

Abstract
ERK-dependent signaling is key to many pathways through which extracellular signals are transduced into cell-fate decisions. One conundrum is the way in which disparate signals induce specific responses through a common, ERK-dependent kinase cascade. While studies have revealed intricate ways of controlling ERK signaling through spatiotemporal localization and phosphorylation dynamics, additional modes of ERK regulation undoubtedly remain to be discovered. We hypothesized that fine-tuning of ERK signaling could occur by cysteine oxidation. We report that ERK is actively and directly oxidized by signal-generated H2O2 during proliferative signaling, and that ERK oxidation occurs downstream of a variety of receptor classes tested in four cell lines. Furthermore, within the tested cell lines and proliferative signals, we observed that both activation loop-phosphorylated and non-phosphorylated ERK undergo sulfenylation in cells and that dynamics of ERK sulfenylation is dependent on the cell growth conditions prior to stimulation. We also tested the effect of endogenous ERK oxidation on kinase activity and report that phosphotransfer reactions are reversibly inhibited by oxidation by as much as 80 to 90%, underscoring the importance of considering this additional modification when assessing ERK activation in response to extracellular signals.
Keywords: Kinase, redox regulation, sulfenic acid, growth factor signaling, reactive cysteine, ERK, MAPK
Graphical Abstract
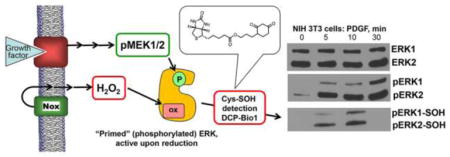
Reactive oxygen species (ROS), while previously considered detrimental byproducts of respiration [1], are now recognized as significant contributors to cellular signaling [2–4]. An important milestone in support of ROS as regulators of cell signaling was the discovery in the mid-1990s by Sundaresan et al. that H2O2 generation was required to elicit appropriate proliferative responses to Platelet-Derived Growth Factor (PDGF) in primary rat vascular smooth muscle cells [5]. Roles for H2O2 as a signaling mediator have now been demonstrated in many cell types and in vivo models [6–8], including our recent work establishing the important role of H2O2 in the proliferative responses to lysophosphatidic acid (LPA), a bioactive lipid synthesized by ovarian and prostate cancer cells and cell lines [9, 10]. ROS have also been implicated in diverse signaling processes such as migration by endothelial cells [11], stem cell pluripotency and differentiation [12], and chondrocyte response to fibronectin fragments [13].
Much work has been done by us and others in the field of redox signaling to identify targets of signal-generated ROS [14–17]. Various ROS, reactive nitrogen species (RNS), and reactive sulfur species (RSS) react with cysteine residues of proteins, modifying the structure, dynamics, and/or function of those proteins. The oxidation of a cysteine residue is often reversible [15, 18], which sets up the oxidation of a target cysteine on a protein to be a molecular switch. This switch-like property of cysteines has enabled them to serve specific functions as redox sensors on proteins [19–22]. For example, a well-established example of a binary cysteine switch is in the protein tyrosine phosphatase (PTP) family of enzymes [23]. In this case, the reactive, low pKa cysteine at the active site can become oxidized by ROS, inactivating the phosphatase, thereby favoring a sustained phosphorylation response to stimuli [3, 4, 24].
Identifying novel proteins whose cysteines are oxidized in response to extracellular signals has been historically difficult due to the generally labile nature of oxidized cysteine products. One class of cysteine probes developed by our research team is based on dimedone, which alkylates sulfenic acid (R-SOH) [15, 25], the intermediate, two-electron oxidized form of cysteine generated upon reaction with ROS molecules such as H2O2 (Fig. 1A). Using these dimedone-based probes to capture proteins undergoing active oxidation in cells responding to external signals can reveal the identity of the proteins oxidized in situ, and in favorable cases the specific cysteine residue undergoing oxidation [16, 26].
Figure 1.

Trapping of sulfenic acids within cellular proteins during lysis. A) Labeling of oxidized proteins with DCP-Bio1. The reactive carbon of DCP-Bio1 selectively reacts with sulfenic acids to form a covalent adduct, thereby biotinylating proteins containing sulfenic acids at the time of lysis. B) Overall experimental approach for labeling of proteins undergoing active oxidation to sulfenic acid. Cells are cultured to ~80% confluency, treated with an exogenous signal of interest such as platelet-derived growth factor (PDGF), and harvested in lysis buffer containing DCP-Bio1 to trap sulfenic acids, N-ethylmaleimide (NEM) and iodoacetamide (IAM) to alkylate free, reduced thiols (to minimize thiol oxidation during lysis), and catalase to remove any H2O2 formed during lysis. After incubation for 30 min on ice, lysates are clarified by centrifugation, excess DCP-Bio1 is removed with a molecular sieve (BioGel P6) spin column, and eluates are subjected to avidin-based affinity capture. Beads are then stringently washed to remove any unlabeled proteins carried over during capture. Captured proteins are eluted by incubation in 2% sodium dodecyl sulfate (SDS) at 100°C for 10 min, followed by electrophoresis and immunoblotting for the protein of interest. To control for differences in capture efficiency and gel loading, a pre-biotinylated bacterial protein, AhpC, is added to lysates at a ratio of 500 μg lysate to 0.5 μg AhpC before affinity capture. Photo courtesy of ATCC.
One of the probes, DCP-Bio1 (Fig. 1A), contains the reactive core of dimedone linked to biotin, allowing for affinity capture of labeled cellular proteins undergoing active oxidation in cells (Fig. 1B) [27, 28]. This probe has been used to identify multiple novel targets of oxidation, such as Akt2 [9, 14, 29], responding to extracellular signals [11, 28, 30, 31]. Based on preliminary data from experiments suggesting that oxidation of mitogen-activated protein kinases ERK1 and ERK2 (designated ERK1/2) occurs during proliferation (unpublished) and migration [11], we undertook a project to further investigate the redox regulation of these proteins during cell signaling. ERK proteins are at the heart of a variety of signaling pathways involved in proliferation, differentiation, survival, migration [32, 33], and at times even apoptosis [34, 35]. This presents a conundrum of specificity – how do disparate signals induce specific responses while sharing the ERK signaling cascade? Evaluating ERK1/2 regulation is vital to understanding diseases from cancer to diabetes to neurological disorders. In fact, various components of the ERK1/2 signaling cascade (particularly Ras and Raf) are the most frequently found oncogenic mutations in human cancers, and an overall hyperactivation of the ERK1/2 pathway is reported in up to 90% of all human cancers [36].
Extensive research to date has revealed much about the ERK1/2 pathway and its regulation. Upon binding of an appropriate ligand to its receptor, a cascade of signaling protein activation occurs: the GTPase Ras activates the Raf kinases, which in turn activate the MAP kinase kinases MEK1 and MEK2 (referred to as MEK1/2). MEK1/2 proteins phosphorylate ERK1/2 at T183 and Y185 [32, 37], hereafter designated as the TEY motif. This induces a conformational change that opens the active site of ERK1/2 [37, 38] and enhances structural flexibility [39, 40], allowing ERK1/2 to phosphorylate substrates. However, with over 300 known substrates in various subcellular compartments [36, 41], multiple layers of regulation are involved, including temporal and spatial control of TEY phosphorylation and dephosphorylation [42, 43]. While we know that dual phosphorylation is a prerequisite for ERK1/2 activation, it is not yet fully clear how cells orchestrate ERK1/2 localization, enable discrimination between substrates, or regulate the dephosphorylation of ERK1/2 by protein phosphatases [44].
