

Abstract
Rationale
Platelet hyperreactivity, which is common in many pathological conditions, is associated with increased atherothrombotic risk. The mechanisms leading to platelet hyperreactivity are complex and not yet fully understood.
Objective
Platelet hyperreactivity and accelerated thrombosis, specifically in dyslipidemia, have been mechanistically linked to accumulation in the circulation of a specific group of oxidized phospholipids (oxPCCD36) that are ligands for the platelet pattern-recognition receptor CD36. In the current manuscript, we tested whether the platelet innate immune system contributes to responses to oxPCCD36 and accelerated thrombosis observed in hyperlipidemia.
Methods and Results
Using in vitro approaches, as well as platelets from mice with genetic deletion of MyD88 or TLRs, we demonstrate that TLR2 and TLR6 are required for the activation of human and murine platelets by oxPCCD36. oxPCCD36 induce formation of CD36/TLR2/TLR6 complex in platelets and activate downstream signaling via TIRAP-MyD88-IRAK1/4-TRAF6, leading to integrin activation via the SFK-Syk-PLCγ2 pathway. Intravital thrombosis studies using ApoE−/− mice with genetic deficiency of TLR2 or TLR6 have demonstrated that oxPCCD36 contribute to accelerated thrombosis specifically in the setting of hyperlipidemia.
Conclusions
Our studies reveal that TLR2 plays a key role in platelet hyperreactivity and the prothrombotic state in the setting of hyperlipidemia by sensing a wide range of endogenous lipid-peroxidation ligands and activating innate immune signaling cascade in platelets.
Keywords: Platelets, TLR2, hyperlipidemia, thrombosis, innate immune system, lipids and lipoproteins, singaling pathways, signal transduction
Subject Terms: Platelets, Oxidative Stress, Thrombosis, Cell Signaling/Signal Transduction, Lipids and Cholesterol
INTRODUCTION
Enhanced platelet reactivity and the risk of thrombotic events are associated with a number of pathophysiological states related to dyslipidemia, including atherosclerosis, diabetes, and metabolic syndrome.1–8 The mechanisms leading to enhanced platelet reactivity are of considerable importance and are the focus of multiple studies.3–10 We have previously demonstrated that scavenger receptor CD36 is the major receptor on platelets promoting platelet reactivity in hyperlipidemic conditions.10, 11 We mechanistically connected platelet hyperreactivity to oxidative stress and specific oxidized phospholipids (oxPCCD36) accumulating in plasma in dyslipidemia.10–13 oxPCCD36 are derived from the free-radical oxidation of phosphatidylcholines containing polyunsaturated fatty acids in the sn-2 position, such as 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine (PAPC) and 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine (PLPC). oxPCCD36 are found in vivo at sites of enhanced oxidative stress, including plasma of hyperlipidemic animals or plasma of patients with low HDL levels.10, 12 oxPCCD36 bind with high affinity to CD36 via an electrostatic interaction of a negatively charged carboxylate at the sn-2 position of oxPCCD36 and CD36 lysines 164 and 166.14, 15 The binding of oxPCCD36 to platelet CD36 leads to platelet activation via the SFK-Syk-PLCγ2 pathway.11 Although significant progress has been made in recent years towards understanding the interaction of CD36 with oxidized phospholipids and downstream signaling in platelets, many details of the cellular and molecular mechanism are still not known.
Platelets express a number of pattern recognition receptors, including toll-like receptors (TLRs).16–21 While TLRs are mostly known to recognize pathogen-associated molecular patterns, it has been proposed that TLRs are the major sensors of “oxidation-specific” epitopes.22 A number of recent studies have demonstrated that lipid peroxidation-derived endogenous ligands could be recognized by TLRs on macrophages and endothelial cells promoting the progression of inflammation and atherosclerosis.22–25 Moreover, we have recently reported that platelets can also respond to oxidation specific epitopes via TLRs.9, 17 Our observation of the strong thromboprotective effect of TLR2 deficiency in hyperlipidemic mice9 prompted us to hypothesize that TLR2 may have a role in oxPCCD36 induced platelet activation.
In this study, we have demonstrated that TLR2 in a complex with TLR6 cooperates with CD36, and that it is essential for platelet activation by oxPCCD36. In vivo studies have demonstrated that both TLR2 and TLR6 genetic deficiencies protect hyperlipidemic mice from accelerated thrombosis. The protective effect of TLR2 or TLR6 deficiency on thrombosis is specific for the setting of hyperlipidemia. In vitro studies have demonstrated that oxPCCD36 induce the innate immune signaling pathway in platelets via TIRAP-MyD88-IRAKs-TRAF6, followed by c-Src and Fyn activation. Our current findings and recent observations9 indicate that TLR2 is involved in the recognition of a wide range of oxidation derived epitopes, and it is a key player in prothrombotic phenotype associated with hyperlipidemia.
METHODS
Detailed methods can be found in the Online Data Supplement.
Materials
Human α-thrombin was purchased from Enzyme Research Laboratories (South Bend, IN) and adenosine diphosphate (ADP) from Chrono-log (Havertown, PA). Phycoerythrin-conjugated anti–P-selectin, FITC-conjugated PAC-1, and anti-human CD61 antibodies were obtained from BD Biosciences (San Jose, CA). Phycoerythrin-conjugated mouse integrin-αIIbβ3 (clone JON/A), FITC-conjugated GPVI, GPV, GPIX and integrin-αIIbβ3 (clone Leo.F2) antibodies and FITC-conjugated rat IgG (negative control) were purchased from Emfret Analytics GmbH & Co. KG (Eibelstadt, Germany). Anti-CD36 (clone FA6-152) antibody was purchased from Beckman Coulter (Brea, CA). Anti-CD36 polyclonal antibody was purchased from Cayman Chemical (Ann Arbor, MI). Anti-hTLR2, non-specific IgG1, and anti-phosphotyrosine antibodies (clone 4G10) were from Millipore (Temecula, CA). Light chain specific HRP-conjugated anti-mouse IgG and anti-rabbit IgG were purchased from Jackson ImmunoResearch (West Grove, PA). FITC-labeled anti-human/mouse-TLR2, PE-conjugated anti-hTLR6 antibodies and isotype negative controls were purchased from BioLegend (San Diego, CA). Anti-TLR3, TLR4 and their isotype control IgGs were purchased from e-Bioscience (San Diego, CA). Anti-TLR1, p-IRAK4, IRAK4, IRAK1, p-TAK1, TAK1, p-Src, Src, p-Syk, p-PLCγ2, and PLCγ2 were obtained from Cell Signaling Technology (Danvers, MA). Anti-hTLR6 antibody was purchased from Hycult Biotech (Plymouth Meeting, PA). Anti-TLR1 antibody was purchased from Abcam (Cambridge, MA). Pam3CSK4, Pam2CSK4, LTA, CpG ODN2006, control DNA, anti-hTLR2, anti-hTLR1, anti-hTLR6, and anti-mTLR2 antibodies, HEK-Blue™-TLR2 cells, and HEK-Blue™-hTLR9 cells were obtained from InvivoGen (San Diego, CA). p-IRAK1 antibody was purchased from GeneTex (Irvine, CA). Anti-integrin β3, anti-MyD88 and Syk antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-mTLR2, anti-mTLR6, and anti-mCD36 antibodies were purchased from R&D systems (Minniapolis, MN). MyD88 homodimerization, TIRAP and TRAF6 inhibitory peptides, and the control peptides were obtained from Imgenex (San Diego, CA). IRAK1/4 inhibitor was purchased from Calbiochem (San Diego, CA). CD45 MicroBeads, magnetic columns and accessories were purchased from Miltenyi Biotec GmbH (San Diego, CA). PLPC (1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine) was purchased from Avanti Polar Lipids (Alabaster, AL). KODA-PC (9-keto-12-oxo-10-dodecenoic acid ester of 2-lyso-phosphocholine), HODA-PC (9-hydroxy-12-oxo-10-dodecenoic acid esters of 2-lyso-PC), and KOOA-PC (5-keto-8-oxo-6-octenoic acid esters of 2-lyso-PC) were synthesized as previously described.11, 26 PLPC and KODA-PC were resuspended in Tyrode’s buffer (without BSA and glucose) to prepare a stock solution just before incubation with platelets, as previously described.11, 12
Experimental animals
Wild type (WT) C57BL/6J, TLR2−/−, MyD88−/−, and ApoE−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). CD36−/− mice were generated as described.27 TLR6−/− mice were obtained from Oriental Yeast Co. LTD (Tokyo, Japan). All strains were of at least 99% C57BL/6J background. Src−/− and Lyn−/− mice were generated as reported earlier.9 We generated ApoE−/−/CD36−/− by crossing ApoE−/− with CD36−/− mice as described10, and ApoE−/−/TLR2−/− and ApoE−/−/TLR6−/− mice by crossing ApoE−/− with TLR2−/− or TLR6−/− mice respectively. We further crossed ApoE−/−/CD36−/− mice to ApoE−/−/TLR2−/− or ApoE−/−/TLR6−/− mice to generate ApoE−/−/CD36−/−/TLR2−/− or ApoE−/−/CD36−/−/TLR6−/− triple knockout mice. Mice were fed a Western diet (TD88137, Harlan Teklad) for 8–10 weeks. Animals were housed in ventilated cages with ad libitum access to food and water, on a 14:10 light dark cycle. Prior to study, the Institutional Animal Care and Use Committee of the Cleveland Clinic approved all animal procedures. Mice were used between 8 and 14 weeks of age.
