

Abstract
Protein misfolding and aggregation are key pathological features of many neurodegenerative diseases including Parkinson’s disease (PD) and other forms of human Parkinsonism. PD is a complex and multifaceted disorder whose etiology is not fully understood. However, several lines of evidence support the multiple hit hypothesis that genetic vulnerability and environmental toxicants converge to trigger PD pathology. Alpha-synuclein (α-Syn) aggregation in the brain is an important pathophysiological characteristic of synucleinopathies including PD. Epidemiological and experimental studies have shown that metals and pesticides play a crucial role in α-Syn aggregation leading to the onset of various neurodegenerative diseases including PD. In this review, we will emphasize key findings of several epidemiological as well as experimental studies of metal- and pesticide-induced α-Syn aggregation and neurodegeneration. We will also discuss other factors such as traumatic brain injury and oxidative insult in the context of α-Syn-related neurodegenerative processes.
Keywords: Alpha-synuclein, pesticides, metals, protein aggregation, traumatic brain injury, oxidative stress, parkinson disease, neurodegenerative disease
Graphical abstract
Introduction
Neurological diseases account for roughly 4% of all deaths worldwide. However, a more concerning aspect is that unlike respiratory and cardiovascular diseases, neurological diseases often pass unrecognized, misdiagnosed or ignored as a minor concern in their early stages. As a result, the number of disease-adjusted life-years for neurological disorders is 1.5 times greater than cardiovascular and respiratory diseases. Genetic and environmental factors have been implicated in many neurological diseases [12, 14, 23]. For instance, the SNCA gene, which encodes for the protein α-synuclein (α-Syn), has a propensity to interact with several known neurotoxicants prior to α-Syn’s misfolding into toxic moieties. Thus, this gene is one among many implicated in familial and sporadic PD. Pesticides, metals, solvents and other chemicals have been shown to elevate the levels of α-Syn expression in neurons and promote α-Syn aggregation in vitro and in vivo [34, 136], In fact, environmental chemicals such as manganese, copper, rotenone, and paraquat can interact with α-Syn, thereby accelerating the fibrillation and aggregation in vitro [135]. Given that aggregated α-Syn mediates neurotoxicity in neurons and glial cells, the role of environmental neurotoxicants in the development of α-Syn aggregation has recently gained much attention. In this review, following a brief introduction to synuclein family proteins, we provide a concise overview of the current neurotoxicological literature linking α-Syn and known environmental neurotoxicants, with particular focus on how they might interact in the central nervous system (CNS) to potentially contribute to progressive neurodegenerative disorders like PD. Along with this we will also shed light on the role of traumatic brain injury as a well as oxidative stress in neurodegeneration and PD.
The synuclein family proteins
Synucleins are a family of small, soluble proteins expressed primarily in neural tissue and in certain tumors. The synuclein family consists of four members: α-, β-, and γ-synuclein and synoretin. Members of the synuclein family have high sequence identity, characterized by the acidic carboxyl terminus and imperfect repeat motifs (KTKEGV) distributed throughout the N-terminal region (Fig. 1). The first synuclein was discovered by Maroteaux et al. [94] in the Pacific electric ray (Torpedo californica) in 1988 and was later isolated from the rat brain. The protein was named synuclein because of its apparent localization in the presynaptic nerve terminals and portions of the nuclear envelop. Subsequent studies failed to confirm its nuclear localization, yet the original name was retained for historical reasons [122]. Synuclein was also named the nonamyloid component (NAC) of plaque precursor protein after the NAC peptide was isolated from amyloid-rich senile plaques sampled from Alzheimer patients [132]. Later studies speculated that the original finding might have been antibody cross-reactivity with the β-amyloid protein antibody [4, 29]. The second member of the synuclein family was discovered by Tobe and colleagues in 1992 and originally termed phosphoneuroprotein-14 [128]. A family of human brain synucleins was recognized for the first time when studies identified two abundant proteins of 140 and 134 amino acids with sequence homology to NAC protein and phosphoneuroprotein-14, thereby unraveling a previously unrecognized homology between these two proteins [67]. Both synucleins are expressed predominantly in the brain, where they are concentrated in presynaptic nerve terminals. Therefore, Jakes and colleagues referred to the 140 and 134 amino acid proteins as α-synuclein and β-synuclein, respectively [67]. The human α-Syn gene maps to chromosome 4q21, whereas the β-synuclein gene maps to chromosome 5q35 [121]. Human α- and β-synucleins are 61% identical in amino acid sequence and share a similar domain organization [122]. The amino-terminal half of each protein is taken up by imperfect amino acid repeats, with the consensus sequence being KTKEGV. An inter-repeat region of 5–8 amino acids separates individual repeats. Depending on the alignment, α-synuclein has 5–7 repeats, whereas β-synuclein has just five repeats [122]. The third member of the synuclein family was isolated from breast cancer tissue and initially termed breast cancer specific gene product 1 (BCSG1) [70]. Because of significant sequence homology to α-synuclein and abundant expression in the peripheral nerves, brain and spinal cord, it was later renamed γ-synuclein. The newest member of synuclein family is synoretin, first identified in retinal cells and the brain by Surguchov and colleagues [125]. Synoretin is an activator of signal transduction through activation of the Elk1 pathway and may play a role in vesicular trafficking [125]. However, with its 87% sequence identity to γ-synuclein and 100% similarity to γ-synuclein’s 5′ untranslated region, synoretin is often ascribed to γ-synuclein rather than a bona fide synuclein. Besides having similarities to γ-synuclein, its complete sequence is not well understood so sequence homology is not presented in Fig. 1.
Fig. 1.
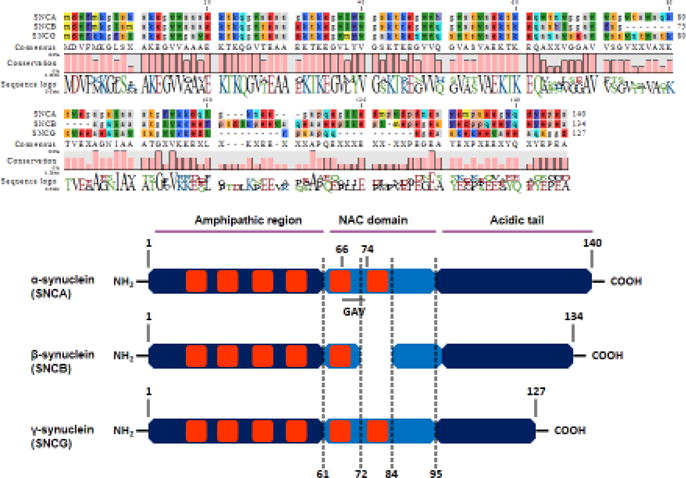
Sequence homology between α-, β-, and γ-synuclein, which consist of 140, 134 and 127 amino acids, respectively. N-terminal region in all three synucleins is conserved. This region consists of a similar number of repeats of the KTKEGV consensus sequence.