Our knowledge of cysteine switches, including the tentative identification of ERK1/2 oxidation resulting from vascular endothelial growth factor (VEGF) signaling [11], led us to hypothesize that ERK1/2 cysteine oxidation could be an additional mode to control cell fates through ERK1/2 regulation. Human ERK1 and ERK2 contain 6 and 7 cysteines, respectively, half of which are solvent exposed and conserved in the MAPK family [37, 45]. Analysis of the literature gives ample evidence that several of these cysteines could be redox sensitive. Under oxidative stress conditions, as mimicked by the bolus addition of H2O2 or nitric oxide donors, others have reported evidence for the modification of ERK1/2 cysteines [11, 26, 46–49]. ERK1/2 cysteine modification can also occur downstream of the physiological signals of vascular endothelial growth factor (VEGF) [11] and tumor necrosis factor α (TNF-α) [49], but it has not definitively been shown that ERK1/2 oxidation commonly occurs as a result of extracellular signals. Moreover, the linkage of signal-mediated oxidation events on ERK1/2 to their functional outputs has not been established. Here, we used the dimedone-based probe DCP-Bio1 in conjunction with immunoprecipitation and assays to test the redox sensitivity of recovered ERK1/2 activity, to assess function-altering ERK1/2 oxidation induced by extracellular stimuli. We report that ERK1/2 cysteine sulfenylation occurs in response to a variety of proliferative signals and that endogenous oxidation strongly modulates ERK1/2 kinase activity in vitro. To our knowledge, this is the first report to document the modification of ERK1/2 cysteines in response non-stress extracellular signals other than VEGF, and to relate the observed sulfenic acid formation to endogenous ERK1/2 kinase activity.
Materials and Methods
Reagents and antibodies
Primary antibodies for Western blots to total ERK [recognizing both p44 (ERK1) and p42 (ERK2), RRID AB_390780], TEY-phosphorylated ERK1/2 (RRID: AB_331646), phospho-Elk1 (RRID: AB_2277933), along with anti-rabbit (RRID: AB_2099233) and anti-mouse (RRID: AB_330924) HRP-conjugated secondary antibodies, and Platelet Derived Growth Factor BB (abbreviated PDGF herein) were from Cell Signaling Technology. AhpC was expressed and purified as described previously [50, 51], and anti-AhpC antibody was purified from rabbit serum [28]. DCP-Bio1 was synthesized as described previously [52]. PEG-Catalase was from Sigma (catalog C4963). DMEM, RPMI, and Fetal Bovine Serum were from Lonza; EMEM was from Gibco. Chemiluminescence reagents for Western blotting (SuperSignal Dura) were from Thermo Scientific. Nitrocellulose was from GE Healthcare - Amersham. Magnetic A beads (Dynabeads) for phospho-ERK1/2 immunoprecipitations were from Novex by Life Technologies, and ATP for in vitro kinase assays was acquired from Promega. Iodoacetamide (IAM) was from Sigma-Aldrich; 1,4-dithiothreitol (DTT) and N-ethylmaleimide (NEM) were from Fisher-Scientific.
Elk1 for ERK1/2 in vitro activity assays was cloned, expressed, and purified as described below. GST-Elk1-Hisx6 was expressed from a pGex-6P-1-derived bacterial expression plasmid. First, the Elk1 DNA sequence corresponding to residues 307–428 of human Elk1 was synthesized by GenScript, with the codon usage modified for expression in E. coli. The gene was subcloned into pGex-6p-1 between BamH1 and XhoI restriction sites. Due to issues with truncated versions of the protein being expressed in E. coli, a 6-His tag was added to the C-terminal end of Elk1 using PCR to reengineer the DNA insert, with a forward primer (5′-CTGGGATCCATTTCGCAACCGC-3′) and a reverse primer encoding the extended His tag at the C-terminus (5′-GCGCTCGAGTTAATGGTGATGGTGATGGTGCGGTTTTTGCGGACCCGGCGAC-3′). After PCR product purification, the DNA fragment was ligated into pGex-6p-1. Successful cloning was verified by a restriction digest followed by gel analysis and was verified in its entirety by sequencing (Eurofins Genomics).
GST-Elk1-Hisx6 was expressed in BL21 Gold cells with growth in TYP media supplemented with 30 mM glucose and 100 μg/mL ampicillin at 37 °C to an optical density of 1 at 600 nm. Expression was then induced by addition of 1 mM IPTG and incubation continued at 24 °C for 4 h, after which cells were harvested by centrifugation and stored at −80 °C until used for purification. Cells were thawed and resuspended in 25 mM potassium phosphate at pH 7.0, with 150 mM NaCl, 10% glycerol, and cOmplete protease inhibitor cocktail (EDTA-free, Roche) and disrupted using an Avestin C5 emulsifier. After centrifugation, the supernatant was applied to a cobalt-NTA column (GE Healthcare) pre-equilibrated in 50 mM sodium phosphate at pH 8.0, with 0.3 mM NaCl and 20 mM imidazole, washed with 4 column volumes of the same buffer, then eluted by increasing the imidazole to 500 mM. Elk1-containing fractions were pooled, bound to Glutathione Sepharose (GE Healthcare) pre-equilibrated and subsequently washed with 50 mM Tris-HCl at pH 8.0 (4 °C), including 150 mM NaCl, 1 mM EDTA and 10% glycerol. Bound protein was then eluted using 50 mM Tris-HCl at pH 8.0 containing 10 mM reduced glutathione and 10% glycerol. GST-Elk1-Hisx6 was further purified on a 15 mL Source Q column using a gradient of 50 mM to 1 M NaCl in 50 mM Tris-HCl pH 7.5, 10% glycerol. Elk1-containing fractions were then pooled, concentrated, exchanged by ultrafiltration into 20 mM Tris-HCl at pH 8.0 with 1 mM EDTA and 10% glycerol, aliquoted, and stored at −80 °C until use.
Cell culture and treatments
Cell lines used (giving Resource Identification Initiative accession numbers), NIH 3T3 (RRID:CVCL_0594), WI-38 (RRID:CVCL_0579), SK-OV-3 (RRID:CVCL_0532), and PC-3 (RRID:CVCL_0035), were obtained from ATCC-derived stocks from the Cell and Viral Vector Laboratory Shared Resource at Wake Forest School of Medicine and cultured at 37 °C in a 5% CO2 incubator in either DMEM (NIH 3T3), EMEM (WI-38) or RPMI (SK-OV-3 and PC-3) supplemented with 10% fetal bovine serum, L-glutamine, penicillin, and streptomycin. Except where indicated, cells were transferred into fresh, serum-free media for 18 h before treatment with PDGF. PDGF-BB was added to the cells to a final concentration of 20 ng/mL and then cells were harvested at indicated time points. Where indicated, PEG-Catalase was added at 400 U/mL 18 h before PDGF stimulation.
DCP-Bio1 labeling and affinity capture
Cells grown to ~80% confluency were harvested and replated (~5 x 105 per 100 mm plate) in FBS-supplemented media for 24 h, then replenished with either new FBS-supplemented media or media lacking FBS for 18 h before treatment with PDGF. Samples were harvested at indicated time points by addition of lysis buffer supplemented with DCP-Bio1 essentially as described previously [27]. Briefly, lysis buffer (50 mM Tris-HCl at pH 8.0, with 100 mM NaCl, 100 μM diethylene triamine pentaacetic acid (DTPA), 20 mM β-glycerophosphate, 0.1% SDS, 0.5% Na Desoxycholate, 0.5% NP-40, and 0.5% Triton-X-100) was freshly prepared with 1 mM PMSF, 10 μg/mL aprotinin, 1 mM Na3VO4, 10 mM NaF, 1 mM DCP-Bio1, 10 mM NEM, 10 mM IAM, and 200 U/mL Catalase (Fig. 1). Seventy-five μL of lysis buffer was added to each dish and cells were scraped from the plates, transferred to micro-centrifuge tubes, incubated on ice for 30 min, and then stored at −80 °C. Before affinity capture and elution of labeled proteins [28], samples were thawed and clarified by centrifugation, then excess DCP-Bio1 was removed via a BioGel P6 spin column. Eluants were assessed for protein concentration using a BCA assay (Pierce), then 500 μg of protein for each sample was diluted into 2 M urea and supplemented with 0.5 μg prebiotinylated AhpC to control for the efficiency of the affinity capture, elution, and gel loading steps. After samples were precleared with Sepharose CL-4B beads (Sigma), they were applied to plugged spin columns containing high capacity streptavidin-agarose beads (Thermo Scientific) and incubated overnight at 4 °C with constant rotation. Samples then underwent a series of stringent washes to remove any contaminating, non-labelled proteins that were co-captured with DCP-Bio1 conjugated proteins. Washes consisted of 2 washes each with 4 column volumes of (in order) 1% SDS, 4 M Urea, 1 M NaCl, 10 mM DTT, 50 mM ammonium bicarbonate and water. Labeled proteins were eluted with 25 mM Tris-HCl at pH 6.8, containing 2% SDS, 50 mM DTT, 0.02% bromophenol blue, and 5% glycerol, incubated at 100 °C for 10 min, then centrifuged to separate protein from beads. Samples were then stored at −80 °C as needed and analyzed by Western blot as described below.