Platelet isolation
Platelet rich plasma (PRP) and gel-filtered platelets were isolated from blood drawn from healthy human donors or mice as described earlier.10, 17
Immunomagnetic separation of leukocytes from platelet rich plasma
Human platelets, used for detecting expression of Toll-like receptors, were further processed according to manufacturer’s protocol (Miltineyi Biotec). Platelet rich plasma was incubated with anti-human CD45 pan-leukocyte antibody bound to magnetic MicroBeads (CD45 MicroBeads) and passed through MACS magnetic columns connected to a midiMACS™ magnet for removal of any residual white blood cells.
Flow cytometry
Human and murine platelet suspensions were prepared by gel filtration as described earlier17 and incubated with various blocking reagents and agonists indicated in figure legends. P-selectin expression and integrin αIIbβ3 activation were assessed as previously described.10, 17 Data were acquired using a FACS Calibur instrument (Becton Dickinson, San Jose, CA) and analyzed using the FlowJo 9.4 software (Tree Star, Ashland, OR).
Immunoblotting
Platelets were isolated by gel filtration (2 × 108 cells) and treated with inhibitors or agonists for the indicated time at 37°C. Platelets were then pelleted down by centrifugation at 931 g for 5 minutes and lysed on ice with RIPA buffer containing protease and phosphatase inhibitors. Lysates were centrifuged at 10000 rpm for 10 minutes at 4°C, and supernatants were collected. Protein concentrations were estimated using BioRad protein assay dye reagent and BSA as standard. Proteins (30 – 50 μg) were resolved on SDS-PAGE, transferred to PVDF membrane, and probed with protein-specific primary antibodies, followed by horseradish peroxidase (HRP)-conjugated specific secondary antibodies. Immunodetection was performed by enhanced chemiluminescence (ECL, GE Healthcare/Amersham, Buckinghamshire, UK) according to the manufacturer’s protocol.
Human TLRs activation assay in HEK-Blue™-hTLR2 and HEK-Blue™-hTLR9 cells
The HEK-Blue™-hTLR2 cells and HEK-Blue™-hTLR9 cells that stably co-express human TLR2 or human TLR9 respectively along with NF-κB-inducible SEAP (secreted embryonic alkaline phosphatase) reporter gene were cultured as described before9 for 16–18 hours in the presence of indicated agonists. NF-κB-induced SEAP activity was assessed in the culture supernatant using QUANTI-Blue™ and reading at OD 630 nm as per manufacturer’s protocol.
Co-immunoprecipitation
Platelets isolated by gel filtration were stimulated with agonist in Tyrode’s buffer (Ca2+, Mg2+) at 37°C for 15 minutes. Platelets were then pelleted down and lysed with RIPA buffer on ice. Protein concentrations were determined and 300 μg of total protein was first pre-cleared with IgG, then incubated with antibody overnight at 4°C with gentle rotation. Protein A/G plus-agarose beads were then added and incubated for 2–4 hours at 4°C with gentle rotation. Beads were then washed three times, boiled with Laemmli protein sample buffer, and subjected to SDS-PAGE and Western blot analysis.
Intravital thrombosis
Intravital thrombosis study was performed using FeCl3-induced carotid artery thrombosis in hyperlipidemic and normolipidemic mice of indicated genotype as previously described.9, 17 The time to occlusion was defined as the moment we removed the FeCl3 soaked filter paper until complete flow cessation lasting for at least 1 minute. The blood vessels were observed under a Leica DM LFS microscope (Leica, Germany) with 10×/0.30 objectives. Images were acquired using a cooled high-speed, digital camera (QImaging Retiga EXi Fast 1394) with Streampix acquisition software.
Bone marrow transplantation
Eight-week-old male or female recipient ApoE−/− mice were lethally irradiated with a single dose of whole-body irradiation (900 rads) on the day of transplantation. Bone marrow cells from the donor seven-week-old male or female ApoE−/− and ApoE−/−/TLR2−/− mice were isolated and intravenously injected into each recipient mouse. Four weeks later, the mice were fed with a Western diet for 8–10 weeks and used for experiments.
Cholesterol and triglycerides estimation
Total plasma cholesterol and triglycerides were enzymatically assessed using the Amplex Red cholesterol assay kit (Molecular Probes/Invitrogen, Eugene, OR) and triglyceride assay kit (Cayman, Ann Arbor, MI), according to the manufacturers’ instructions.
Statistical analysis
Data are presented as mean ±SD. The statistical significance of differences was evaluated using the two-tailed nonparametric Mann Whitney test for in vivo thrombosis experiments and Student’s t-test for others. The p values less than 0.05 were considered to be statistically significant.
RESULTS
TLR2 plays a role in platelet activation induced by oxPCCD36
To test whether TLRs participate in platelet activation by oxPCCD36, we first tested if the universal adaptor for TLRs, myeloid-differentiation factor 88 (MyD88), plays a role in platelet activation by oxPCCD36. We observed that platelet integrin activation by the representative member of oxPCCD36, KODA-PC, was significantly reduced in MyD88−/− mice as compared to the platelets of wild type (WT) mice (Figure 1A). On the other hand, MyD88 did not play role in platelet activation induced by G-protein coupled receptor agonists ADP nor thrombin (Figure 1A and Supplemental Figure IA). We also observed that the MyD88 blocking peptide significantly inhibited the P-selectin expression induced by KODA-PC in human platelets, while no effect of the control peptide was observed (Figure 1B, 1C). The MyD88 blocking peptide had no effect on human platelet activation induced by ADP nor thrombin (Figure 1C). These finding demonstrate that MyD88 is required for the signaling induced by oxPCCD36 in human and murine platelets and strongly suggest the involvement of TLRs.
Figure 1. TLR2 and TLR6 play a role in platelet activation induced by oxPCCD36.