α-Synuclein
Alpha-Synuclein (α-Syn) is a small, 14.5-kDa acidic protein comprising 140 amino acids. It is highly conserved in vertebrates and predominantly expressed presynaptically in neurons throughout the mammalian brain as well as in the cerebrospinal fluid (CSF). Physiological functions of α-Syn are poorly understood, but evidence has suggested a role for it in synaptic plasticity, dopamine regulation, and membrane trafficking. Structurally, α-Syn is a natively unfolded protein that lacks a defined secondary structure and therefore belongs to the intrinsically unstructured protein family.
As shown in Figure 2, α-Syn protein has three distinct fundamental domains. The amphipathic N-terminal region (residues 1 to 60) contains 11 amino acid repeats including the consensus sequence KTKEGV, which is important to the α-helix formation. Point mutations in the N-terminal region are identified as A30P, A53T, E46K, G51D and H50Q, which are associated with familial PD [83]. In a study done by Lazaro et al. [83] these mutants demonstrated similar inclinations to form oligomeric intermediates but distinguishable capabilities to form inclusions. Mutations in this region, especially those that result in the loss of positively charged lysine (K) residues, result in weaker binding of α-Syn to phospholipids [148]. Indeed some animal models of PD, as well as patients suffering from familial PD with an A30P point mutation in the N-terminal region, exhibit significantly reduced vesicular binding activity [69]. Another well-characterized mutation is the A53T point mutation. This mutation generates more β-sheet structures and aggregated protein species enriched with β-sheet structure [110]. Conversion of α-helical structure into β-sheet structure is one of the hallmarks of fibrillation and aggregation processes [87]. This missense mutation at position 53 causes substitution of Thr for Ala, whereas in the A30P mutation, Ala is replaced by Pro [21, 110]. Substitutions that occur in these mutations reduce hydrophobicity, which heightens aggregation propensity [87]. Another important point mutation, E46K, is in the fourth KTKEGV repeat where it causes Glu to be substituted by Lys [149]. Since Glu residues are known to prevent β-sheets formation, this replacement should result in higher rates of fibrillization. H50Q and G51D are newly discovered familial α-Syn mutations, which have been linked to the early onset of PD pathogenesis, dementia (for H50Q) [75] and multiple-system atrophy (for G51D) [51]. H50Q, an α-Syn missense mutation, significantly stimulates α-Syn aggregation as well as amyloid formation and fibrillization in vitro [51]. H50Q can increase α-Syn secretion into the culture media of an SHSY5Y neuroblastoma cell line and it stimulates further mitochondrial fragmentation and toxicity in hippocampal neurons [75]. The G51D familial mutation attenuates α-Syn aggregation in vitro [43]. Fares et al, [43] also show that mutant G51D acts like the A30P mutant, forming few inclusions, which are non-toxic and exhibit a weakened membrane association. All these mutations, except A30P and G51D, result in higher incidences of intermediate α-Syn oligomers, fibril formation and aggregation, possibly because these amino acid substitutions prevent assembly of the protein into regular helical structures. Interestingly, Lazaro et al. [83] demonstrated that different mutations were associated with varying degrees of α-Syn oligomerization as well as inclusion generation.
Fig. 2.
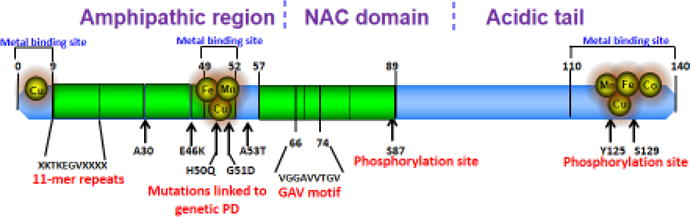
Structure of α-Syn and the binding sites for various divalent metals such as Manganese (Mn), Copper (Cu), Iron (Fe), Cobalt (Co). The three regions are N-terminus (amphipathic region), NAC domain and C-terminus (Acidic tail). N-terminus region consists of 11 amino acid repeats that includes the consensus sequence KTKEGV. The mutations A30P, E46K, H50Q, G51D and A53T are present in amphipathic region as shown. Phosphorylation sites on α-Syn are S87 on the NAC domain, and Y125 and S129 on the acidic tail. Glycine, alanine and valine residuals containing the consensus sequence also known as GAV motif are present on the NAC domain.
The central hydrophobic region (residues 61 to 91) contains the NAC domain, which is important in protein aggregation. Phosphorylation of Ser 87 in this region results in the conformational change from helix to fibrils [103]. The hydrophobic GAV motif (residues 66–74), mainly consisting of glycine, alanine and valine residues, has been identified as the critical core for the fibrillization and cytotoxicity of α-Syn [37].
Finally, the C-terminal region (residues 91 to 140), which is highly acidic and proline-rich, is responsible for the intrinsically disordered nature of α-Syn [57]. Its acidic residues impart a strong negative charge [50]. During protein folding, residues aa109–140 in this region are hypothesized to form intra-disulfide bonds favoring helix formation over β-sheets. Various C-terminal truncation studies show that loss of this region increases α-Syn aggregation [64, 89]. C-terminal truncation and the presence of truncated α-Syn protein inclusions in Lewy bodies and Lewy neurites have been reported [28, 89]. Multiple sites on this terminal can be phosphorylated, although the precise outcomes of these phosphorylations are not known [101]. Perhaps the best-known site of C-terminal phosphorylation is at S129. Its phosphorylation by G-protein coupled receptor protein kinases increases fibrillization [48]. Synuclein gene (SNCA) multiplication has also been implicated in familial PD. SNCA duplication, or two copies of SNCA on one allele, increases the amount of synuclein being produced by 50%, whereas SNCA triplication, or 3 copies of SNCA on the same allele, increases synuclein by 100% [65]. These increased doses of α-Syn lead to proteasomal dysfunction and clinical manifestations of PD and DLB.
The link between α-Syn and PD pathogenesis is based on case studies of familial PD and the observation that misfolded α-Syn is a major constituent of Lewy bodies and Lewy neurites in both familial and sporadic PD. The idea that α-Syn can pathologically propagate throughout the CNS recently gained much attention with the finding of α-Syn species circulating in human plasma and CSF [40, 77] and host-to-graft propagation of α-Syn-positive Lewy bodies in fetal ventral mesencephalic and embryonic nigral neurons transplanted in human PD patients [77, 88]. Even though several models have postulated the cell-to-cell transmission of pathological α-Syn species [33, 38, 86] as a potential mechanism in PD pathogenesis and related synucleinopathies, the precise cellular and molecular signaling pathways remain unknown. Growing in vitro evidence indicates that extracellular α-Syn induces pathogenic actions by activating neuroinflammatory and neurodegenerative responses [41, 123].