Western blotting
For immunoblots of proteins labeled with DCP-Bio1 (after affinity capture), 40–80 μg of protein per sample was separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes for 1 h at 100 V and 4 °C (or overnight at 100 mA). For immunoblots of cell lysates, 10 μg of total protein was typically loaded per lane. For phospho-Elk1 immunoblots after kinase assays (described below), 1.2–2.4 μg of ERK1/2-treated GST-Elk1-Hisx6 was added to each lane. In all cases, after transfer to nitrocellulose, membranes were blocked with 5% Nonfat dry milk, probed with protein-specific antibodies, and visualized using SuperSignal Dura chemiluminescence reagent. Exposed film was then scanned on an Epson Perfect V30, where original image integrity was maintained by adjusting scanner settings to prevent auto-adjustments in contrast and gamma. Band density was measured using ImageJ software. To analyze data from DCP-Bio1 experiments, each sample was first normalized to AhpC band density to control for variations in affinity-capture efficiency, and then each replicate was normalized to the most intense ERK1/2 band, which was set to 1. For quantitation of Elk1 phosphorylation after in vitro kinase assays of immunoprecipitated ERK1/2, each lysate sample was normalized to phospho-Elk1 intensity from kinase reactions including DTT. For DCP-Bio1 labeling experiments, p-values were calculated using Student’s t-tests assuming 2-tailed distributions with two-sample equal variance. For activity assays, p-values were calculated using Student’s t-test for paired samples, 2-tailed distribution.
In vitro kinase assays of ERK1/2 immunoprecipitated from cells grown in culture
NIH 3T3 cells were treated and harvested as described above, but without the addition of DCP-Bio1, IAM, or NEM to the lysis buffer. Lysates were subsequently clarified by centrifugation and protein content of the supernatants was measured via BCA assay. Samples containing 50 μg of protein were added to 4 μL of anti-phospho-p42/44 (i.e., phospho-ERK1/2) rabbit antibody into a total volume 200 μL of lysis buffer supplemented with protease and phosphatase inhibitors and 200 U/mL of catalase. Twenty-five μL of protein A magnetic beads were suspended in the lysate plus antibody mix and incubated overnight at 4 °C with constant rotation. The following morning, beads were washed twice with 5 column volumes of lysis buffer with phosphatase and protease inhibitors, followed by 2 washes with 5 volumes of kinase buffer (25 mM Tris-HCl at pH 7.5, with 5 mM β-glycerophosphate, 0.1 mM Na3VO4, and 10 mM MgCl2). After washes, beads were re-suspended in 100 μL of 40 mM Tris-HCl at pH 7.5, with 0.1 mM Na3VO4; samples were diluted five-fold with either DTT (to give a final concentration of 2 mM DTT) or HPLC water, then incubated with 2.21 μM GST-Elk1-Hisx6 fusion protein substrate, 200 μM ATP, and kinase buffer at 30°C for 30 min with constant shaking. Reactions were quenched by the addition of protein gel loading buffer (25 mM Tris-HCl at pH 6.8, with 2% SDS, 50 mM DTT, 0.2 % bromophenol blue, and 5% glycerol) and incubated at 100 °C for 5 min. Samples were either stored at −80 °C or directly loaded onto a 10% SDS-polyacrylamide gel, transferred to nitrocellulose, and probed for phospho-Elk1 as described above.
Results
To test whether ERK1 and/or ERK2 are direct targets of oxidation in cells responding to proliferative signals, we used the dimedone based probe DCP-Bio1, as described in Fig. 1 and Materials and Methods. Various cell lines were treated with cell-specific growth factors over a time course of 5 to 30 min before harvest. Cells were harvested in the presence of DCP-Bio1, catalase, and cysteine alkylators (NEM and IAM) to minimize artifactual oxidation of cysteines during lysis [27]. After clarification, lysates were supplemented with pre-biotinylated AhpC protein as an internal control for differences in capture efficiency and gel loading, then subjected to biotin-dependent affinity capture as described in Materials and Methods. Captured proteins were resolved by SDS-PAGE and immunoblotted using antibodies that either recognize total ERK1/2 (with ERK1 at ~44 kDa and ERK2 at ~42 kDa) or dual (TEY) phosphorylated ERK1/2 at T183 and Y185 (referred to here as phospho-ERK1/2) [28]. Quantitation of relative changes in oxidized ERK band intensities followed normalization to the AhpC internal control in each case to account for potential differences in capture affinity. Using these approaches, we observed that ERK1/2 proteins were labeled by DCP-Bio1 in response to PDGF in NIH 3T3 mouse fibroblasts (Fig. 2) as well as other treated cell types (see below). Considering the observations that ERK1/2 proteins were not endogenously biotinylated and that biotinylation could be detected in immunoprecipitated phospho-ERK after the above treatments (Supplemental Fig. S1), we conclude that the biotinylation of ERK1/2 when DCP-Bio1 is added to lysis buffer indicates active sulfenic acid formation occurring on phospho-ERK1/2 after PDGF addition. In NIH 3T3 cells, we observed a consistent increase in sulfenic acid formation of ERK1/2 10 min after stimulation with a return to basal levels of sulfenylation 30 min after stimulation (Fig. 2), confirming our hypothesis that ERK1/2 oxidation occurs as a result of physiologically-relevant extracellular signals like growth factors.
Figure 2.

ERK1/2 cysteines are oxidized to sulfenic acid in response to PDGF in NIH 3T3 cells. ERK oxidation was monitored during proliferative signaling as described in Materials and Methods. Briefly, cells were treated with 20 ng/mL PDGF after 18 h in serum-free media and lysed in the presence of the sulfenic acid trap DCP-Bio1, thereby biotinylating proteins undergoing cysteine oxidation. After addition of prebiotinylated AhpC as an internal control (see Figure 1 legend and Methods), DCP-Bio1 labeled proteins were captured via streptavidin-agarose beads, resolved by SDS-PAGE, and subjected to Western blot for total and dual-(TEY) phosphorylated ERK (ppERK). Upper panels of (A) and (B) show representative blots of total (A) and phosphorylated (B) ERK oxidation, as well as corresponding blots of whole cell lysates, detected in NIH 3T3 cells treated with PDGF. Lower panels summarize ERK band intensity from multiple experiments (n=4) of captured proteins following normalization to AhpC and then to the band intensity at the 10 min time point (set to 1) for each replicate. *, p<0.05; ***, p<0.001; ****, p<2x10−19.