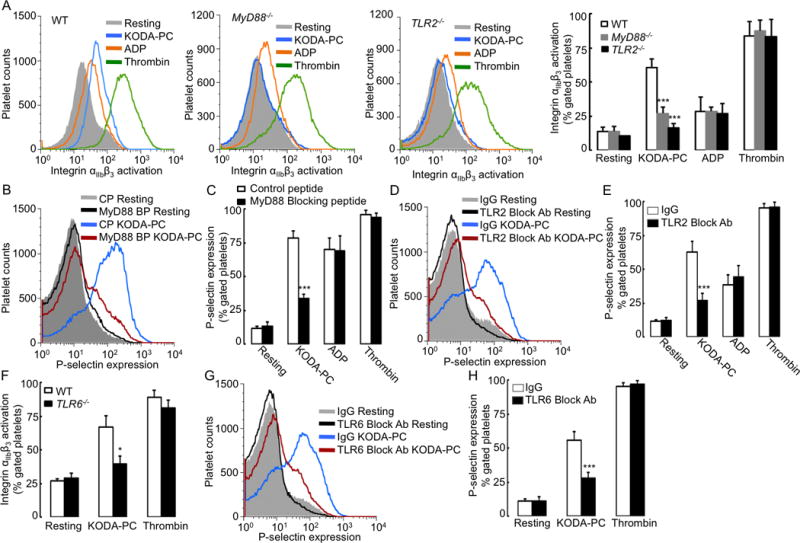
(A, F) Murine platelets of indicated genotypes (WT, MyD88−/−, TLR2−/−, and TLR6−/−) were isolated by gel filtration and incubated with representative member of oxPCCD36 (KODA-PC, 20 μM), ADP (10 μM), or thrombin (0.05 U/ml), and integrin αIIbβ3 activation was determined by FACS analysis using PE-conjugated integrin αIIbβ3 (JON/A) antibody. (A) Left panels present FACS analysis data as histograms, (A) right panel and (F) present data as mean ± SD of 3 independent experiments. (B–E, G–H) Human platelets were isolated by gel filtration and pre-incubated with either (B, C) 2 μM MyD88 blocking peptide (BP), or control peptide (CP) or (D, E) 4 μg/ml TLR2 neutralizing antibody, or (G, H) 4 μg/ml TLR6 neutralizing antibody or non-immune isotype matched control IgG followed by stimulation with oxPCCD36 (KODA-PC, 20 μM). P-selectin expression was assessed by FACS analysis. FACS analysis data are presented as histograms and as mean ± SD of 3 independent experiments. * p<0.05, *** p<0.001, and NS p>0.05 vs control.
In the next experiment, we assessed the effect of blocking antibodies to several TLRs on platelet activation by oxPCCD36. While blocking antibodies to TLR3, TLR4, and TLR9 had no effect (Supplemental Figure IB, IC), the blocking antibody to TLR2 significantly inhibited P-selectin expression induced by oxPCCD36 in human platelets (Figure 1D, 1E). Importantly, this antibody had no effect on platelet activation induced by physiological agonists (Figure 1E). We then assessed the effect of oxPCCD36 on the integrin αIIbβ3 activation in platelets isolated from WT and TLR2−/− mice. Genetic deficiency of TLR2 significantly reduced integrin αIIbβ3 activation in response to KODA-PC in murine platelets (Figure 1A). In contrast, activation of murine platelets by ADP and thrombin was not affected by the lack of TLR2 expression, indicating specificity of this effect (Figure 1A, Supplemental Figure IE). The TLR2 blocking antibody also significantly inhibited the platelet P-selectin expression in response to two other members of oxPCCD36 family - HODA-PC and KOOA-PC, as well as oxLDL, a lipoprotein which contains all of the members of oxPCCD36 family (Supplemental Figure IF). This result indicates that TLR2 plays a general role in platelet activation by oxPCCD36.
TLR2 functions as a heterodimer, either with TLR1 or TLR6. We observed a significant reduction of integrin αIIbβ3 activation induced by oxPCCD36 in platelets of TLR6−/− mice as compared to WT platelets (Figure 1F). In agreement with this result, the blocking antibody to TLR6 significantly inhibited P-selectin expression induced by oxPCCD36 in human platelets (Figure 1G, 1H), indicating that in these settings, TLR2 cooperates with TLR6. This conclusion is further supported by the lack of an inhibitory effect of a TLR1 blocking antibody on oxPCCD36-induced P-selectin expression in human platelets (Supplemental Figure ID). We verified the activity and specificity of blocking antibodies and blocking peptides in a series of control experiments using platelet activation by synthetic and bacterial ligands for TLRs, as well as in HEK-Blue™-hTLR2/9 cells reporter assay (Supplemental Figure IIA–IID). In agreement with previous reports,16–18 we confirmed the presence of TLR2 and TLR6 in both human and murine platelets by flow cytometry and by Western blotting (Supplemental figure IIIA–IIIE). TLR2 expression was absent in platelets of TLR2−/− mice, as anticipated (Supplemental Figure IIIF, IIIG), while TLR1 and TLR6 expression was similar to platelets of WT mice (Supplemental Figure IIIG).
oxPCCD36 induce formation of TLR2/TLR6/CD36/MyD88 complex
TLRs are known to induce signaling via the assembly of multiprotein complexes. We next tested whether CD36 and TLR2 in platelets form a complex in response to oxPCCD36. A co-immunoprecipitation assay revealed that oxPCCD36 stimulation induced the formation of a TLR2/CD36 complex in human platelets (Figure 2A). We also detected TLR6 and MyD88 in this complex (Figure 2B, 2C). A co-immunoprecipitation with the anti-MyD88 antibody confirmed the induction of CD36/TLR2/TLR6/MyD88 multiprotein complex formation by oxPCCD36 in platelets (Figure 2D). We verified the specificity of the complex formation by using synthetic bacterial ligands for TLR1/TLR2 and TLR2/TLR6 (Pam3CSK4 and Pam2CSK4 respectively), for platelet activation (Supplemental Figure IIE, IIF). We detected increased association of CD36 specifically in the complex with TLR2/TLR6.
Figure 2. oxPCCD36 induce formation of CD36/TLR2/TLR6/MyD88 complex.
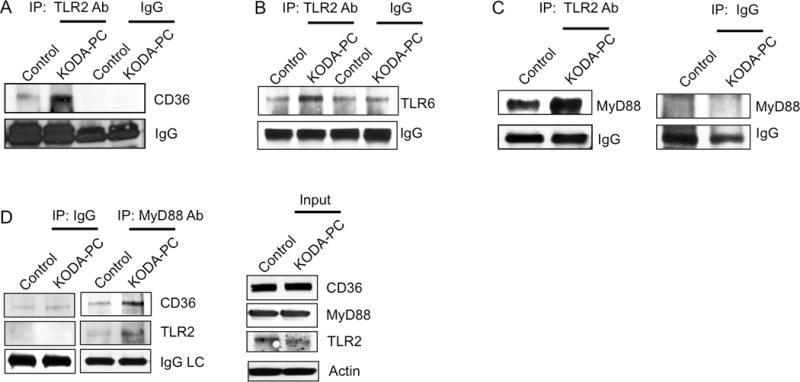
Human platelets were isolated by gel filtration and stimulated with 20 μM oxPCCD36 (KODA-PC) for 15 minutes, lysed with RIPA buffer and 300 μg of lysate protein was immuno-precipitated with TLR2 antibody. CD36 (A), TLR6 (B) and MyD88 (C) were then detected in precipitate by immunoblotting. (D) MyD88 was immunoprecipitated from lysate of human platelets and TLR2 and CD36 were detected in precipitate by immunoblotting. As a control TLR2, CD36, and MyD88 were detected in platelet lysates by immunoblotting.
Innate immune signaling cascade is activated in platelets by oxPCCD36
Toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP), an adaptor protein in the TLR pathway, facilitates the association of TLRs with MyD88. The TIRAP blocking peptide significantly inhibited platelet P-selectin expression induced by oxPCCD36, while control peptide had no effect (Figure 3A, 3B), demonstrating the involvement of TIRAP in the signaling pathway induced by oxPCCD36 in platelets. The effect was specific for oxPCCD36, since this peptide did not affect platelet activation by physiological agonists ADP and thrombin (Figure 3B).
Figure 3. Innate immune signaling cascade is activated in platelets by oxPCCD36.