Although genetic mutations are an important risk factor, at least in many familial cases, a link has been established between environmental neurotoxicant exposure and PD and PD-related disorders [14, 34, 57], stemming from overexposure to metals, pesticides and other neurotoxicants. Importantly, α-Syn protein has three metal-binding sites: one at its N-terminus, one at its central region and one at its C-terminus. The metal-binding sites near 49–52 and 110–140 are known to interact with divalent metals, including copper and manganese [135], leading to α-Syn aggregation. Several other mechanisms have been identified in α-Syn neurotoxicity, including mitochondrial impairment, oxidative and nitrative damage, astroglia- and microglia-mediated inflammation, and dopamine metabolism impairment which are reviewed elsewhere [23, 99].
Synucleinopathies and Parkinson’s disease
Synucleinopathies are a subset of neurodegenerative disorders characterized by pathological lesions composed primarily of fibrillary aggregates of insoluble α-Syn in selective populations of neurons and glia. The most common synucleinopathy is PD, but other neurodegenerative diseases, including dementia with Lewy bodies (DLB), glial cytoplasmic inclusions in multiple system atrophy (MSA) and neurodegeneration with brain iron accumulation type I (NBIIA) formerly known as Hallervorden-Spatz disease, also manifest abnormal α-Syn deposition as a major pathologic feature. Although these synucleinopathies share some clinical symptoms and the presence of Lewy bodies, they also have many clinical and pathological differences. As an example, the predominant clinical symptoms of PD are indistinguishable from those of DLB and MSA patients with α-Syn accumulation in neurons and glia. However, hallucinations and psychosis manifest much earlier in DLB patients compared to PD cases where it is seen only in the late stages of the disease. Similarly, while MSA patients also show bradykinesia and decreased striatal dopamine levels as seen in PD, they respond poorly to levodopa and have pronounced autonomic dysfunction. In the case of Mn-induced Parkinsonism, which does not display abnormal accumulations of α-Syn in the brain, neurons in the globus pallidus seem to be preferentially affected and patients seem to be resistant to levodopa treatment despite sharing many common clinical characteristics seen in PD, including chronic and progressive declines in motor, cognitive, behavioral, and non-motor symptoms. However, given the number of epidemiological studies indicating that exposure to some metals, including manganese [53, 54, 112], is associated with a higher risk of PD, case-controlled characterization of α-Syn aggregation in brain tissues from human manganism cases is urgently needed. Because of these clinical and pathological overlaps, differential diagnosis of synucleinopathies is very difficult. In addition, several rare neuroaxonal dystrophies, such as striatonigral degeneration, olivopontocerebellar atrophy, Shy-Drager syndrome and Pick’s disease, also show α-Syn immunoreactive inclusions.
PD is the second most common neurodegenerative disorder after Alzheimer’s disease and is the most common movement disorder in people over age 65, causing progressive disability characterized by severe motor symptoms including uncontrollable tremor, postural imbalance, slowness of movement and rigidity. PD is also recognized as one of the most common neurologic disorders, affecting approximately 1% of individuals older than 60 years [92], and thus aging appears to be the single most important risk factor for developing PD. Pathologically, this disease is characterized by a progressive degeneration of dopaminergic neurons projecting from the substantia nigra pars compacta (SNpc) to the striatum, resulting in pronounced loss of the neurotransmitter dopamine, thereby leading to the above-mentioned extrapyramidal deficits. The disease is often associated with abnormal accumulations of misfolded proteins in cytoplasmic inclusions called Lewy bodies (LB) and Lewy neurites. However, the association between Lewy pathology and pathogenesis of the disease is poorly understood. Recent evidence has implicated several genes associated with early-onset of PD, such as α-Syn (PARK1), Parkin (PARK-2), PINK1 (PARK6), DJ-1 (PARK7), ATP13A2 (PARK9), SLC30A10 as well as those genes linked with late onset of the disorder, LRRK2 (PARK8) and VPS35 (PARK-17) [31, 111]. For clarification, loss-of-function mutations in SLC30A10 lead to Mn accumulation in the basal ganglia, loss of neurons in the globus pallidus and disease pathology consistent with Mn-induced Parkinsonism, but not with classical PD [84]. While major emphasis has been given to familial PD caused by gene mutations, more than 90% of PD occurrences represent the sporadic form of PD, which is likely caused by a complex interplay of genetic and environmental factors. Although PD is classically defined as a movement disorder associated with degeneration of neurons in the nigrostriatal system, non-motor symptoms have been recognized in recent years. During the initial presymptomatic phase of this movement disorder, patients develop non-motor deficits including cognitive changes, behavioral/neuropsychiatric changes, autonomic nervous system failure, olfactory impairment and sleep disturbances. Non-motor symptoms represent some of the greatest challenges to quality of life and appropriate management in PD since they are usually less responsive than motor symptoms to dopamine therapy.
Epidemiological and clinical studies have identified potential environmental risk factors for PD, including repeated head injury, heavy metal exposure, excess body weight, pesticide exposure and some surrogate measures such as rural living, drinking well water and farming [35, 107]. Although neurotoxicants have diverse chemical properties, they share some common mechanisms by which they cause either destruction or disruption of important constituents of the CNS. Based on their mechanism of action, neurotoxicants can be classified into various channel inhibitors, receptor inhibitors, receptor agonists, synaptic vesicle inhibitors and many more. In the next section, we will focus on the extensive PD-related research into how metals and pesticides interact with α-Syn.
Environmental neurotoxicants and neurodegeneration
Research has demonstrated strong links between long-term exposure to environmental neurotoxicants, such as pesticides, heavy metals, and endocrine disrupters, and a range of dopamine-associated neurological diseases. MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a major impurity in the synthesis of certain opioid drugs, can cause neurodegeneration ultimately producing features pathologically similar to PD. MPTP passes through the blood brain barrier and gets metabolized into its active toxic form MPP+ by the MAO-B enzyme of glial cells. Once it is released into the synaptic cleft, it enters neurons through the dopamine transporter and accumulates in the mitochondria where it inhibits complex I function leading to ATP depletion and oxidative stress [68, 82].
Metals are essential for very basic cellular processes, such as cell structure maintenance, gene expression, antioxidant response and neurotransmission. However, overexposure leading to the excessive accumulation of metals can damage the nervous system through induction of oxidative stress, mitochondrial dysfunction and disrupting the usual activities of certain enzymes [18]. Epidemiological and clinical studies have linked excessive metal exposure to neurological diseases such as Alzheimer disease (AD), PD, spinocerebellar ataxia, Huntington disease, and amyotrophic lateral sclerosis. A well-characterized example involves chronic lead exposure increasing the risk of developing AD. Used in the production of gasoline, paints and toys until 1971, lead was shown to induce AD like pathology in animal models including primates and rodents [8, 39]. In the 19th century, it was discovered that manganese ore miners exposed to manganese (Mn) dust developed speech and movement deficits. This poisoning was irreversible and lead to psychosis and ultimately death. In recent years, overexposure to metals such as Iron (Fe), Copper (Cu), Aluminum (Al), has been found to affect CNS functioning [142]. Numerous epidemiological and clinical studies have also linked pesticide exposure to the etiology of neurodegenerative diseases, primarily PD [12, 20]. We will return to metals and pesticides in the following sections.