To further investigate the source and nature of the ROS responsible for ERK1/2 oxidation, we utilized the cell-permeable ROS modulator PEG-Catalase, which scavenges H2O2. NIH 3T3 cells were treated with PDGF for 10 min with or without PEG-Catalase pre-treatment. We observed that PEG-Catalase significantly dampened the amount of total and phosphorylated ERK1/2 labeled by DCP-Bio1 (Fig. 3). Thus, intracellular H2O2, or a derivative thereof, is implicated as the ROS molecule responsible for observed ERK1/2 oxidation in NIH 3T3 cells.
Figure 3.

H2O2 generated in response to PDGF in NIH 3T3 cells is responsible for observed sulfenylation of ERK1/2. NIH 3T3 cells were grown as in Fig. 2 and treated or not with PDGF for 10 min with or without preincubation with PEG-Catalase as described in Materials and Methods. PEG-Catalase significantly reduced the amount of sulfenic acid formation on total ERK (A) and phosphorylated ERK (B) in response to PDGF, but had no effect on total or phosphorylated ERK content in whole lysates, indicating that sulfenic acid formation on ERK is a result of cysteine reaction with H2O2 (or a derivative of H2O2). Lower panels summarize ERK band intensity from multiple experiments (n=4) normalized as described in Figure 2. Images are representative of four biological replicates. *, p<0.05; **, p< 0.01; ***, p<0.001; ****, p<1x10−8
We also investigated the temporal dynamics of ERK1/2 sulfenic acid formation in another fibroblast cell line, WI-38, in response to PDGF stimulation. WI-38 cells were serum starved for 18 h before PDGF stimulation, as was done with NIH 3T3 cells in Fig. 2. Following PDGF stimulation, we observed a very similar pattern in sulfenic acid formation among phospho-ERK1/2 proteins, with an initial abrupt increase followed by a decline in observed sulfenic acids on ERK1/2 (Fig. 4B). However, when probing DCP-Bio1 labeled proteins with an antibody measuring total-ERK1/2, we observed a modest amount of total-ERK1/2 undergoing active oxidation before PDGF treatment. Upon PDGF stimulation, sulfenylation of the total population of ERK1/2 decreased and then considerably increased by 30 min after treatment (Fig. 4A).
Figure 4.

ERK1/2 cysteines are oxidized to sulfenic acid in response to PDGF in WI-38 fibroblasts. Cells were serum-starved for 18 h, treated with PDGF, and lysed in the presence of DCP-Bio1 as described in Figures 1 and 2. Affinity-captured proteins were immunoblotted for total (A) or phospho-ERK1/2 (B). Lower panels summarize ERK band intensity from multiple experiments (n=4) normalized to AhpC, then to the sample with highest band intensity. *, p<0.05; **, p< 0.01; ****, p<5x10−10
We also tested the hypothesis that oxidation patterns vary with cell growth conditions by repeating the experiments with WI-38 cells under serum-replete conditions (including 10% Fetal Bovine Serum). In contrast to the PDGF-induced oxidation pattern observed in serum-starved cells, serum-supplemented WI-38 cells exhibited very different dynamics of ERK1/2 oxidation (Supplemental Fig. S2). We observed relatively high levels of total-ERK1/2 oxidation occurring before PDGF treatment that significantly decreased 10 min after stimulation and only modestly increased again by 30 min (Supplemental Fig. S2A). Phospho-ERK1/2, however, had a delayed oxidation response when compared to the response observed in serum-starved cells, with a maximal increase in sulfenic acid formation in serum-supplemented cells occurring 30 min after PDGF stimulation. These data confirm that basal growth conditions of cells significantly modulate the temporal dynamics of ERK1/2 oxidation in response to a stimulus.
Using this same sulfenic acid labeling technique, we also investigated ERK1/2 cysteine oxidation in SK-OV-3 ovarian cancer cells (Fig. 5) and PC-3 prostate cancer cells (Supplemental Fig. S3) during responses to LPA, a lipid growth factor produced by prostate cancers and found in the ascites fluid of many ovarian cancers [53, 54]. Our group has previously shown that LPA induces the intracellular formation of H2O2 in these cell lines and that the H2O2 is necessary for the proliferative response of these cell lines to LPA [9, 10]. In serum-starved SK-OV-3 cells, there is no detectable pattern for changes in sulfenic acid formation in the total population of ERK1/2 in response to LPA; instead, it appears that total ERK1/2 is continuously oxidized across the time course (Fig. 5A). Assessing just the population of phospho-ERK1/2, however, relatively high levels of active sulfenylation are observed five min post-stimulus, followed by a drop in relative levels of sulfenylation 10 min post-stimulus (Fig. 5B). There may be an additional increase in phospho-ERK1/2 sulfenylation 30 min after LPA stimulation in SK-OV-3 cells, but the intensities of phospho-ERK1/2 sulfenylation at 30 min are variable between biological replicates and therefore we cannot conclude whether phospho-ERK1/2 undergoes active oxidation 30 min post stimulus.
Figure 5.

ERK1/2 oxidation in ovarian cancer-derived SK-OV-3 cells treated with lysophosphatidic acid (LPA). SK-OV-3 cells depleted of serum for 18 h were treated with 100 nM LPA and harvested in the presence of DCP-Bio1 as described in Figures 1 and 2. The upper panels show representative immunoblots of DCP-Bio1 labeled proteins for total ERK (A) and TEY-phosphorylated ERK1/2 (B). The lower panels depict averaged relative intensity for each sample after LPA treatment, normalized as in Figure 4 (n=6 for total ERK, n=3 for phospho-ERK). *** p<0.001
Within serum-supplemented PC-3 cells, sulfenylation of total ERK1/2 is highly variable (Supplemental Fig. S3A), but it appears to be relatively low at 10 min post-stimulus and peak 30 min post-stimulus. With phospho-ERK1/2, an increase in sulfenylation is observed 10–30 min after LPA stimulation (Supplemental Fig. S3B).
Sulfenic acid formation on proteins can be detected using chemical probes like DCP-Bio1, but the observation of active oxidation does not address whether or not the observed oxidation changes the activity of the protein. Furthermore, the sulfenic acid oxoform trapped chemically by DCP-Bio1 may be stabilized within the protein prior to attack by a reductant, but sulfenic acid can often serve as a transient species subject to subsequent disulfide bond formation either with another cysteine, or with glutathione (another potentially meta-stable oxoform of the protein). To determine the effect and extent of reversible cysteine oxidation on ERK1/2 kinase activity, NIH 3T3 cells treated with PDGF (Fig. 6) and SK-OV-3 cells treated with LPA (Supplemental Fig. S4) were harvested at various time points after addition of the stimulus and phospho-ERK1/2 was immunoprecipitated. Catalase was included in the immunoprecipitation buffer to minimize post-lysis oxidation by H2O2. Each sample of immunoprecipitated phospho-ERK1/2 was divided, with one part incubated with 2 mM DTT to reverse endogenous oxidation, and the other half lacking DTT (to preserve the endogenous oxidation state from cells). Each sample was assayed in vitro by the addition of recombinant Elk1 and ATP for 30 min at 30 °C as described in Materials and Methods. Samples were then immunoblotted for phosphorylation of S383 of Elk1 (Fig. 6 and Supplemental Fig. S4). At each time point tested, ERK1/2 kinase activity towards Elk1 increased by as much as 5- to 10-fold after reduction by DTT. While we cannot definitively rule out air or oxidant-mediated, post-lysis oxidation of ERK1/2 occurring in samples during the immunoprecipitation and assay procedures, catalase has in the past demonstrably protected against post-lysis oxidation occurring during lysis and DCP-Bio1 labeling of oxidized proteins [27]. Furthermore, it is notable that the percent inhibition is different between SK-OV-3 and NIH-3T3 cells, which indicates that the extent of inhibition is cell specific and not likely due to post-lysis oxidation. Taken together, we interpret our results as revealing that ERK1/2 kinase activity, at least towards the substrate Elk1, is 80–90% inhibited by endogenous ERK1/2 oxidation in NIH 3T3 cells (Fig. 6) and ~50% inhibited in SK-OV-3 cells (Supplemental Fig. S4). These data strongly indicate that the activity of ERK1/2 is modulated by endogenous oxidative cysteine modifications not post-lysis effects, and that the observed increase in sulfenic acid formation, and potentially subsequent oxidation products like disulfides, strongly modulate ERK1/2 kinase activity.