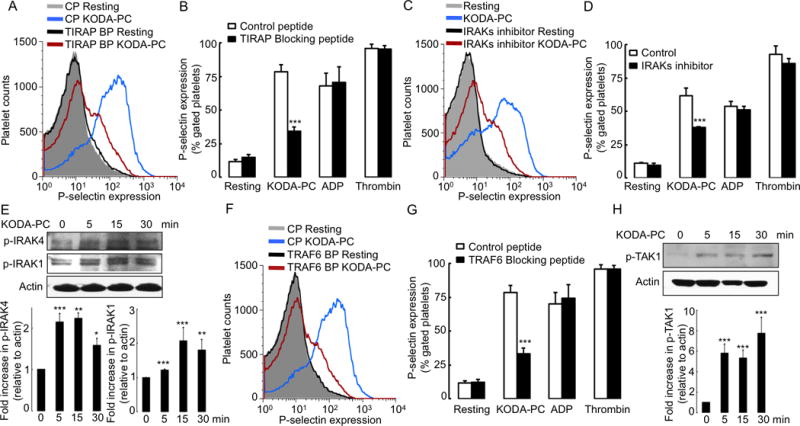
Human platelets were isolated by gel filtration, and pre-incubated with (A, B) 2 μM TIRAP blocking peptide (BP) or control peptide (CP), (C–D) IRAK1/4 inhibitor (IRAKs inhibitor), (F–G) TRAF6 blocking peptide (BP) or control peptide (CP), followed by stimulation with oxPCCD36 (KODA-PC, 20 μM) or ADP (10 μM) or thrombin (0.05 U/ml). P-selectin expression was assessed by FACS analysis. FACS analysis data are presented as histograms as well as bar graphs (mean ± SD) from 3 independent experiments. (E, H) Human platelets were stimulated with oxPCCD36 (KODA-PC, 20 μM) for increasing time and phosphorylation of IRAK4 and IRAK1 (E), and (H) TAK1 were assessed by Western blot analysis using phosphospecific antibodies for respective proteins. *** p<0.001, ** p<0.01, * p<0.05 vs control.
Interleukin-1 receptor associated kinases (IRAKs) are involved in the innate immune signaling cascade downstream of MyD88. A pharmacological inhibitor of IRAKs (IRAK-1/4 Inhibitor) significantly suppressed platelet activation induced by oxPCCD36 (Figure 3C, 3D). IRAK4 and IRAK1 were phosphorylated in response to oxPCCD36 (Figure 3E), indicating their activation by oxPCCD36. As previously shown, MyD88 activates the cGMP dependent protein kinase pathway during LPS induced platelet aggregation via TLR4.28 However, we found that pharmacological inhibitors of cGMP dependent pathway had no effect on oxPCCD36 induced platelet activation (data not shown).
E3 ubiquitin-protein ligase TNF receptor associated factor 6 (TRAF6) is a crucial signaling molecule downstream to IRAKs. The TRAF6 blocking peptide significantly inhibited platelet activation by oxPCCD36, whereas the control peptide had no effect (Figure 3F, 3G). TRAF6 activates a number of downstream kinases in the innate immune signaling pathway, including transforming growth factor beta-activated kinase 1 (TAK1), which in turn activates several key intracellular pathways.29–36 We observed increased TAK1 phosphorylation in platelets in response to oxPCCD36 (Figure 3H). Thus, multiple elements of innate immune signaling cascade are activated in platelets by oxPCCD36.
oxPCCD36 activate Src and Fyn downstream of innate immune signaling in platelets
We have recently shown that oxPCCD36-induced platelet activation is Src family kinase (SFK) dependent.11 The TRAF6 blocking peptide significantly inhibited SFK phosphorylation induced in platelets by oxPCCD36 (Figure 4A), demonstrating that SFK activation is downstream of TRAF6 activation. To identify SFK members that are activated downstream of CD36/TLR2/TLR6, we assessed platelet activation in Src−/− and Lyn−/− mice. We observed reduced activation of platelets of Src−/− mice by oxPCCD36 as compared to WT platelets (Figure 4B), while we detected no effect of Lyn deficiency (Figure 4C). The expressions of major platelet receptors were similar in Src−/− and Lyn−/− mice as compared to WT mice (Supplemental Figure VA–VD). We also assessed the phosphorylation of various members of SFK by performing immunoprecipitation using p-tyrosine antibody and subsequent immunoblotting for SFKs. This approach revealed an induction of phosphorylation of c-Src and Fyn (Figure 4D–4E), but not Lyn (data not shown). c-Src phosphorylation in response to oxPCCD36 was confirmed using a reverse IP approach (Figure 4F).
Figure 4. Src and Fyn are involved in platelet activation induced by oxPCCD36 downstream of TRAF6.
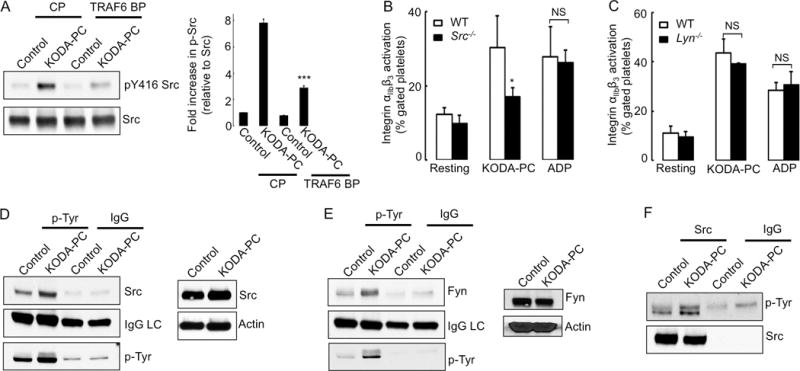
(A) Human platelets were pre-incubated with 2 μM TRAF6 blocking peptide (BP) or control peptide (CP) followed by stimulation with oxPCCD36 (KODA-PC, 20 μM) for 5 minutes. Phosphorylation of Src was determined by Western blot analysis using phosphospecific antibody. Quantified data (mean ± SD) of at least 3 independent experiments are shown in the right panel. (B, C) Platelets were isolated from WT, Src−/−, Lyn−/− mice and stimulated with oxPCCD36 (KODA-PC, 20 μM) or ADP (10 μM). Integrin αIIbβ3 activation was assessed by FACS analysis and presented as mean ± SD of at least 3 independent experiments. (D–F) Human platelets were isolated by gel filtration and stimulated with oxPCCD36 (KODA-PC, 20 μM) for 5 minutes, lysed and 200 μg of protein lysate was immunoprecipitated with (D, E) phospho-tyrosine (p-Tyr) antibody or (F) c-Src antibody then c-Src (D), Fyn (E), p-Tyr (F) were detected by immunoblotting using Src and Fyn or p-Tyr primary antibody and light chain specific secondary antibody. IgG light chain (IgG LC) and p-Tyr, c-Src are shown as control. c-Src and Fyn were detected by Western blot analysis (D, E). * p<0.05, *** p<0.001, and NS p>0.05 vs control.
We have previously shown that oxPCCD36 induce platelet activation in a CD36 dependent manner via the SFK/Syk/PLCγ2 pathway.11 We now show that the TLR2 blocking antibody prevented the phosphorylation of Syk and PLCγ2 (Figure 5A) induced by oxPCCD36. Furthermore, both TLR2 deficiency and TLR6 deficiency blunted PLCγ2 phosphorylation in response to oxPCCD36 (Figure 5B). These findings demonstrate that both TLR2 and TLR6 are required for the phosphorylation of Syk and PLCγ2 induced by oxPCCD36.
Figure 5. TLR2 and TLR6 are required for activation of PLCg2 and Syk by oxPCCD36 in human and murine platelets.