Metals
Manganese
Manganese (Mn) is an essential trace metal that is important in various processes such as bone, fat and carbohydrate metabolism and blood sugar regulation [63]. It is essential both in the normal functioning of various enzymes such as hydrolases, glutamine synthetase and lyases [17, 63] and as a cofactor for arginase, MnSOD and other metalloproteins. Its primary route of entrance is through food consumption [17, 97], and secondarily through inhalation, which poses an occupational hazard for people who work in mining and welding industries. Chronic overexposure to Mn can cause devastating neurological effects. Mn not only leads to manganism, which is a form of Parkinsonism, but it is also proposed to play a crucial role in PD etiology [80]. Multiple mechanisms are involved in Mn-induced neurotoxicity, including autophagy [55], oxidative stress[44], altered cAMP signaling [91], reactive oxygen species (ROS) generation [95], mitochondrial dysfunction [44], altered iron homeostasis [81] and altered acetylcholinesterase (AChE) activity [146]. Mn exposure induces α-Syn aggregation in non-human primates, and may also play a role in α-Syn protein fibrillation and oligomerization [138]. When Bates et al, [3] examined how Mn exposure alters α-Syn expression and transport at the blood-CSF (cerebrospinal fluid) barrier, they found that Mn induces the uptake of α-Syn by rat primary choroid epithelial cells from the extracellular medium. Another interesting result was that Mn exposure significantly increased intracellular α-Syn without changing α-Syn mRNA expression. They also observed Mn-induced α-Syn aggregation in a cell-free system. Their study indicates that Mn exposure can escalate α-Syn accumulation at the blood-CSF barrier by accelerating its uptake and aggregation. Other studies show that Mn exposure induces α-Syn expression in vitro [13, 90], and stimulates α-Syn oligomerization, oxidative stress and ROS generation in organotypic brain slice cultures [145].
In our recent study of α-Syn’s capacity for Mn storage, we demonstrated α-Syn’s role as a neuroprotective mechanism against Mn-induced neurotoxicity in a dopaminergic neuronal cell line stably expressing human wild-type α-Syn at physiological levels [57], which agrees with our previous findings that α-Syn plays a neuroprotective role by repressing the transcription of the oxidative stress-sensitive kinase PKCδ [71]. Specifically, α-Syn expression attenuates the Mn-induced mitochondria-dependent apoptotic-signaling pathway by inhibiting caspase-9 and -3 activation and subsequent proteolytic activation of PKCδ. However, during prolonged Mn exposures, the protein becomes increasingly susceptible to aggregation and the accumulation of oligomeric α-Syn protein [57]. Our study uncovered a novel, neuroprotective role for human wild-type α-Syn in attenuating acute Mn toxicity, which may be a direct effect of its metal-binding capability, thereby providing much needed insight into the physiological role of wild-type α-Syn protein.
Iron
Iron (Fe) is a major trace element in the human body, and its importance stems from its ability to bind oxygen for transport throughout the body and for redox reactions [118]. It also serves as a cofactor for many proteins like hemoglobin and various enzymes [7]. However, disrupting Fe metabolism and homeostasis can lead to excessive Fe accumulation in the brain [16, 118] where it can stimulate α-Syn aggregation typically associated with P [46, 108]. Tyrosine fluorescence quenching shows only moderate binding of Fe (II) to α-Syn [52]. Another study demonstrated that binding sites for Fe (II) are Asp-121, Asn-122 and Glu-123 at the C-terminus on α-Syn [10]. In a study done by Kostka [78], the addition of Fe (III) induced larger oligomer formation following DMSO-stimulated oligomer formation in vitro in unilamellar vesicles. These larger oligomers become toxic by binding to the lipid bilayer where they form pores. Some researchers hypothesize that Fe cytotoxicity arises through synergistic interaction with α-Syn [15]. He et al. [62] reported that Fe (II) stimulates α-Syn aggregation by inhibiting Nrf2/heme oxygenase-1 (HO-1), which generates a neurotoxic cycle of Fe accumulation and more α-Syn aggregation [62].
Copper
The trace element copper (Cu) serves as a cofactor for many enzymes such as cytochrome c oxidase, SODs (super oxide dismutase) and certain proteins, thereby playing a crucial role in the transportation of oxygen and electrons, neurotransmitter synthesis and protein modification [18]. Elevated Cu stimulates ROS generation, mitochondrial dysfunction and DNA damage [15]. Occupational exposure to Cu increases the risk of developing PD, most probably because it can induce α-Syn aggregation [49, 53, 54, 112]. Cu binds with strong affinity to α-Syn, which has multiple metal-binding sites for Cu [9, 66, 150]. This strong affinity for Cu could contribute to the etiology of PD by inducing Lewy body formation [32, 135]. With the help of NMR studies conducted at physiological pH, researchers have identified two different Cu binding sites on the α-Syn sequence: His-50 [109] and Met-1 [11]. NMRs conducted at a lower pH of 5.0 revealed additional α-Syn binding sites for Cu (II), including Asp-119, Asp-121, and Glu-123 [36]. Importantly, α-Syn can bind to both oxidation states, Cu (II) and Cu (I) [137]. It was also reported that Cu exposure becomes increasingly cytotoxic as α-Syn expression increases [141]. Zhang et al, [150] showed that incubating α-Syn with Cu (II) for 60 h produced protofibrillar, annular and fibrillar species; protofibrils are especially cytotoxic. Interestingly, more α-Syn aggregation was seen in dopaminergic neurons following Cu exposure, suggesting that Cu and dopamine work synergistically on the misfolding and aggregation of α-Syn [56, 127].
Other metals such as Zinc (Zn), Al, and Cadmium (Cd) were also found to induce α-Syn fibril formation. One recent study [114] demonstrated a neurotoxic role for α-Syn in Al-mediated oxidative stress and cell death. Tsunemi et al. [131] demonstrated that silencing PARK9 (putative cation transporter) leads to intracellular dyshomeostasis of Zn2+, causing lysosomal dysfunction and ultimately α-Syn accumulation. Uversky et al. [135] showed that metals such as Al, Zn, Fe, Co can efficiently induce fibril formation and they suggest that metal ion-induced ligand bridging plays a crucial role in stimulating conformational modifications in α-Syn structure, which could lead to its aggregation and/or fibrillation.
Pesticides
Pesticides are used worldwide in commercial farming and home gardening. In 1962, Rachel Carson’s book Silent Spring raised public awareness about pesticides and their toxic effects in our environment. The role of pesticides in neurodegenerative diseases has long been suspected, and now published studies are linking exposure to pesticides such as paraquat, maneb, rotenone, dieldrin, etc., to the etiopathogenesis of major neurodegenerative diseases such as AD and PD. In general, these pesticides exert their neurotoxic effects by inducing oxidative stress, α-Syn fibrillization, mitochondrial dysfunction and neuronal death.