Figure 6.

Endogenous ERK oxidation inhibits kinase activity towards Elk1. Dual (TEY) phosphorylated ERK1/2 was immunoprecipitated from NIH 3T3 cells treated with PDGF for various time points as in Fig. 2 and as described in Materials and Methods. Each sample of immunoprecipitated ERK1/2 was then split into two: one sample was diluted into dithiothreitol (DTT)-containing buffer while the other was left in its native redox state (diluted into buffer lacking DTT). Recombinant Elk1 and ATP were added and samples were incubated at 30°C for 30 min. An immunoblot of S383-phosphorylated Elk1 was conducted to measure relative activities of immunoprecipitated ERK1/2. (A) Representative immunoblot of phosphorylated Elk1. (B) Normalized Western blot data from 5 independent replicates. DTT-treated samples are at least 5-fold more active toward Elk1 than non-reduced immunoprecipitates at all time points tested after PDGF addition. ***p< 0.001, ****p< 0.000005. (C) Representative immunoblot for dually (TEY) phosphorylated (top) and total (bottom) ERK1/2 from cell lysates.
Discussion
ERK1/2 proteins are extensively studied serine/threonine kinases central to the signal transduction of a variety of pathways. Due to their ubiquitous nature, understanding ERK1/2 regulation is key to identifying how ERK1/2 signaling induces varying, and even contradictory, responses to particular signals [44]. To date, several modes of regulation have been identified, including the activating dual phosphorylation on the TEY motif of the activation loop by MEK [37], interactions with anchoring proteins and scaffolds which regulate substrate/enzyme colocalization and modulate activity [33, 41, 55], control over localization via phosphorylation on the SPS motif [56], crosstalk with other signaling cascades [44], and modulation of signal duration as controlled by phosphatases [57–59]. This current work identifies an additional layer of regulation through the post-translational modification of cysteine oxidation, which we demonstrate to be a modification on ERK1 and ERK2 proteins that is not restricted to a particular class of receptors or cell types.
Regulation of kinases by oxidation is becoming increasingly recognized as an important aspect of their activity modulation. Unlike protein tyrosine phosphatases, where the primary oxidation target is the catalytic cysteine at the active site such that oxidation is always inhibitory, kinase families tend to have non-catalytic cysteine residues in unique locations specific to individual kinase families. Thus, function-altering oxidation of kinases can impose a spectrum of changes in structure and function [20, 60] thereby impacting activity, stability or interaction with partner proteins. This is why kinase oxidation can lead to either inhibition or activation depending on the specific protein. In some cases such as Src, particular cysteine residues and/or oxoforms involved in ROS responses can lead to either inhibition or activation of even an individual protein [61, 62].
In the present work, it is evident in multiple cell lines that ERK1/2 is labeled by DCP-Bio1 upon activation of the receptor tyrosine kinase PDGF receptor or the G-Protein coupled LPA receptor (Fig. 2, 4, 5, S3). DCP-Bio1 labeling of ERK1/2 is indicative that at least one of the cysteine residues on ERK1/2 is reacting with oxidants to form sulfenic acid in response to the extracellular signal. Although this does not rule out the possibility that ERK1/2 oxidation can occur through a disulfide-transfer or “redox relay” mediated by an oxidized protein such as a peroxiredoxin (as recently reported for STAT3 [63]), the fact that dimedone-based probes are able to react directly with ERK1/2 cysteines reveals that ERK1/2 can react directly with oxidants produced by the cell (i.e., a redox relay would generate a disulfide on the target protein through thiol-disulfide interchange instead of an initial sulfenic acid) [18]. Furthermore, no biotinylation of ERK1/2 is observed without the addition of DCP-Bio1 to lysis buffer, indicating that biotinylation is due to sulfenic acid formation on ERK1/2 and not endogenous biotinylation (Supplemental Fig. S1A). Moreover, direct labeling of ERK by the chemical probe is confirmed by the complementary approach of phospho-ERK immunoprecipitation followed by Western blot for biotin (Supplemental Fig. S1B). A strongly positive aspect of using DCP-Bio1 to detect protein oxidation is that it labels cysteines undergoing direct and active oxidation. However, it should be noted that, because DCP-Bio1 detects only the likely transient sulfenic acid (or potentially sulfenylamide [16, 17]) intermediate, it does not report on the total pool of oxidized protein.
In NIH 3T3 mouse fibroblasts, there is very low basal sulfenic acid formation on ERK1/2 in the absence of serum (Fig. 2). Upon PDGF treatment, sulfenic acid formation significantly increases 5–10 min after stimulation among both total and phospho-ERK1/2 populations (Fig 2). By 30 min after PDGF stimulation, ERK1/2 sulfenylation decreased to basal levels even though the relative extent of phosphorylation did not decrease, indicating that active oxidation of ERK1/2 and phosphorylation of the activation loop are not coupled. This conclusion is further corroborated in Figure 3, where scavenging of H2O2 by PEG-Catalase decreased sulfenic acid formation on ERK1/2 but did not decrease phosphorylation. While DCP-Bio1 labeling and subsequent immunoblotting is unable to report on absolute ratios of sulfenic acid-oxidized ERK1/2 to total or phosphorylated forms of ERK1/2, the temporal patterns of oxidation of total ERK1/2 and phospho-ERK1/2 are identical under this condition and in this cell line. This suggests that in NIH 3T3 cells stimulated with PDGF, phosphorylation and oxidation are co-temporal events, or that primarily phospho-ERK1/2 undergoes active reaction with H2O2 to form sulfenic acids in these cells.
Serum-depleted NIH 3T3 and WI-38 cells, both being embryonic, mesenchymal-derived fibroblast cell lines from mice and humans, respectively [64, 65], exhibited very similar patterns of phospho-ERK1/2 oxidation to sulfenic acid upon PDGF stimulation; both exhibited rapid increases in oxidation 5–10 min after stimulation followed by a substantial drop in the active oxidation detected by DCP-Bio1. Although a direct comparison has not been made in the kinetics of cell responses between these two cell lines, it is not surprising that they exhibit a similar profile of ERK1/2 cysteine oxidation given the fact that both cell lines require PDGF for sustained growth, survival, and chemotactic responses [66, 67]. It is notable, however, that the patterns of oxidation in WI-38 cells detected using antibodies against total ERK1/2 differ from those observed using antibodies against phospho-ERK1/2 in both serum depleted and serum supplemented conditions. Before PDGF stimulation, serum-depleted cells have almost no observable phospho-ERK1/2 (Fig. 4B), indicating that the oxidized ERK1/2 observed before PDGF treatment in Fig. 4A is largely non-phosphorylated ERK1/2. Furthermore, there is little sulfenylation of phospho-ERK1/2 30 min post stimulus, indicating that the sharp increase in sulfenylation detected at this time point using total ERK1/2 antibodies is occurring on non-phosphorylated ERK1/2. Thus, it appears that WI-38 cells are differentially targeting sub-populations of ERK1/2 for active sulfenylation. Interestingly, serum-supplemented WI-38 cells treated with PDGF exhibited a very different pattern of ERK1/2 sulfenic acid formation (Supplemental Fig. S2). The fact that the same cell line exhibited such a remarkable difference in sulfenylation dynamics for ERK1/2 in the presence or absence of serum indicates that the basal state of the cells has a significant impact on how the cells utilize ROS as second messengers upon stimulation.
ERK1/2 oxidation also occurred in the ovarian cancer-derived SK-OV-3 (Fig. 5) and prostate cancer-derived PC-3 cell lines (Supplemental Fig. S3). Based on these data, we can conclude that oxidation of ERK1/2 does not only occur in a small subset of cell types or in response to only a certain class of receptors. Furthermore, it is notable that these cancer-derived cell lines exhibited either continuous, as in SK-OV-3 (Fig. 5A), or somewhat erratic patterns of ERK1/2 oxidation (Supplemental Fig. S3A) compared to the non-transformed cell lines used in this study.
While ERK1/2 perform a number of cell functions unconnected to kinase activity [68, 69], the primary mode of ERK1/2 action is through its ATP-dependent phosphorylation of over 300 known substrates [36, 70]. One of the early response transcription factors phosphorylated by ERK1/2 during proliferative signaling is Elk1 [70]. Figures 6 and S4 demonstrate that endogenous ERK1/2 kinase activity towards Elk1 is significantly dampened in the absence of reductant compared to levels measured in the presence of DTT (included in standard kinase assays), even though ERK1/2 is fully phosphorylated on the activation loop. Because DTT is a broad spectrum reductant, the observed oxidative inhibition may reflect sulfenylation, disulfide bond formation, or both (at later time points, disulfide bond formation most likely predominates given observed sulfenylation time courses). These results, at first glance, seem to contradict the known role of ERK1/2 in proliferative signaling, where ERK1/2 kinase activity is required for the cell response to the proliferative stimuli [36]. However, ERK1/2 regulation by cysteine oxidation could be a mode of regulation more complex than simply switching the enzyme activity between on and off states. As depicted in Figure 7, there are multiple possible ways for oxidation to modulate ERK1/2 cellular activity. For example, with its vast array of known substrates spanning both the cytosol and nucleus, a model that is consistent with our data and the important role of activated ERK in signaling is that ERK1/2 cysteine oxidation occurs to prevent phosphorylation of substrates in the cytosol while phospho-ERK1/2 is en route to the nucleus; pERK1/2 in the nucleus would then be subjected to a more reducing environment than in the cytosol, potentially undergoing “unmasking” of its activity by reduction (Fig. 7) [71, 72]. Alternatively, oxidation of ERK in a docking domain could affect interactions with other kinases, phosphatases, scaffold or anchor proteins, or substrates, thereby altering substrate specificity, phosphorylation status and/or localization.
Figure 7.

Model for ERK phosphorylation and oxidation controlling activity in response to extracellular signals. Receptor (R) activation due to stimulus (S) binding (1) leads to MEK1/2 activation (2p) as well as concurrent NADPH oxidase (Nox) activation to produce H2O2 (2o). Activated MEK1/2 phosphorylates inactive ERK1/2 (gray or red) on Thr and Tyr residues (3p), and ERK1/2 is reversibly oxidized by H2O2 or a derivative thereof (3o) (our data suggest that neither modification precludes the other). Our data support a strong inhibitory effect of this oxidation on phospho-ERK. Note that dually phosphorylated AND oxidized ERK (yellow) is “primed” to be active once reduced (e.g., by thioredoxin or other dithiol). Note that this diagram does not emphasize the high degree of localization that is expected to be important in the posttranslational modification of ERK due to signal-mediated receptor activation.
The mapping of regulatory cysteine oxidation sites in ERK will require further investigation, but of particular interest given their locations and previous findings are two Cys residues, C159 and C164. C159 sits within the “D-recruitment site” (DRS), an important region supporting protein-protein interactions between ERK1/2 and its regulatory proteins such as the phosphatase MKP3 [73–75], MEK1/2 [76, 77], anchors and other scaffolds [78]. Many substrates also interact with ERK1/2 in this binding region. Structural studies reveal that hydrophobic residues of interacting proteins insert into hydrophobic pockets on the surface of ERK1/2 on either side of C159 [79–82]. Thus, oxidation of this residue could sterically prevent ERK1/2 from interacting with DRS partners. Indeed, DRS-binding peptides protect C159 from alkylation [83, 84] and this cysteine has been shown to be sensitive to oxidation by H2O2 [26] and nitrosylation [48], and has been shown to be conjugated to β-mercaptoethanol in a crystal structure [85]. Just five residues farther down the chain is C164, a site of nitrosylation on ERK1 (C183 in human ERK1 numbering) in cells treated with GSNO, a NO+ donor that participates in transnitrosylation [49]. Nitrosylation of this cysteine appears to direct cells into apoptosis. Based on the location of this Cys proximal to the ATP binding site, oxidation or nitrosylation of this cysteine could prevent, or slow, ATP binding and exchange, thereby inhibiting kinase activity. One or both (or additional) cysteine residues could undergo active oxidation during the early phase of receptor activation and modulate ERK activity via changes in protein-protein interactions or ATP binding. It should be considered that one or multiple cysteine residues in ERK1/2 could be reactive toward oxidants in given conformations or complexes of the ERK protein, as well, and the scenarios under which each cysteine undergoes oxidation could vary depending on the specific stimulus and on other factors influencing the strength and localization of ROS production.
In conclusion, similar to how phosphorylation of ERK1/2 on key residues affects ERK1/2 structure and function, oxidation of ERK cysteines could affect the surface, structure, and dynamics of ERK1/2 in key regions of ERK1/2 functionality. We have observed that sulfenylation can occur to both non-phosphorylated and TEY-phosphorylated ERK1/2, and that this additional post-translational modification that affects kinase activity is reversible. Irreversible modification may also ensue at these or other cysteine residues, under alternative and/or potentially pathological conditions. Based on the location of solvent-exposed cysteines, ERK1/2 oxidation could be a significant mode of regulating ERK1/2 activity through protein-protein interactions, spatiotemporal localization, and modulation of kinase activity. Further studies to understand how ERK1/2 is regulated by oxidation are essential to illuminate modes of ERK1/2 signaling specificity, potentially leading to the development of novel therapeutics to target ERK1/2 in signal-specific redox states.
Supplementary Material
Highlights.
Sulfenylation of ERK in cells is observed based on DCP-Bio1 labeling of lysates
Function-altering ERK oxidation occurs in PDGF-treated NIH 3T3 and WI38 fibroblasts
Most ERK from PDGF-treated cells is reversibly oxidized given Elk1 kinase activity
ERK phosphorylation assessment is insufficient as a proxy for activation status
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 GM050389 and R01 GM119227 to L.B.P., and R33 CA177461 to L.B.P. and C.M.F.), and by support to J.D.K. from the Wake Forest Center for Molecular Signaling and from T32 AI007401. The authors thank S. Bruce King and colleagues for providing the DCP-Bio1, as well as Natalie Ahn and Rony Seger for suggestions and discussions, and Rob Newman, Larry Daniel, Doug Lyles and Todd Lowther for helpful suggestions during development of the project and preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 2.Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. 2015;33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 4.Russell EG, Cotter TG. New Insight into the Role of Reactive Oxygen Species (ROS) in Cellular Signal-Transduction Processes. International review of cell and molecular biology. 2015;319:221–254. doi: 10.1016/bs.ircmb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 6.Back P, Braeckman BP, Matthijssens F. ROS in Aging Caenorhabditis elegans: Damage or Signaling? Oxidative medicine and cellular longevity. 2012;2012:1–14. doi: 10.1155/2012/608478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morimoto H, Iwata K, Ogonuki N, Inoue K, Atsuo O, Kanatsu-Shinohara M, Morimoto T, Yabe-Nishimura C, Shinohara T. ROS Are Required for Mouse Spermatogonial Stem Cell Self-Renewal. Cell stem cell. 2013;12:774–786. doi: 10.1016/j.stem.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Oakley FD, Abbott D, Li Q, Engelhardt J. Signaling Components of Redox Active Endosomes: The Redoxosomes. Antioxid Redox Signal. 2009;11:1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klomsiri C, Rogers LC, Soito L, McCauley AK, King SB, Nelson KJ, Poole LB, Daniel LW. Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radic Biol Med. 2014;71C:49–60. doi: 10.1016/j.freeradbiomed.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saunders JA, Rogers LC, Klomsiri C, Poole LB, Daniel LW. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic Biol Med. 2010;49:2058–2067. doi: 10.1016/j.freeradbiomed.2010.10.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaplan N, Urao N, Furuta E, Kim SJ, Razvi M, Nakamura Y, McKinney RD, Poole LB, Fukai T, Ushio-Fukai M. Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free radical research. 2011;45:1124–1135. doi: 10.3109/10715762.2011.602073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamalainen RH, Ahlqvist KJ, Ellonen P, Lepisto M, Logan A, Otonkoski T, Murphy MP, Suomalainen A. mtDNA Mutagenesis Disrupts Pluripotent Stem Cell Function by Altering Redox Signaling. Cell reports. 2015;11:1614–1624. doi: 10.1016/j.celrep.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood ST, Long DL, Reisz JA, Yammani RR, Burke EA, Klomsiri C, Poole LB, Furdui CM, Loeser RF. Cysteine-Mediated Redox Regulation of Cell Signaling in Chondrocytes Stimulated With Fibronectin Fragments. Arthritis & rheumatology (Hoboken, NJ) 2016;68:117–126. doi: 10.1002/art.39326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, Furdui CM. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci U S A. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devarie-Baez NO, Silva Lopez EI, Furdui CM. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free radical research. 2016;50:172–194. doi: 10.3109/10715762.2015.1090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furdui CM, Poole LB. Chemical approaches to detect and analyze protein sulfenic acids. Mass spectrometry reviews. 2014;33:126–146. doi: 10.1002/mas.21384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta V, Paritala H, Carroll KS. Reactivity, Selectivity, and Stability in Sulfenic Acid Detection: A Comparative Study of Nucleophilic and Electrophilic Probes. Bioconjug Chem. 2016;27:1411–1418. doi: 10.1021/acs.bioconjchem.6b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 2014;1844:1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol. 2010;404:902–916. doi: 10.1016/j.jmb.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Molecular biology and evolution. 2000;17:1232–1239. doi: 10.1093/oxfordjournals.molbev.a026406. [DOI] [PubMed] [Google Scholar]
- 23.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 24.Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- 25.Allison WS. Formation and reactions of sulfenic acids in proteins. Acc Chem Res. 1976;9:293–299. [Google Scholar]
- 26.Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nature communications. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu SE, King SB, Furdui CM, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection: evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol. 2010;473:77–94. doi: 10.1016/S0076-6879(10)73003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection; methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wani R, Bharathi NS, Field J, Tsang AW, Furdui CM. Oxidation of Akt2 kinase promotes cell migration and regulates G1-S transition in the cell cycle. Cell Cycle. 2011;10:3263–3268. doi: 10.4161/cc.10.19.17738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nolin JD, Tully JE, Hoffman SM, Guala AS, van der Velden JL, Poynter ME, van der Vliet A, Anathy V, Janssen-Heininger YM. The glutaredoxin/S-glutathionylation axis regulates interleukin-17A-induced proinflammatory responses in lung epithelial cells in association with S-glutathionylation of nuclear factor kappaB family proteins. Free Radic Biol Med. 2014;73:143–153. doi: 10.1016/j.freeradbiomed.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshikawa J, Urao N, Kim HW, Kaplan N, Razvi M, McKinney R, Poole LB, Fukai T, Ushio-Fukai M. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PloS one. 2010;5:e10189. doi: 10.1371/journal.pone.0010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods in molecular biology (Clifton, NJ) 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 33.Roskoski R. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, Keyomarsi K, Yarden Y, Seger R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20:147–155. doi: 10.1038/sj.onc.1204062. [DOI] [PubMed] [Google Scholar]
- 35.Davey L, Cohen A, LeBlanc J, Halperin SA, Lee SF. The disulfide oxidoreductase SdbA is active in Streptococcus gordonii using a single C-terminal cysteine of the CXXC motif. Mol Microbiol. 2016;99:236–253. doi: 10.1111/mmi.13227. [DOI] [PubMed] [Google Scholar]
- 36.Maik-Rachline G, Seger R. The ERK cascade inhibitors: Towards overcoming resistance. Drug Resistance Updates. 2016;25:1–12. doi: 10.1016/j.drup.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Zhang F, Ebert D, Cobb MH, Goldsmith EJ. ACTIVITY OF THE MAP KINASE ERK2 IS CONTROLLED BY A FLEXIBLE SURFACE LOOP (VOL 3, PG 299, 1995) Structure. 1995;3:1126–1126. doi: 10.1016/s0969-2126(01)00160-5. [DOI] [PubMed] [Google Scholar]
- 39.Sours KM, Xiao Y, Ahn NG. Extracellular-Regulated Kinase 2 Is Activated by the Enhancement of Hinge Flexibility. Journal of Molecular Biology. 2014;426:1925–1935. doi: 10.1016/j.jmb.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao Y, Lee T, Latham MP, Warner LR, Tanimoto A, Pardi A, Ahn NG. Phosphorylation releases constraints to domain motion in ERK2. Proceedings of the National Academy of Sciences. 2014;111:2506–2511. doi: 10.1073/pnas.1318899111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao Z, Seger R. The ERK signaling cascade-Views from different subcellular compartments. Biofactors. 2009;35:407–416. doi: 10.1002/biof.52. [DOI] [PubMed] [Google Scholar]