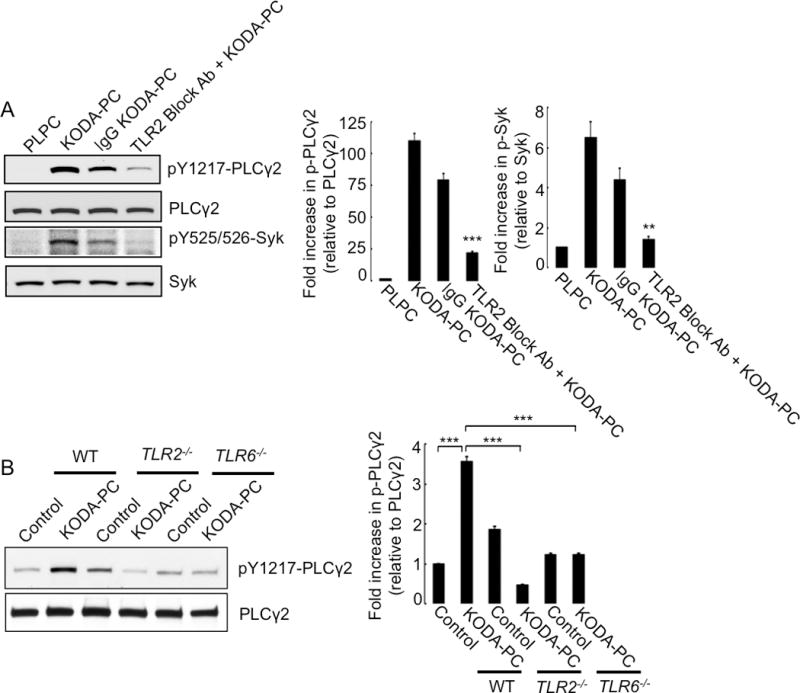
(A) Human platelets were pre-incubated with 4 μg TLR2 blocking antibody or control non-immune IgG, followed by stimulation with oxPCCD36 (KODA-PC, 20 μM) or control phospholipid PLPC for 5 minutes. Phosphorylations of Syk and PLCg2 were determined by Western blotting using phosphospecific antibodies of respective proteins. (B) Platelets were isolated from WT, TLR2−/−, and TLR6−/− mice, stimulated with oxPCCD36 (KODA-PC, 20 μM) for 5 minutes, and PLCg2 phosphorylation was assessed by Western blot analysis using phosphospecific antibody. Quantified data are shown as bar graphs (mean ± SD of at least 3 independent experiments) in the right panels. *** p<0.001, ** p<0.01 vs corresponding control.
TLR2 plays a critical role in platelet hyperreactivity induced by hyperlipidemia
CD36 mediated recognition of oxPCCD36 plays a critical role in platelet hyperreactivity and in prothrombotic phenotype associated with hyperlipidemia.10 We next tested if TLR2 deficiency plays a role in this process. While platelet activation by suboptimal concentrations of ADP was significantly higher in platelets of ApoE−/− mice fed a Western diet, as compared to ApoE−/− mice fed a chow diet, this hyperreactivity was abolished in platelets isolated from hyperlipidemic ApoE−/−/TLR2−/− mice (Figure 6A). Importantly, TLR2 deficiency had no effect on platelet activation in response to increasing concentrations of ADP (1–10μM), as compared to WT mice (Supplemental Figure IVA). Furthermore, the TLR2 blocking antibody suppressed platelet activation induced by preincubation of isolated human platelets with human plasma containing increased levels of endogenous oxPCCD36, yet the blocking antibody had no effect on platelets pre-incubated with human plasma containing low levels of endogenous oxPCCD36 (Figure 6B). These results demonstrate that TLR2 contributes to platelet activation both in human and murine platelets. We also tested the role of TLR2 on the background of CD36 deficiency by comparing platelets from ApoE−/−/CD36−/− and ApoE−/−/CD36−/−/TLR2−/− mice, which were fed either a chow diet or a Western diet. We observed that the lack of TLR2 expression suppressed platelet hyperreactivity (albeit to a lesser degree), even in the absence of CD36 (Figure 6A). Interestingly, responses of platelets from hyperlipidemic ApoE−/−/TLR2−/− mice were similar to platelets from ApoE−/−/CD36−/−/TLR2−/− mice (Figure 6A). This finding suggests that in addition to cooperation with CD36, TLR2 also contributes to platelet hyperreactivity independently of CD36, as we recently proposed.9 Importantly, plasma cholesterol levels were similar in ApoE−/−/CD36−/− and ApoE−/−/CD36−/−/TLR2−/− mice (Supplemental Figure IVB), indicating that the extent of hyperlipidemia is not affected by TLR2 deficiency.
Figure 6. TLR2 and TLR6 play a critical role in platelet hyperreactivity and accelerated thrombosis induced by hyperlipidemia.
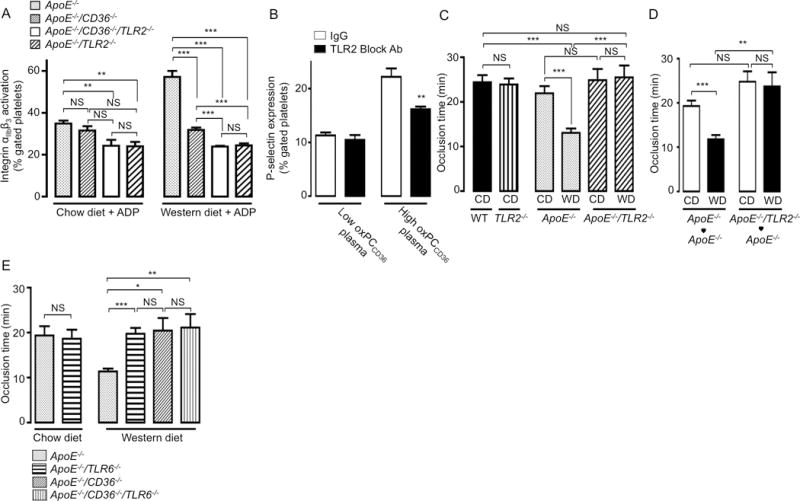
(A) Platelet rich plasma (PRP) was isolated from ApoE−/−, ApoE−/−/TLR2−/−, ApoE−/−/CD36−/−, and ApoE−/−/CD36−/−/TLR2−/− mice fed either a chow diet or a Western diet. Integrin αIIbβ3 activation after stimulation with suboptimal concentration of (3 μM) ADP was determined by FACS analysis. Data presented as mean ± SD of at least 3 independent experiments. (B) Human platelets were isolated by gel filtration from healthy control donors and pre-incubated for 30 min either with (4 μg/ml) TLR2 blocking antibody or isotype matched control IgG followed by 30 min incubation with citrated human plasma samples containing either low endogenous levels of oxPCCD36 (<1 μM, N=5) or relatively high endogenous levels of oxPCCD36 (3.1±0.9 μM, N=3). P-selectin expression was then assessed by FACS analysis and expressed as mean ± SD. (C–E) FeCl3-induced carotid artery thrombosis times were assessed as described in Methods section, in (C) WT and TLR2−/− mice fed a chow diet (CD), and ApoE−/− and ApoE−/−/TLR2−/− mice, fed either a chow (CD) or a Western diet (WD); (D) ApoE−/− bone marrow chimeric mice with ApoE−/− or ApoE−/−/TLR2−/− bone marrow fed either a chow diet (CD) or a Western diet (WD); N≥6 (E) ApoE−/−, ApoE−/−/TLR6−/−, ApoE−/−/CD36−/−, and ApoE−/−/CD36−/−/TLR6−/− mice fed either a chow diet (CD) or a Western diet (WD), N≥6. * p<0.05, ** p<0.01, *** p<0.001, and NS p>0.05.
TLR2 deficiency protects hyperlipidemic mice from accelerated thrombosis
The contribution of TLR2 to platelet hyperreactivity suggests that the prothrombotic phenotype of hyperlipidemic mice may depend on TLR2. To test this hypothesis, we used a FeCl3-induced carotid artery thrombosis assay and compared the vessel occlusion times in sex- and age-matched groups of ApoE−/− and ApoE−/−/TLR2−/− mice, fed either a chow diet or a Western diet. We observed no significant difference in occlusion time between the groups fed a chow diet (Figure 6C). We also observed no difference in the occlusion time of FeCl3-induced carotid artery thrombosis between WT and TLR2−/− mice (Figure 6C), indicating that TLR2 has no significant role in thrombosis in normolipidemic conditions. However, the time to complete thrombotic occlusion was significantly extended in ApoE−/−/TLR2−/− mice fed a Western diet as compared to ApoE−/− mice fed a Western diet (Figure 6C), and was similar to that in mice on chow diet. We observed a similar effect using ApoE−/− chimeras with ApoE−/− or ApoE−/−/TLR2−/− bone marrow, indicating that the phenotype depends on TLR2 expression in bone marrow derived (presumably platelets) cells (Figure 6D). Surface expression of TLR2 did not changed in hyperlipidemic ApoE−/− mice (Supplemental Figure IVC), and plasma concentrations of total cholesterol and triglycerides were similar in hyperlipidemic ApoE−/− and ApoE−/−/TLR2−/− mice (Supplemental Figure IVD, IVE). We also found no effect of TLR2 deficiency on the expression of major platelet receptors (Supplemental Figure IIIG, IVF, VA–VD).