Paraquat
Paraquat is one of the most widely used herbicides. Due to its ability to induce neurodegeneration, it is used experimentally in in vitro and in vivo mechanistic studies. It is an extremely toxic herbicide that has been linked to PD based on epidemiological studies and clinical work done in rodents. Experimental studies have shown that paraquat exposure causes lesions in the SNpc in certain mouse strains as well as rats. In in vitro studies, paraquat induces conformational changes in α-Syn and also stimulates α-Syn aggregation [134]. Manning-Bog et al, [93] reported that not only did α-Syn expression increase in the brains of mice exposed to paraquat, but also that it was aggregated. Paraquat induces α-Syn overexpression by reducing proteasomal degradation activity, leading to the accumulation of α-Syn aggregates [143]. Feng et al. [47] demonstrated that dopaminergic cell death occurs when paraquat treatment, combined with dopamine (DA), triggers α-Syn-induced increases in membrane conductivity through leak channels.
Rotenone
Rotenone is a widely used colorless, odorless insecticide found to induce a Parkinsonian phenotype such as Lewy body formation and nigrostriatal neurodegeneration [6]. Lee and Lee [85] reported that rotenone induces α-Syn aggregation and causes nigrostriatal degeneration in clinical models of PD. The mechanism by which rotenone stimulates α-Syn aggregation remains unclear, but Yuan et al. [147] demonstrated that rotenone increases intracellular calcium and stimulates phosphorylation and aggregation of α-Syn. They not only showed that phosphorylation and aggregation of α-Syn were calcium-dependent, but that rotenone also impaired autophagy, which would normally help degrade aggregated α-Syn. Betarbet et al. [5] showed that rotenone modifies DJ-1 and induces α-Syn accumulation as well as dysfunction of the proteasomal system, leading to oxidative stress and mitochondrial dysfunction, thus showing a pathology similar to that of familial PD. Another study demonstrated that subcutaneous exposure to rotenone stimulated cytoplasmic aggregates, which were positive for α-Syn in nigral neurons [119]. In addition to their other findings, Yuan et al. [147] found that the GSK3β/calcium signaling pathway mediates α-Syn aggregation following rotenone exposure.
Dieldrin
Dieldrin is an organochlorine insecticide that was banned in the 1970s in many developed countries. However, its lipophilic properties and less volatile nature makes dieldrin environmentally persistent, allowing it to bio-accumulate and bio-magnify in the soil as well as in non-target organisms. Humans still get exposed to dieldrin through contaminated ground water, food and residuals in the environment [72]. Post-mortem reports indicate higher dieldrin levels in the caudate nucleus of PD patients than in age-matched control brains [24–26]. Dieldrin is selectively neurotoxic to dopaminergic neurons in in vitro studies, showing comparatively lower toxicity to GABAergic neurons [115]. When tested in mouse and rat dopaminergic cell lines, dieldrin causes dopamine depletion [61], ROS generation [19], altered mitochondrial membrane potential and apoptosis [73, 76]. More importantly, dieldrin stimulates conformational change as well as fibrillization in α-Syn. Sun and colleagues [73, 124] have shown that exposing dopaminergic neurons to dieldrin increases α-Syn levels and disrupts the ubiquitin proteasomal system, making dopaminergic neuronal cells more susceptible to apoptotic cell death. Interestingly, even though dieldrin can induce ROS generation, mitochondrial dysfunction, oxidative stress, α-Syn aggregation and DA depletion, it still fails to stimulate motor deficits commonly seen in PD patients.
Traumatic brain injury and α-synuclein aggregation
Traumatic brain injury (TBI) encompasses injuries ranging from a penetrating injury to the cranium to a succession of mild, subconcussive blows to the head. With the increased incidence of armed conflict as well as vehicular and contact sports-related accidents, the cases of TBI have steadily increased over the years. According to a report by Faul et al, [45], the leading cause of TBI in the US is falls (35%), followed by a similar combined incidence of vehicular (17%) and sports-related injuries (17%). Almost all deployed military personnel experience mild to severe forms of TBI due to exposure to IEDs (improvised explosive devices).
Because TBI describes a broad spectrum of injuries that range from mild and diffuse to severe and focal, it can also involve multiple pathophysiological mechanisms that can alter behavior and/or motor skills. The Glasgow Comma Scale (GCS) is widely used by physicians to determine the severity of the injury with a score between 13–15 being considered a mild head injury, 9–12 is a moderate injury, while a score <8 is considered severe. Researchers classify TBI based on the mechanism of injury (penetrative head trauma vs. superficial non-invasive head injury), pathology (whether the injury is focal or diffuse) or clinical diagnosis (axonal damage, hematomas, contusion or subdural hemorrhage) [106]. It is necessary to classify the type of brain injury since computed tomography (CT) scans reveal that, despite similarities in severity, the way in which the insult occurred leads to symptoms and pathophysiology that vary between subjects [113]. Thus, to develop targeted treatments for TBI, it is necessary to understand the mode of injury, its regional extent as well as the mechanism behind the developing neuroinflammation and neurodegeneration.
In recent years, much effort has been focused on understanding the pathophysiology and longterm effects of mild to severe TBI due to reports that have attributed the rapid development of mood disorders, movement disorders and cognitive decline to TBI [42, 96]. Following primary mechanical insult, there is a delayed cellular and physiological change that results in the early development and accelerated progression of various neuropsychiatric, movement and cognitive disorders. One of the early and prolonged responses is neuroinflammation at the site of injury. Following a blow to the head, the neurons and glia at the primary site of impact undergo shear and strain stress owing to the stretching of cells caused by accelerating, decelerating or rotational forces. There is local or diffuse ischemia and hypoxia resulting in increased permeability of the blood brain barrier (BBB). In a normal brain, the BBB restricts the influx of large proteins, peripheral immune cells such as leukocytes as well as many toxins into the CNS. However following TBI, there is breakdown of junctional proteins such as claudin-1 and zona occludin [27]. An increased presence of leukocyte adhesion receptors and a concomitant decrease in integrin expression leads to increased permeability, leukocyte extravasation and subsequent development of edema and inflammation [105]. Also, traumatized neurons experience a rapid release of ATP. This results in the recruitment and activation of both microglia and astrocytes to the site of damage often within hours of neurons sending stress signals [30]. Recruitment of activated microglia results in the production of pro-inflammatory factors such as IL-1β, TNFα as well as inducible nitric oxide species (iNOS) [74]. Microglia activation and cytokine production is an expected and indeed beneficial response. Microglia that infiltrate the site of injury help remove dead and dying neurons as well as promote healing by the production of neurotrophic factors such as brain-derived neurotrophin (BDNF), glia-derived neurotrophin (GDNF) and anti-inflammatory cytokines such as IL-10 and TGFβ. However, it has been observed in various in vivo models of TBI as well as in post-mortem analyses of patients who had previously suffered head injury that the microglia are in a state of constant activation resulting in chronic neuroinflammation. Persistent activation of microglia results in the production of neurotoxic factors such superoxide, nitric oxide and the continued presence of pro-inflammatory cytokines and chemokines. Together, this secondary insult is chronic and largely perseveres resulting in neurodegeneration and subsequent neuronal dysfunction [58, 79].