- 42.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends in Biochemical Sciences. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 43.Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Molecular and Cellular Biology. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochimica Et Biophysica Acta-Molecular Cell Research. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Zhang FM, Strand A, Robbins D, Cobb MH, Goldsmith EJ. ATOMIC-STRUCTURE OF THE MAP KINASE ERK2 AT 2.3-ANGSTROM RESOLUTION. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 46.Galli S, Antico Arciuch VG, Poderoso C, Converso DP, Zhou Q, Bal de Kier Joffe E, Cadenas E, Boczkowski J, Carreras MC, Poderoso JJ. Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PloS one. 2008;3:e2379. doi: 10.1371/journal.pone.0002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luanpitpong S, Chanvorachote P, Nimmannit U, Leonard SS, Stehlik C, Wang L, Rojanasakul Y. Mitochondrial superoxide mediates doxorubicin-induced keratinocyte apoptosis through oxidative modification of ERK and Bcl-2 ubiquitination. Biochemical pharmacology. 2012;83:1643–1654. doi: 10.1016/j.bcp.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paige JS, Xu G, Stancevic B, Jaffrey SR. Nitrosothiol Reactivity Profiling Identifies S-Nitrosylated Proteins with Unexpected Stability. Chemistry & Biology. 2008;15:1307–1316. doi: 10.1016/j.chembiol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng XJ, Sun TZ, Bei YC, Ding S, Zheng W, Lu Y, Shen PP. S-nitrosylation of ERK inhibits ERK phosphorylation and induces apoptosis. Scientific reports. 2013;3 doi: 10.1038/srep01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parsonage D, Nelson KJ, Ferrer-Sueta G, Alley S, Karplus PA, Furdui CM, Poole LB. Dissecting peroxiredoxin catalysis: Separating binding, peroxidation, and resolution for a bacterial AhpC. Biochemistry. 2015;54:1567–1575. doi: 10.1021/bi501515w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 52.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bian D, Su S, Mahanivong C, Cheng RK, Han Q, Pan ZK, Sun P, Huang S. Lysophosphatidic acid stimulates ovarian cancer cell migration via a Ras-MEK kinase 1 pathway. Cancer Res. 2004;64:4209–4217. doi: 10.1158/0008-5472.CAN-04-0060. [DOI] [PubMed] [Google Scholar]
- 54.Umezu-Goto M, Tanyi J, Lahad J, Liu S, Yu S, Lapushin R, Hasegawa Y, Lu Y, Trost R, Bevers T, Jonasch E, Aldape K, Liu J, James RD, Ferguson CG, Xu Y, Prestwich GD, Mills GB. Lysophosphatidic acid production and action: validated targets in cancer? J Cell Biochem. 2004;92:1115–1140. doi: 10.1002/jcb.20113. [DOI] [PubMed] [Google Scholar]
- 55.Liang Y, Sheikh F. Scaffold Proteins Regulating Extracellular Regulated Kinase Function in Cardiac Hypertrophy and Disease. Frontiers in Pharmacology. 2016;7:8. doi: 10.3389/fphar.2016.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wainstein E, Seger R. The dynamic subcellular localization of ERK: mechanisms of translocation and role in various organelles. Current Opinion in Cell Biology. 2016;39:15–20. doi: 10.1016/j.ceb.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Bendetz-Nezer S, Seger R. Role of non-phosphorylated activation loop residues in determining ERK2 dephosphorylation, activity, and subcellular localization. Journal of Biological Chemistry. 2007;282:25114–25122. doi: 10.1074/jbc.M703120200. [DOI] [PubMed] [Google Scholar]
- 58.Perlson E, Michaelevski I, Kowalsman N, Ben-Yaakov K, Shaked M, Seger R, Eisenstein M, Fainzilber M. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. Journal of Molecular Biology. 2006;364:938–944. doi: 10.1016/j.jmb.2006.09.056. [DOI] [PubMed] [Google Scholar]
- 59.Yao Z, Dolginov Y, Hanoch T, Yung YV, Ridner G, Lando Z, Zharhary D, Seger R. Detection of partially phosphorylated forms of ERK by monoclonal antibodies reveals spatial regulation of ERK activity by phosphatases. Febs Letters. 2000;468:37–42. doi: 10.1016/s0014-5793(00)01191-1. [DOI] [PubMed] [Google Scholar]
- 60.Heppner DE, Janssen-Heininger YM, van der Vliet A. The role of sulfenic acids in cellular redox signaling: Reconciling chemical kinetics and molecular detection strategies. Arch Biochem Biophys. 2017;616:40–46. doi: 10.1016/j.abb.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci U S A. 2009;106:5070–5075. doi: 10.1073/pnas.0806117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol. 2015;11:64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 64.Hayflick L, Moorhead PS. SERIAL CULTIVATION OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 1961;25:585. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 65.Jainchill JL, Aaronson SA, Todaro GJ. Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. Journal of virology. 1969;4:549–553. doi: 10.1128/jvi.4.5.549-553.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clark JG, Madtes DK, Raghu G. EFFECTS OF PLATELET-DERIVED GROWTH-FACTOR ISOFORMS ON HUMAN LUNG FIBROBLAST PROLIFERATION AND PROCOLLAGEN GENE-EXPRESSION. Experimental lung research. 1993;19:327–344. doi: 10.3109/01902149309064350. [DOI] [PubMed] [Google Scholar]
- 67.Hannink M, Donoghue DJ. STRUCTURE AND FUNCTION OF PLATELET-DERIVED GROWTH-FACTOR (PDGF) AND RELATED PROTEINS. Biochimica Et Biophysica Acta. 1989;989:1–10. doi: 10.1016/0304-419x(89)90031-0. [DOI] [PubMed] [Google Scholar]
- 68.McReznolds AC, Karra AS, Li Y, Lopez ED, Turjanski AG, Dioum E, Lorenz K, Zaganjor E, Stippec S, McGlynn K, Earnest S, Cobb MH. Phosphorylation or Mutation of the ERK2 Activation Loop Alters Oligonucleotide Binding. Biochemistry. 2016;55:1909–1917. doi: 10.1021/acs.biochem.6b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rodriguez J, Crespo P. Working Without Kinase Activity: Phosphotransfer-Independent Functions of Extracellular Signal-Regulated Kinases. Science signaling. 2011:4. doi: 10.1126/scisignal.2002324. [DOI] [PubMed] [Google Scholar]
- 70.Yoon S, Seger R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth factors (Chur, Switzerland) 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 71.Go YM, Jones DP. Redox control systems in the nucleus: mechanisms and functions. Antioxid Redox Signal. 2010;13:489–509. doi: 10.1089/ars.2009.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radical Biology and Medicine. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang JL, Zhou B, Zheng CF, Zhang ZY. A bipartite mechanism for ERK2 recognition by its cognate regulators and substrates. Journal of Biological Chemistry. 2003;278:29901–29912. doi: 10.1074/jbc.M303909200. [DOI] [PubMed] [Google Scholar]
- 74.Zhao Y, Zhang ZY. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. Journal of Biological Chemistry. 2001;276:32382–32391. doi: 10.1074/jbc.M103369200. [DOI] [PubMed] [Google Scholar]
- 75.Zhou B, Wu L, Shen K, Zhang JL, Lawrence DS, Zhang ZY. Multiple regions of MAP kinase phosphatase 3 are involved in its recognition and activation by ERK2. Journal of Biological Chemistry. 2001;276:6506–6515. doi: 10.1074/jbc.M009753200. [DOI] [PubMed] [Google Scholar]
- 76.Bardwell AJ, Flatauer LJ, Matsukuma K, Thorner J, Bardwell L. A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. Journal of Biological Chemistry. 2001;276:10374–10386. doi: 10.1074/jbc.M010271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu BE, Stippec S, Robinson FL, Cobb MH. Hydrophobic as well as charged residues in both MEK1 and ERK2 are important for their proper docking. Journal of Biological Chemistry. 2001;276:26509–26515. doi: 10.1074/jbc.M102769200. [DOI] [PubMed] [Google Scholar]
- 78.Chuderland D, Seger R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Molecular Biotechnology. 2005;29:57–74. doi: 10.1385/MB:29:1:57. [DOI] [PubMed] [Google Scholar]
- 79.Critton DAy, Tortajada A, Stetson G, Peti W, Page R. Structural Basis of Substrate Recognition by Hematopoietic Tyrosine Phosphatase. Biochemistry. 2008;47:13336–13345. doi: 10.1021/bi801724n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Francis DM, Koveal D, Tortajada A, Page R, Peti W. Interaction of Kinase-Interaction-Motif Protein Tyrosine Phosphatases with the Mitogen-Activated Protein Kinase ERK2. PloS one. 2014:9. doi: 10.1371/journal.pone.0091934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min XS, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Molecular Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 82.Piserchio A, Warthaka M, Devkota AK, Kaoud TS, Lee S, Abramczyk O, Ren PY, Dalby KN, Ghose R. Solution NMR Insights into Docking Interactions Involving Inactive ERK2. Biochemistry. 2011;50:3660–3672. doi: 10.1021/bi2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abramczyk O, Rainey MA, Barnes R, Martin L, Dalby KN. Expanding the repertoire of an ERK2 recruitment site: Cysteine Footprinting identifies the D-recruitment site as a mediator of Ets-1 binding. Biochemistry. 2007;46:9174–9186. doi: 10.1021/bi7002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Callaway K, Abramczyk O, Martin L, Dalby KN. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment sitet. Biochemistry. 2007;46:9187–9198. doi: 10.1021/bi700206u. [DOI] [PubMed] [Google Scholar]
- 85.Zhang J, Shapiro P, Pozharski E. Structure of extracellular signal-regulated kinase 2 in complex with ATP and ADP. Acta Crystallogr F-Struct Biol Cryst Commun. 2012;68:1434–1439. doi: 10.1107/S1744309112042972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.