Effect of TLR6 deficiency on thrombosis is similar to that of CD36 deficiency
As we have shown that TLR6 is required for oxPCCD36 induced platelet activation, we next tested its contribution to accelerated thrombosis in hyperlipidemia. We compared sex- and age-matched ApoE−/−, ApoE−/−/CD36−/−, ApoE−/−/CD36−/−/TLR6−/−, and ApoE−/−/TLR6−/− mice fed a Western diet using FeCl3 induced carotid artery thrombosis assay. While control experiments demonstrated that TLR6 does not contribute to thrombosis in mice on a chow diet (Figure 6E), we observed that the time to complete occlusion was significantly extended in TLR6 deficient ApoE−/− mice on a Western diet (Figure 6E). TLR6 deficiency had no significant effect on thrombosis in the ApoE−/−/CD36−/− background, as the occlusion times in hyperlipidemic ApoE−/−/CD36−/−, ApoE−/−/CD36−/−/TLR6−/−, and ApoE−/−/TLR6−/− mice were similar (Figure 6E). This result indicates that TLR6 promotes thrombosis in hyperlipidemic condition exclusively in cooperation with CD36.
DISCUSSION
Patients with enhanced platelet reactivity are at increased risk for cardiovascular events.4, 37–39 Enhanced platelet reactivity is associated with chronic and acute inflammation, infections, diabetes, and a number of pathophysiological states related to dyslipidemia, including atherosclerosis, diabetes, and metabolic syndrome.3–8 Previously we have linked platelet hyperreactivity in dyslipidemia to accumulation in circulation of specific oxidized phospholipids, oxPCCD36, which activate platelets via the scavenger receptor CD36. In the current manuscript, we established that signaling via the platelet innate immune system, specifically via TLR2/TLR6, is an integral part of platelet responses to oxPCCD36. We further demonstrated that TLR2 and TLR6 are required for accelerated thrombosis induced by hyperlipidemia.
We used a combination of multiple complementary in vivo and in vitro approaches and tools to demonstrate the general role of TLRs, and specifically TLR2 and TLR6, in platelet function in the setting of hyperlipidemia. In vitro results are based on studies using platelets from mice with genetic deficiencies of MyD88, TLR2, or TLR6 on wild type or ApoE−/− background. To demonstrate the role of TLRs in human platelets, we used a number of blocking antibodies and peptides, with activity and specificity verified in multiple control experiments using bacterial ligands for TLRs. Our conclusions on the key role of TLR2 and contribution of TLR6 in thrombosis in vivo, specifically in the setting of hyperlipidemia, are supported by in vivo thrombosis assay using TLR2−/− or TLR6−/− on wild type or ApoE−/− background mice fed a chow or Western type diet. The specific role of bone marrow derived TLR2 is supported by the bone marrow transplantation approach. To compare the role of TLR2 and TLR6 to that of CD36, we generated and performed experiments in ApoE−/−/CD36−/−/TLR2−/− and ApoE−/−/CD36−/−/TLR6−/− triple knockout mice. These experiments revealed that platelet TLR2/TLR6 is required for platelet activation by oxidized phospholipids, and for platelet hyperreactivity and accelerated thrombosis induced by hyperlipidemia. These experiments and our previous studies9 further suggest that the role of TLR6 is restricted to cooperation with CD36, while TLR2 contributes to thrombosis in cooperation with CD36, as well as independently of CD36. In vitro experiments showed that responses of platelets isolated from normolipidemic mice to physiologically relevant agonists such as ADP, thrombin, and the GPVI ligand convulxin are TLR2 independent. In agreement with this result, we observed no TLR contribution to platelet function or thrombosis in normolipidemic conditions.
Toll like receptors play a key role in innate and adaptive immunity and initiate inflammatory responses to pathogen associated molecular patterns, mostly by regulating gene expression. Recent studies have also implicated TLRs in recognition of the endogenous ligands, including so-called “altered-self” ligands.23, 24 Many components of the innate immune signaling machinery are expressed and functional in platelets, despite the absence of a nucleus.9, 17, 20, 21 In this manuscript, we confirmed the expression of TLR2 and TLR6 in murine and human platelets using Western blotting and their surface expression using flow cytometry. While the role of platelet TLRs in response to pathological ligands in the circulation has started to emerge21, 28, 40, studies of the endogenous ligands that may regulate thrombosis by engaging TLRs on platelets are very limited. We have recently reported that TLR2 in complex with TLR1 recognizes carboxyalkylpyrrole-phosphatidylethanolamine derivatives, a novel group of end products of lipid peroxidation that are present in hyperlipidemic plasma.9 Engagement of TLR2/TLR1 by these derivatives leads to platelet activation in vitro and accelerates thrombosis in vivo. The current study demonstrates that by collaborating with CD36, and mediating response to oxPCCD36, TLR2 plays an even broader role in platelet responses to oxidative stress than previously thought. A deficiency of TLR2 in hyperlipidemic ApoE−/− mice abolished accelerated thrombosis phenotype in these mice.
CD36 (or SR-BII) is an abundant platelet glycoprotein that belongs to a family of class B scavenger receptors.41 CD36 is involved in the recognition of a diverse array of pathological and physiological ligands, including members of a family of specific oxidized phospholipids, oxPCCD36.42 CD36 mediated the recognition of oxPCCD36 in oxidized LDL and subsequent binding and internalization of oxidized LDL by macrophages in the arterial wall contributes to atherogenesis in hyperlipidemic mice.43−45 Platelet CD36 serves as a sensor of oxPCCD36 in circulation. Interaction of oxPCCD36 and CD36 has been characterized in a number of studies. oxPCCD36 bind via an electrostatic mechanism to region aa157–171 of CD36.14, 15 This binding is required for inducing platelet activation, as oxidized phospholipids that bind weakly to CD36, such as oxovaleroyl-PC, do not activate platelets.10, 12 Thus binding to CD36 seems to be a first and required step in the induction of CD36/TLR2/TLR6 complex and the activation of signaling downstream of TLR2/TLR6.
oxPCCD36 are relatively early products of phospholipid peroxidation, and they accumulate in the circulation in hyperlipidemic animals and subjects with low HDL.10 The plasma concentration of individual oxPCCD36 is 0.5–3.0 μM, and combined concentrations of all oxPCCD36 can reach up to 20 μM or higher in individual animals.10 In relatively normolipidemic human samples, combined oxPCCD36 concentration can reach 4.6 μM.10 These levels are sufficient to induce mild platelet activation and promote platelet hyperreactivity in vivo and in vitro.
The interaction of scavenger receptors with other pattern recognition receptors, including TLRs, attracted significant attention.23, 24, 46, 47 Such interactions potentiate the function of pattern recognition receptors and augment the inflammatory response to microbial ligands.46, 48 It has been suggested that epitopes generated by peroxidation of lipids and lipoproteins resemble microbial wall epitopes, and recognition of such epitopes by scavenger receptors plays a role in homeostasis and immunity.46, 49 TLR2/TLR6 complex is responsible for responses to gram-positive bacteria and fungi,50, 51 and contribution of CD36 expressed on immune cells to this process has been documented.52 Our current study demonstrates that the “side effect” of such collaboration between CD36 and TLR2/TLR6 is an excessive platelet responses to physiological agonists in hyperlipidemic conditions.
Previous studies have shown that macrophage CD36 cooperates with TLR2 upon binding of oxidized phospholipids to induce apoptosis.23 Our current study demonstrates that these two receptors also cooperate in platelets, but the downstream events are different. While the activation of macrophages requires ERK and is independent of SFK, activation of platelets by oxidized phospholipids is independent of ERK and proceeds via the SFK-Syk-PLCγ2 pathway.11 We have now shown that in addition to CD36, TLR2 and TLR6 are also required for activation of the SFK-Syk-PLCγ2 pathway in platelets by oxPCCD36. Signaling events downstream of CD36/TLR receptor complex include TIRAP-MyD88 dependent activation of TRAF6, leading to activation of c-Src and Fyn kinases (Figure 7). The critical role of TRAF6 in SFK activation is demonstrated by the inhibitory effect of the TRAF6 blocking peptide on SFK phosphorylation induced by oxPCCD36. This finding identifies a new connection between CD36 and integrin activation pathway via the innate immune signaling pathway.
Figure 7. Proposed model of platelet activation induced by oxPCCD36.
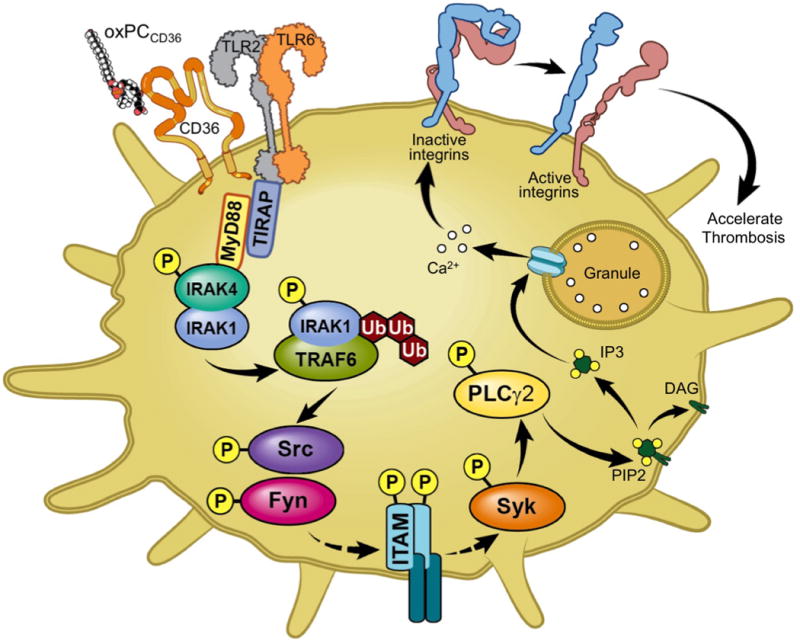
oxPCCD36 accumulate in the circulation in hyperlipidemia, bind to platelet CD36 and induce formation of CD36/TLR2/TLR6 complex and activate downstream signaling via innate immune signaling cascade, TIRAP-MyD88-IRAK1/4-TRAF6, leading to platelet integrin αIIbβ3 activation via the SFK-Syk-PLCg2 pathway.
In summary, our study demonstrates the critical contribution of the platelet innate immune system and, specifically, TLR2 to the platelet hyperreactivity and prothrombotic state associated with hyperlipidemia. Furthermore, our findings identify the cooperation between CD36 and TLR2/TLR6 as an attractive therapeutic target.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
A specific group of oxidized phospholipids accumulate in the circulation in conditions of dyslipidemia and oxidative stress and promote platelet reactivity and thrombosis via platelet scavenger receptor CD36.
Platelets express functional Toll-like receptors and other components of the innate immune system and can sense pathogens in circulation.
What New Information Does This Article Contribute?
In vitro studies showed that platelet TLR2 and TLR6 collaborate with CD36 and engage the innate immune signaling cascade to induce platelet activation by oxidized phospholipids.
In vivo studies demonstrated that platelet TLR2 and TLR6 play a key role in the enhance platelet reactivity and prothrombotic state associated with hyperlipidemia.
Enhanced platelet reactivity, common in many pathophysiological conditions, is associated with increased atherothrombotic risk. Enhanced platelet reactivity, specifically in dyslipidemia, has previously been mechanistically linked to the plasma accumulation of oxidized phospholipids (oxPCCD36), which bind to and activate platelets via pattern-recognition receptor CD36. Although significant progress has been made in recent years towards understanding the interaction of CD36 with oxidized phospholipids and downstream signaling in platelets, many details of the cellular and molecular mechanism are still not known. In this study, we demonstrate that TLR2/TLR6 and the platelet innate immune signaling cascade are playing the critical role in platelet activation by oxPCCD36. We further established that platelet TLR2 and TLR6 are required for induction of prothrombotic phenotype in hyperlipidemia, but play no significant role in thrombosis in normolipidemic conditions. The role of TLR6 is restricted to cooperation with CD36, while TLR2 contributes to thrombosis in cooperation with CD36, as well as independently of CD36. Our study is the first to show the role of platelet innate immune system in sensing endogenous lipid-peroxidation ligands and the key role of this system in prothrombotic phenotype associated with hyperlipidemia. These findings establish a novel connection between the immune system and thrombosis.
Acknowledgments
The authors thank Lifang Zhang, Jessica Altemus, and Miroslava Tischenko for technical assistance, and Jessica Altemus, Emelye Crehore, and Samantha Stefl for proof reading of the manuscript.
SOURCES OF FUNDING.
This work was supported in part by National Institutes of Health grants HL077213, HL073311, HL126738, HL071625 and AHA postdoctoral fellowship grant 14POST20230027.
Nonstandard Abbreviations and Acronyms
- ADP
Adenosine diphosphate
- cGMP
Cyclic guanosine monophosphate
- ERK
Extracellular signal-regulated kinases
- GPVI
Glycoprotein VI
- HDL
High density lipoprotein
- HODA-PC
9-hydroxy-12-oxo-10-dodecenoic acid esters of 2-lyso-PC
- IRAK
Interleukin-1 receptor associated kinases
- KODA-PC
9-keto-12-oxo-10-dodecenoic acid ester of 2-lyso-phosphocholine
- KOOA-PC
5-keto-8-oxo-6-octenoic acid esters of 2-lyso-PC
- LDL
Low density lipoprotein
- MyD88
Myeloid-differentiation factor 88
- oxLDL
Oxidized low density lipoprotein
- PAPC
1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine
- PLPC
1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine
- PLCγ2
Phospholipase Cγ2
- PRP
Platelet rich plasma
- SFK
Src family kinase
- SR-BII
Scavenger receptor B II
- Syk
Spleen tyrosine kinase
- TAK1
Transforming growth factor beta-activated kinase 1
- TIRAP
Toll-interleukin 1 receptor (TIR) domain containing adaptor protein
- TRAF6
TNF receptor associated factor 6
Footnotes
AUTHOR CONTRIBUTIONS
S.B. designed the research, performed the experiments, analyzed the data, and wrote the manuscript. A.Z. and D.G. performed experiments and analyzed the data. T.V.B and E.A.P. designed the research, analyzed the data, and wrote the manuscript.
DISCLOSURES
The authors have no conflict of interest to disclose.