The protein α-Syn is mainly associated with neurons although it is found to some extent in epicardial fat tissue, muscles, platelets and hematopoietic cells [2, 60, 100, 120]. In individuals that have experienced moderate to severe TBI, aggregated α-Syn has been shown to accumulate in swollen axons and in the cerebrospinal fluid (CSF) [98, 102, 133]. Histopathological analysis of post-mortem brains from boxers with a known history of dementia showed neurofibrillary tangles, amyloid β deposits and dystrophic neurons [130]. Despite these clinical observations, the pathophysiological mechanism behind brain trauma and the aggregation of α-Syn has not yet been well studied. In 2014, Acosta et al. [1] reported the behavioral and pathological changes in rats that were subjected to TBI via a controlled cortical impactor. Following TBI, microglia became chronically activated. Even after 60 days post-TBI, the site of impact showed increased α-Syn with a concomitant loss of dopaminergic neurons [1]. The study also found that increased α-Syn levels correlated with MHCII-positive microglia and were negatively associated with a healthy dopaminergic neuron population.
Repeated mild TBI or a single severe impact to the brain leads to axonal damage, BBB permeability, glutamate excitotoxicity and calcium influx. Axonal injury leads to caspase and calpain enzyme activation, which in turn cleaves cytoskeletal proteins such as α-spectrin II, neurofilaments, α-Syn and microtubule-associated proteins [117, 126]. Breakdown of these proteins and the presence of activated microglia result in the continued presence of neuroinflammation. This leads to the development of ROS and reactive nitrogen species (RNS), which would possibly oligomerize α-Syn, present mainly in the neuronal terminals, via oxidation or nitration [144]. Oligomerized α-Syn can subsequently form fibrillar structures - the highly pathogenic form of α-Syn. Recent evidence from a number of in vitro studies indicates a prionlike propagation of this protein, resulting in its increased and unchecked transcellular spread to various parts of the brain following oxidative insult [140].
Thus, even mild brain injury can lead to chronic neuroinflammation and diffusion of pathogenic α-Syn from the focal area of impact to other regions of the brain. Elevated levels of misfolded α-Syn maintain microglia in a chronic state of activation, thus accelerating neuronal dysfunction with more inflammation and neurodegeneration. More research needs to be conducted to understand the exact cellular mechanism by which α-Syn phosphorylation/nitration can be avoided to prevent the development of various synucleinopathies following TBI.
Oxidative stress and its role in α-Syn aggregation
Various synucleinopathies such as PD, DLB and MSA show not only the presence of aggregated α-Syn, but also chronically activated microglia. Under normal cellular conditions, a small pool of ROS and RNS is present in every cell without causing cellular toxicity and might even play important physiological roles in the cell [116]. Post-translational modification of α-Syn in the presence of ROS and RNS can result in the formation of HNE-α-Syn, o-α-Syn or n-α-Syn. These modified forms can now oligomerize, promoting the formation of protofibrils and β-sheet structures [144]. Misfolded α-Syn causes microglial activation, leading to the release of pro-inflammatory cytokines and inflammation. When microglia attempt to clear up the misfolded protein, they activate NADPH oxidase that in turn results in the production of more ROS. If the concentration of ROS is persistently greater than the cell’s ability to counteract it by producing antioxidants, this results in mitochondrial dysfunction and subsequently cellular apoptosis [151].
Misfolded α-Syn and Aβ can translocate to the mitochondrial membrane, causing release of cytochrome C and activation of the apoptosis cascade [59]. The cell requires mitochondria to pump a continuous supply of ATP for its survival, but this process continuously produces ROS contributing to more oxidative stress. The ubiquitin-proteasome system (UPS) is a complex process wherein the proteins to be degraded are tagged with ubiquitin. This highlights proteins for the 26S proteasome that then degrades the tagged protein via its proteolytic activity [22]. The rate limiting aspect of this process is the degradation of ubiquitin-tagged proteins. It is thought that in disorders such as PD, DLB and MS, protein degradation is much slower than the development of misfolded proteins. Indeed, post-mortem histopathological analysis shows that aggregated proteins are also ubiquitinated [129].
Environmental stressors such as exposure to toxins, pesticides, herbicides and industrial effluents also promote oxidative stress. In a study conducted by Pan-Montojo and colleagues [104], mice given rotenone via intragastric gavage showed the release of α-Syn by enteric neurons through exosomes. These exosomes propagated from one neuron to another thus transporting α-Syn across large distances. However, resection of autonomic neurons prevented the PD-like pathology progression in these mice. In another study, mice given dextran sodium sulfate (DSS) in drinking water experienced not only intestinal inflammation but also greater BBB permeability. The increased BBB permeability and/or colitis induced pro-inflammatory transcripts of IL-1β and TNFα in the substantia nigra of these animals [139]. Taken together, these results indicate that environmental neurotoxins can promote neuroinflammation in both the enteric and central nervous systems. Chronic neuroinflammation can lead to mitochondrial and UPS dysfunction thus leading to the development and persistent presence of aggregated proteins including α-Syn.
Conclusion
As experimental and epidemiological data suggest, many neurodegenerative disorders, including PD, are the result of the complex interaction between an individual’s underlying genetic composition, lifestyle and cumulative, life-long exposure to environmental stressors. In light of recent discoveries about synucleinopathies, aggregated α-Syn-mediated oxidative stress and mitochondrial dysfunction have been identified as key pathological processes that contribute to neuronal death, leading to functional impairments. Nevertheless, the key environmental toxicants described in this review have also been shown to induce protein aggregation and oxidative stress and to exacerbate neuronal vulnerability caused by gene mutations. However, despite extensive efforts to define the molecular mechanisms underlying protein misfolding, protein aggregation, and neurodegeneration, many aspects about the role of environmental neurotoxicants in these pathologies remain elusive.
Fig 3.
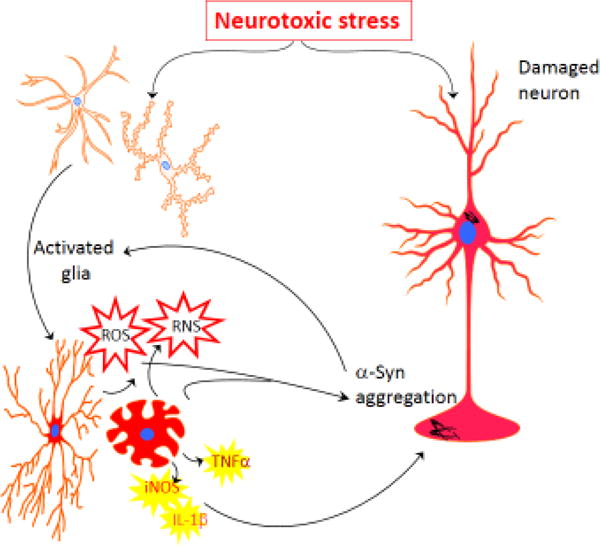
Trauma to the head can result in direct mechanical injury to the neurons as well as glia. Injured neurons send stress signals such as caspase-3 that activate microglia and astrocytes that in turn produce pro-inflammatory factors such as iNOS, TNFα and IL-1β. These factors help to recruit microglia to the site of damage for clearance of cellular debris and to produce neurotrophic factors. However, chronic upregulation of pro-inflammatory factors ultimately leads to continued generation of ROS and RNS leading to mitochondrial stress and aggregation of proteins, such as α-Syn in neurons. The presence of aggregated a-Syn further increases the number of activated microglia and astrocytes. This cycle of neuroinflammation continues ultimately leading to neuronal death.
Highlights.
Principle features of various Synuclein protein family members are highlighted.
New findings on metal-induced α-Syn misfolding & consequences are emphasized.
Important effects of pesticides on α-Syn and neurodegeneration are illustrated.
Role of TBI and oxidative stress on α-Syn and neurodegeneration is highlighted.
Acknowledgments
This work was supported by the National Institutes of Health R01 grants ES019267, ES026892, NS074443 and ES010586 awarded to A.G.K, and NS088206 to A.K. The W. Eugene and Linda Lloyd Endowed Chair to A.G.K, the Dean Endowed Professorship to AK and the Syngenta Fellowship Award in Human Health Applications of New Technologies to D.S.H. are also acknowledged. We also thank Mr. Gary Zenitsky for assistance in preparing this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests:
The authors declare they have no competing financial interests.
References
- 1.Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J Cell Physiol. 2015;230:1024–1032. doi: 10.1002/jcp.24830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Askanas V, Engel WK, Alvarez RB, McFerrin J, Broccolini A. Novel immunolocalization of alpha-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J Neuropathol Exp Neurol. 2000;59:592–598. doi: 10.1093/jnen/59.7.592. [DOI] [PubMed] [Google Scholar]
- 3.Bates CA, Fu S, Ysselstein D, Rochet JC, Zheng W. Expression and Transport of alpha-Synuclein at the Blood-Cerebrospinal Fluid Barrier and Effects of Manganese Exposure. ADMET DMPK. 2015;3:15–33. doi: 10.5599/admet.3.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayer TA, Jakala P, Hartmann T, Havas L, McLean C, Culvenor JG, Li QX, Masters CL, Falkai P, Beyreuther K. Alpha-synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies but not in Alzheimer’s disease beta-amyloid plaque cores. Neuroscience letters. 1999;266:213–216. doi: 10.1016/s0304-3940(99)00311-0. [DOI] [PubMed] [Google Scholar]
- 5.Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinefelter G, Cookson MR, Greenamyre JT. Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiology of disease. 2006;22:404–420. doi: 10.1016/j.nbd.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 7.Biasiotto G, Di Lorenzo D, Archetti S, Zanella I. Iron and Neurodegeneration: Is Ferritinophagy the Link? Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9473-y. [DOI] [PubMed] [Google Scholar]
- 8.Bihaqi SW, Zawia NH. Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb) Neurotoxicology. 2013;39:95–101. doi: 10.1016/j.neuro.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binolfi A, Lamberto GR, Duran R, Quintanar L, Bertoncini CW, Souza JM, Cervenansky C, Zweckstetter M, Griesinger C, Fernandez CO. Site-specific interactions of Cu(II) with alpha and beta-synuclein: Bridging the molecular gap between metal binding and aggregation. Journal of the American Chemical Society. 2008;130:11801–11812. doi: 10.1021/ja803494v. [DOI] [PubMed] [Google Scholar]
- 10.Binolfi A, Rasia RM, Bertoncini CW, Ceolin M, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO. Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J Am Chem Soc. 2006;128:9893–9901. doi: 10.1021/ja0618649. [DOI] [PubMed] [Google Scholar]
- 11.Q L, Binolfi A, Bertoncini CW, Griesinger C, F.a.C. O. Retraction for Ramos et al., The second RNA chaperone, Hfq2, is also required for survival under stress and full virulence of Burkholderia cenocepacia J2315. J Bacteriol. 2014;196:3980. doi: 10.1128/JB.02242-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS. Pesticides and Parkinson’s disease–is there a link? Environ Health Perspect. 2006;114:156–164. doi: 10.1289/ehp.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai T, Yao T, Zheng G, Chen Y, Du K, Cao Y, Shen X, Chen J, Luo W. Manganese induces the overexpression of alpha-synuclein in PC12 cells via ERK activation. Brain Res. 2010;1359:201–207. doi: 10.1016/j.brainres.2010.08.055. [DOI] [PubMed] [Google Scholar]
- 14.Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci. 2011;124:225–250. doi: 10.1093/toxsci/kfr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carboni E, Lingor P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics. 2015;7:395–404. doi: 10.1039/c4mt00339j. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee S, Sarkar S, Bhattacharya S. Toxic metals and autophagy. Chem Res Toxicol. 2014;27:1887–1900. doi: 10.1021/tx500264s. [DOI] [PubMed] [Google Scholar]
- 17.Chen P, Chakraborty S, Mukhopadhyay S, Lee E, Paoliello MM, Bowman AB, Aschner M. Manganese homeostasis in the nervous system. J Neurochem. 2015;134:601–610. doi: 10.1111/jnc.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen P, Miah MR, Aschner M. Metals and Neurodegeneration. F1000Res. 2016;5 doi: 10.12688/f1000research.7431.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun HS, Gibson GE, DeGiorgio LA, Zhang H, Kidd VJ, Son JH. Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem. 2001;76:1010–1021. doi: 10.1046/j.1471-4159.2001.00096.x. [DOI] [PubMed] [Google Scholar]
- 20.Cicchetti F, Drouin-Ouellet J, Gross RE. Environmental toxins and Parkinson’s disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci. 2009;30:475–483. doi: 10.1016/j.tips.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:120–129. [PubMed] [Google Scholar]
- 22.Cook C, Stetler C, Petrucelli L. Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009423. doi: 10.1101/cshperspect.a009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corrigan FM, French M, Murray L. Organochlorine compounds in human brain. Hum Exp Toxicol. 1996;15:262–264. doi: 10.1177/096032719601500314. [DOI] [PubMed] [Google Scholar]
- 25.Corrigan FM, Murray L, Wyatt CL, Shore RF. Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson’s disease. Exp Neurol. 1998;150:339–342. doi: 10.1006/exnr.1998.6776. [DOI] [PubMed] [Google Scholar]
- 26.Corrigan FM, Wienburg CL, Shore RF, Daniel SE, Mann D. Organochlorine insecticides in substantia nigra in Parkinson’s disease. J Toxicol Environ Health A. 2000;59:229–234. doi: 10.1080/009841000156907. [DOI] [PubMed] [Google Scholar]
- 27.Couraud PO. Infiltration of inflammatory cells through brain endothelium. Pathol Biol (Paris) 1998;46:176–180. [PubMed] [Google Scholar]
- 28.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS letters. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 29.Culvenor JG, McLean CA, Cutt S, Campbell BC, Maher F, Jakala P, Hartmann T, Beyreuther K, Masters CL, Li QX. Non-Abeta component of Alzheimer’s disease amyloid (NAC) revisited. NAC and alpha-synuclein are not associated with Abeta amyloid. Am J Pathol. 1999;155:1173–1181. doi: 10.1016/s0002-9440(10)65220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 31.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desai V, Kaler SG. Role of copper in human neurological disorders. Am J Clin Nutr. 2008;88:855S–858S. doi: 10.1093/ajcn/88.3.855S. [DOI] [PubMed] [Google Scholar]
- 33.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Monte DA, Lavasani M, Manning-Bog AB. Environmental factors in Parkinson’s disease. Neurotoxicology. 2002;23:487–502. doi: 10.1016/s0161-813x(02)00099-2. [DOI] [PubMed] [Google Scholar]
- 35.Dick FD, De Palma G, Ahmadi A, Scott NW, Prescott GJ, Bennett J, Semple S, Dick S, Counsell C, Mozzoni P, Haites N, Wettinger SB, Mutti A, Otelea M, Seaton A, Soderkvist P, Felice A, g. Geoparkinson study Environmental risk factors for Parkinson’s disease and parkinsonism: the Geoparkinson study. Occup Environ Med. 2007;64:666–672. doi: 10.1136/oem.2006.027003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drew SC, Leong SL, Pham CL, Tew DJ, Masters CL, Miles LA, Cappai R, Barnham KJ. Cu2+ binding modes of recombinant alpha-synuclein–insights from EPR spectroscopy. J Am Chem Soc. 2008;130:7766–7773. doi: 10.1021/ja800708x. [DOI] [PubMed] [Google Scholar]
- 37.Du HN, Li HT, Zhang F, Lin XJ, Shi JH, Shi YH, Ji LN, Hu J, Lin DH, Hu HY. Acceleration of alpha-synuclein aggregation by homologous peptides. FEBS letters. 2006;580:3657–3664. doi: 10.1016/j.febslet.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 38.Dunning CJ, George S, Brundin P. What’s to like about the prion-like hypothesis for the spreading of aggregated alpha-synuclein in Parkinson disease? Prion. 2013;7:92–97. doi: 10.4161/pri.23806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eid A, Bihaqi SW, Renehan WE, Zawia NH. Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of Alzheimer’s disease. Alzheimers Dement (Amst) 2016;2:123–131. doi: 10.1016/j.dadm.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003;17:1945–1947. doi: 10.1096/fj.03-0098fje. [DOI] [PubMed] [Google Scholar]
- 41.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Enzenauer RW, Montrey JS, Enzenauer RJ, Mauldin WM. Boxing-related injuries in the US Army, 1980 through 1985. JAMA. 1989;261:1463–1466. [PubMed] [Google Scholar]
- 43.Fares MB, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovicic A, Kiely A, Holton JL, Lee SJ, Gitler AD, Eliezer D, Lashuel HA. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet. 2014;23:4491–4509. doi: 10.1093/hmg/ddu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farina M, Avila DS, da Rocha JB, Aschner M. Metals, oxidative stress and neurodegeneration: a focus on iron, manganese and mercury. Neurochem Int. 2013;62:575–594. doi: 10.1016/j.neuint.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faul MLX, Wald MM, Coronado V, Dellinger AM. Traumatic brain injury in the United States: national estimates of prevalence and incidence, 2002–2006. Injury Prevention. 2010;16:A268. [Google Scholar]
- 46.Febbraro F, Giorgi M, Caldarola S, Loreni F, Romero-Ramos M. alpha-Synuclein expression is modulated at the translational level by iron. Neuroreport. 2012;23:576–580. doi: 10.1097/WNR.0b013e328354a1f0. [DOI] [PubMed] [Google Scholar]
- 47.Feng LR, Maguire-Zeiss KA. Dopamine and paraquat enhance alpha-synuclein-induced alterations in membrane conductance. Neurotox Res. 2011;20:387–401. doi: 10.1007/s12640-011-9255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 49.Fukushima T, Tan X, Luo Y, Kanda H. Serum Vitamins and Heavy Metals in Blood and Urine, and the Correlations among Them in Parkinson’s Disease Patients in China. Neuroepidemiology. 2011;36:240–244. doi: 10.1159/000328253. [DOI] [PubMed] [Google Scholar]
- 50.George JM. The synucleins. Genome Biol. 2002;3 doi: 10.1186/gb-2001-3-1-reviews3002. REVIEWS3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, Ghosh S, Jha NN, Kumar A, Maji SK. The Parkinson’s disease-associated H50Q mutation accelerates alpha-Synuclein aggregation in vitro. Biochemistry. 2013;52:6925–6927. doi: 10.1021/bi400999d. [DOI] [PubMed] [Google Scholar]
- 52.Golts N, Snyder H, Frasier M, Theisler C, Choi P, Wolozin B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J Biol Chem. 2002;277:16116–16123. doi: 10.1074/jbc.M107866200. [DOI] [PubMed] [Google Scholar]
- 53.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology. 1997;48:650–658. doi: 10.1212/wnl.48.3.650. [DOI] [PubMed] [Google Scholar]
- 54.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- 55.Gorojod RM, Alaimo A, Porte Alcon S, Pomilio C, Saravia F, Kotler ML. The autophagic- lysosomal pathway determines the fate of glial cells under manganese-induced oxidative stress conditions. Free Radic Biol Med. 2015;87:237–251. doi: 10.1016/j.freeradbiomed.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 56.Ha Y, Yang A, Lee S, Kim K, Liew H, Lee SH, Lee JE, Lee HI, Suh YH, Park HS, Churchill DG. Dopamine and Cu+/(2+) Can Induce Oligomerization of alpha-Synuclein in the Absence of Oxygen: Two Types of Oligomerization Mechanisms for alpha-Synuclein and Related Cell Toxicity Studies. Journal of Neuroscience Research. 2014;92:359–368. doi: 10.1002/jnr.23323. [DOI] [PubMed] [Google Scholar]
- 57.Harischandra DS, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. alpha-Synuclein protects against manganese neurotoxic insult during the early stages of exposure in a dopaminergic cell model of Parkinson’s disease. Toxicol Sci. 2015;143:454–468. doi: 10.1093/toxsci/kfu247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hart J, Jr, Kraut MA, Womack KB, Strain J, Didehbani N, Bartz E, Conover H, Mansinghani S, Lu H, