Redox Signaling. Antioxid Redox Signaling. 2013;19:1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lobanov AV, Hatfield DL, Gladyshev VN. Eukaryotic selenoproteins and selenoproteomes. Biochim Biophys Acta. 2009;1790:1424–1428. doi: 10.1016/j.bbagen.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snider GW, Dustin CM, Ruggles EL, Hondal RJ. A Mechanistic investigation of the C-terminal redox motif of thioredoxin reductase from plasmodium falciparum. Biochemistry. 2014;53:601–609. doi: 10.1021/bi400931k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cenas N, Nivinskas H, Anusevicius Z, Sarlauskas J, Lederer F, Arnér ESJ. Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein. J Biol Chem. 2004;279:2583–2592. doi: 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- 14.Bradshaw TD, Matthews CS, Cookson J, Chew EH, Shah M, Bailey K, Monks A, Harris E, Westwell AD, Wells G, Laughton CA, Stevens MFG. Elucidation of thioredoxin as a molecular target for antitumor quinols. Cancer Res. 2005;65:3911–3919. doi: 10.1158/0008-5472.CAN-04-4141. [DOI] [PubMed] [Google Scholar]
- 15.Powis G, Wipf P, Lynch SM, Birmingham A, Kirkpatrick DL. Molecular pharmacology and antitumor activity of palmarumycin based inhibitors of thioredoxin reductase. Mol Cancer Ther. 2006;5:630–636. doi: 10.1158/1535-7163.MCT-05-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chew E-H, Lu J, Bradshaw TD, Holmgren A. Thioredoxin reductase inhibition by antitumor quinols: a quinol pharmacophore effect correlating to antiproliferative activity. The FASEB Journal. 2008;22:2072–2083. doi: 10.1096/fj.07-101477. [DOI] [PubMed] [Google Scholar]
- 17.Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 18.Fang J, Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-Hydroxy-2-nonenal in vitro and in vivo. J Am Chem Soc. 2006;128:1879–1885. doi: 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]
- 19.Gan FF, Kaminska KK, Yang H, Liew CY, Leow PC, So CL, Tu LNL, Roy A, Yap CW, Kang TS, Chui WK, Chew EH. Identification of michael acceptor-centric pharmacophores with substituents that yield strong thioredoxin reductase inhibitory character correlated to antiproliferative activity. Antioxid Redox Signaling. 2013;19:1149–1165. doi: 10.1089/ars.2012.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radwan FFY, Wang Z, Ramsdell JS. Identification of a rapid detoxification mechanism for brevetoxin in rats. Toxicol Sci. 2005;85:839–846. doi: 10.1093/toxsci/kfi138. [DOI] [PubMed] [Google Scholar]
- 21.Walsh CJ, Leggett SR, Henry MS, Blum PC, Osborn S, Pierce RH. Cellular metabolism of brevetoxin (PbTx-2) by a monocyte cell line (U-937) Toxicon. 2009;53:135–145. doi: 10.1016/j.toxicon.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 22.Baden DG, Mende TJ, Lichter W, Wellham L. Crystallization and toxicology of T34: A major toxin from Florida’s red tide organism (Ptychodiscus brevis) Toxicon. 1981;19:455–462. doi: 10.1016/0041-0101(81)90003-9. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B, Ge C, Yao J, Liu Y, Xie H, Fang J. Selective selenol fluorescent probes: design, synthesis, structural determinants, and biological applications. J Am Chem Soc. 2015;137:757–769. doi: 10.1021/ja5099676. [DOI] [PubMed] [Google Scholar]
- 24.Zhong L, Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J Biol Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 25.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 26.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Boligon A, Machado M, Athayde M. Technical evaluation of antioxidant activity. Med chem. 2014;4:517–522. [Google Scholar]
- 29.Montano SJ, Lu J, Gustafsson TN, Holmgren A. Activity assays of mammalian thioredoxin and thioredoxin reductase: Fluorescent disulfide substrates, mechanisms, and use with tissue samples. Anal Biochem. 2014;449:139–146. doi: 10.1016/j.ab.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 30.Chen M, Cook KD. Oxidation artifacts in the electrospray mass spectrometry of Aβ peptide. Anal Chem. 2007;79:2031–2036. doi: 10.1021/ac061743r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem, Int Ed. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 32.Lothrop AP, Ruggles EL, Hondal RJ. No Selenium Required: Reactions Catalyzed by Mammalian Thioredoxin Reductase That Are Independent of a Selenocysteine Residue. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckenroth BE, Lacey BM, Lothrop AP, Harris KM, Hondal RJ. Investigation of the c-terminal redox center of high M(r) thioredoxin reductase by protein engineering and semisynthesis. Biochemistry. 2007;46:9472–9483. doi: 10.1021/bi7004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Q, Sandalova T, Lindqvist Y, Arnér ESJ. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J Biol Chem. 2009;284:3998–4008. doi: 10.1074/jbc.M807068200. [DOI] [PubMed] [Google Scholar]
- 35.Lacey BM, Eckenroth BE, Flemer S, Hondal RJ. Selenium in thioredoxin reductase: a mechanistic perspective. Biochemistry. 2008;47:12810–12821. doi: 10.1021/bi800951f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fritz-Wolf K, Kehr S, Stumpf M, Rahlfs S, Becker K. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat Commun. 2011;2:383. doi: 10.1038/ncomms1382. [DOI] [PubMed] [Google Scholar]
- 37.Martin JL. Thioredoxin —a fold for all reasons. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 38.Netto LES, de Oliveira MA, Tairum CA, da Silva Neto JF. Conferring specificity in redox pathways by enzymatic thiol/disulfide exchange reactions. Free Radical Res. 2016;50:206–245. doi: 10.3109/10715762.2015.1120864. [DOI] [PubMed] [Google Scholar]
- 39.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH. The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc Natl Acad Sci U S A. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radical Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 41.Cheng Q, Antholine WE, Myers JM, Kalyanaraman B, Arnér ESJ, Myers CR. The selenium- independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. J Biol Chem. 2010;285:21708–21723. doi: 10.1074/jbc.M110.117259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anestål K, Prast-Nielsen S, Cenas N, Arnér ESJ. Cell Death by SecTRAPs: Thioredoxin Reductase as a Prooxidant Killer of Cells. PLoS ONE. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C, Liu Z, Li M, Li X, Wong Y-S, Ngai S-M, Zheng W, Zhang Y, Chen T. Enhancement of auranofin- induced apoptosis in MCF-7 human breast cells by selenocystine, a synergistic inhibitor of thioredoxin reductase. PLoS ONE. 2013;8:e53945. doi: 10.1371/journal.pone.0053945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prast-Nielsen S, Cebula M, Pader I, Arnér ESJ. Noble metal targeting of thioredoxin reductase — covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radical Biol Med. 2010;49:1765–1778. doi: 10.1016/j.freeradbiomed.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Lu J, Chew E-H, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anestål K, Prast-Nielsen S, Cenas N, Arnér ES. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PloS one. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poli MA, Musser SM, Dickey RW, Eilers PP, Hall S. Neurotoxic shellfish poisoning and brevetoxin metabolites: a case study from Florida1. Toxicon. 2000;38:981–993. doi: 10.1016/s0041-0101(99)00191-9. [DOI] [PubMed] [Google Scholar]
- 48.Walsh CJ, Butawan M, Yordy J, Ball R, Flewelling L, de Wit M, Bonde RK. Sublethal red tide toxin exposure in free-ranging manatees (Trichechus manatus) affects the immune system through reduced lymphocyte proliferation responses, inflammation, and oxidative stress. Aquat Toxicol. 2015;161:73–84. doi: 10.1016/j.aquatox.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 49.Walsh CJ, Leggett SR, Carter BJ, Colle C. Effects of brevetoxin exposure on the immune system of loggerhead sea turtles. Aquat Toxicol. 2010;97:293–303. doi: 10.1016/j.aquatox.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 50.Walsh CJ, Stuckey JE, Cox H, Smith B, Funke C, Stott J, Colle C, Gaspard J, Manire CA. Production of nitric oxide by peripheral blood mononuclear cells from the Florida manatee, Trichechus manatus latirostris. Vet Immunol Immunopathol. 2007;118:199–209. doi: 10.1016/j.vetimm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 51.Ross C, Ritson-Williams R, Pierce R, Bullington JB, Henry M, Paul VJ. Effects of the Florida red tide dinoflagellate, Karenia brevis, on oxidative stress and metamorphosis of larvae of the coral Porites astreoides. Harmful Algae. 2010;9:173–179. [Google Scholar]
- 52.Woo SPS, Liu W, Au DWT, Anderson DM, Wu RSS. Antioxidant responses and lipid peroxidation in gills and erythrocytes of fish (Rhabdosarga sarba) upon exposure to Chattonella marina and hydrogen peroxide: Implications on the cause of fish kills. J Exp Mar Biol Ecol. 2006;336:230–241. [Google Scholar]
- 53.Walsh C, Leggett S, Strohbehn K, Pierce R, Sleasman J. Effects of in vitro Brevetoxin Exposure on Apoptosis and Cellular Metabolism in a Leukemic T Cell Line (Jurkat) Mar Drugs. 2008;6:291. doi: 10.3390/md20080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leighfield TA, Muha N, Ramsdell JS. Brevetoxin B is a clastogen in rats, but lacks mutagenic potential in the SP-98/100 Ames test. Toxicon. 2009;54:851–856. doi: 10.1016/j.toxicon.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 55.Sayer A, Hu Q, Bourdelais A, Baden D, Gibson J. The effect of brevenal on brevetoxin-induced DNA damage in human lymphocytes. Archives of Toxicology. 2005;79:683–688. doi: 10.1007/s00204-005-0676-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murrell R, Gibson J. Brevetoxins 2, 3, 6, and 9 show variability in potency and cause significant induction of DNA damage and apoptosis in Jurkat E6-1 cells. Archives of Toxicology. 2009;83:1009–1019. doi: 10.1007/s00204-009-0443-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.