References
- 1.Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5:150–159. doi: 10.1038/ncpendmet1066. [DOI] [PubMed] [Google Scholar]
- 2.Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J. 2016;37:1113–1121. doi: 10.1093/eurheartj/ehv718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacoste L, Lam JY, Hung J, Letchacovski G, Solymoss CB, Waters D. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation. 1995;92:3172–3177. doi: 10.1161/01.cir.92.11.3172. [DOI] [PubMed] [Google Scholar]
- 4.Carvalho AC, Colman RW, Lees RS. Platelet function in hyperlipoproteinemia. N Engl J Med. 1974;290:434–438. doi: 10.1056/NEJM197402212900805. [DOI] [PubMed] [Google Scholar]
- 5.Cipollone F, Mezzetti A, Porreca E, Di Febbo C, Nutini M, Fazia M, Falco A, Cuccurullo F, Davi G. Association between enhanced soluble CD40L and prothrombotic state in hypercholesterolemia: Effects of statin therapy. Circulation. 2002;106:399–402. doi: 10.1161/01.cir.0000025419.95769.f0. [DOI] [PubMed] [Google Scholar]
- 6.Davi G, Averna M, Catalano I, Barbagallo C, Ganci A, Notarbartolo A, Ciabattoni G, Patrono C. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation. 1992;85:1792–1798. doi: 10.1161/01.cir.85.5.1792. [DOI] [PubMed] [Google Scholar]
- 7.Davi G, Romano M, Mezzetti A, Procopio A, Iacobelli S, Antidormi T, Bucciarelli T, Alessandrini P, Cuccurullo F, Bittolo Bon G. Increased levels of soluble P-selectin in hypercholesterolemic patients. Circulation. 1998;97:953–957. doi: 10.1161/01.cir.97.10.953. [DOI] [PubMed] [Google Scholar]
- 8.Stuart MJ, Gerrard JM, White JG. Effect of cholesterol on production of thromboxane b2 by platelets in vitro. N Engl J Med. 1980;302:6–10. doi: 10.1056/NEJM198001033020102. [DOI] [PubMed] [Google Scholar]
- 9.Biswas S, Xin L, Panigrahi S, Zimman A, Wang H, Yakubenko VP, Byzova TV, Salomon RG, Podrez EA. Novel phosphatidylethanolamine derivatives accumulate in circulation in hyperlipidemic ApoE−/− mice and activate platelets via TLR2. Blood. 2016;127:2618–2629. doi: 10.1182/blood-2015-08-664300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimman A, Titz B, Komisopoulou E, Biswas S, Graeber TG, Podrez EA. Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PLoS One. 2014;9:e84488. doi: 10.1371/journal.pone.0084488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 13.Zimman A, Podrez EA. Regulation of platelet function by class B scavenger receptors in hyperlipidemia. Arterioscler Thromb Vasc Biol. 2010;30:2350–2356. doi: 10.1161/ATVBAHA.110.207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao D, Ashraf MZ, Kar NS, Lin D, Sayre LM, Podrez EA. Structural basis for the recognition of oxidized phospholipids in oxidized low density lipoproteins by class B scavenger receptors CD36 and SR-BI. J Biol Chem. 2010;285:4447–4454. doi: 10.1074/jbc.M109.082800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kar NS, Ashraf MZ, Valiyaveettil M, Podrez EA. Mapping and characterization of the binding site for specific oxidized phospholipids and oxidized low density lipoprotein of scavenger receptor CD36. J Biol Chem. 2008;283:8765–8771. doi: 10.1074/jbc.M709195200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, Ejiri J, Kobayashi S, Hirata K, Kawashima S, Yokoyama M. Expression of Toll-like receptors on human platelets. Thromb Res. 2004;113:379–385. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 17.Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, Byzova TV, Podrez EA. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ Res. 2013;112:103–112. doi: 10.1161/CIRCRESAHA.112.274241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83:196–198. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 19.D’Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, Yin H, Schattner M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J Thromb Haemost. 2015;13:839–850. doi: 10.1111/jth.12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beaulieu LM, Freedman JE. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb Res. 2010;125:205–209. doi: 10.1016/j.thromres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nature reviews Immunology. 2011;11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 22.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, Montano E, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. Cd36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curtiss LK, Tobias PS. Emerging role of Toll-like receptors in atherosclerosis. J Lipid Res. 2009;50:S340–345. doi: 10.1194/jlr.R800056-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun M, Deng Y, Batyreva E, Sha W, Salomon RG. Novel bioactive phospholipids: Practical total syntheses of products from the oxidation of arachidonic and linoleic esters of 2-lysophosphatidylcholine(1) J Org Chem. 2002;67:3575–3584. doi: 10.1021/jo0105383. [DOI] [PubMed] [Google Scholar]
- 27.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 28.Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. 2009;182:7997–8004. doi: 10.4049/jimmunol.0802884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 30.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem. 2007;282:4102–4112. doi: 10.1074/jbc.M609503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Besse A, Lamothe B, Campos AD, Webster WK, Maddineni U, Lin SC, Wu H, Darnay BG. Tak1-dependent signaling requires functional interaction with TAB2/TAB3. J Biol Chem. 2007;282:3918–3928. doi: 10.1074/jbc.M608867200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamothe B, Webster WK, Gopinathan A, Besse A, Campos AD, Darnay BG. TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun. 2007;359:1044–1049. doi: 10.1016/j.bbrc.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, Wang PX, Huang Z, Li H. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1-dependent signalling. Nat Commun. 2016;7:11267. doi: 10.1038/ncomms11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moon G, Kim J, Min Y, Wi SM, Shim JH, Chun E, Lee KY. Phosphoinositide-dependent kinase-1 inhibits TRAF6 ubiquitination by interrupting the formation of TAK1-TAB2 complex in TLR4 signaling. Cell Signal. 2015;27:2524–2533. doi: 10.1016/j.cellsig.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 35.He A, Ji R, Shao J, He C, Jin M, Xu Y. TLR4-MyD88-TRAF6-TAK1 complex-mediated NF-kB activation contribute to the anti-inflammatory effect of V8 in LPS-induced human cervical cancer SiHa cells. Inflammation. 2016;39:172–181. doi: 10.1007/s10753-015-0236-8. [DOI] [PubMed] [Google Scholar]
- 36.Chen IT, Hsu PH, Hsu WC, Chen NJ, Tseng PH. Polyubiquitination of transforming growth factor beta-activated kinase 1 (TAK1) at lysine 562 residue regulates TLR4-mediated JNK and p38 MAPK activation. Sci Rep. 2015;5:12300. doi: 10.1038/srep12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabbani SS, Watkins MW, Ashikaga T, Terrien EF, Holoch PA, Sobel BE, Schneider DJ. Platelet reactivity characterized prospectively: A determinant of outcome 90 days after percutaneous coronary intervention. Circulation. 2001;104:181–186. doi: 10.1161/01.cir.104.2.181. [DOI] [PubMed] [Google Scholar]
- 38.Everett CJ, Mainous AG, 3rd, Koopman RJ, Diaz VA. Predicting coronary heart disease risk using multiple lipid measures. Am J Cardiol. 2005;95:986–988. doi: 10.1016/j.amjcard.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs I, Frossard M, Spiel A, Riedmuller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost. 2006;4:2547–2552. doi: 10.1111/j.1538-7836.2006.02239.x. [DOI] [PubMed] [Google Scholar]
- 40.Garraud O, Berthet J, Hamzeh-Cognasse H, Cognasse F. Pathogen sensing, subsequent signalling, and signalosome in human platelets. Thromb Res. 2011;127:283–286. doi: 10.1016/j.thromres.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 41.PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M, Herz J, Kobzik L, Krieger M, Loike J, McVicker B, Means TK, Moestrup SK, Post SR, Sawamura T, Silverstein S, Speth RC, Telfer JC, Thiele GM, Wang XY, Wright SD, El Khoury J. A consensus definitive classification of scavenger receptors and their roles in health and disease. J Immunol. 2017;198:3775–3789. doi: 10.4049/jimmunol.1700373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, Cohen PA, Frazier WA, Hoff HF, Hazen SL. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–1108. doi: 10.1172/JCI8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kennedy DJ, Kuchibhotla SD, Guy E, Park YM, Nimako G, Vanegas D, Morton RE, Febbraio M. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:1481–1487. doi: 10.1161/ATVBAHA.109.191940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nature reviews Immunology. 2013;13:621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 47.Kim YW, Yakubenko VP, West XZ, Gugiu GB, Renganathan K, Biswas S, Gao D, Crabb JW, Salomon RG, Podrez EA, Byzova TV. Receptor-mediated mechanism controlling tissue levels of bioactive lipid oxidation products. Circ Res. 2015;117:321–332. doi: 10.1161/CIRCRESAHA.117.305925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukhopadhyay S, Varin A, Chen Y, Liu B, Tryggvason K, Gordon S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood. 2011;117:1319–1328. doi: 10.1182/blood-2010-03-276733. [DOI] [PubMed] [Google Scholar]
- 49.Hartvigsen K, Chou MY, Hansen LF, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. The role of innate immunity in atherogenesis. J Lipid Res. 2009;50:S388–393. doi: 10.1194/jlr.R800100-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.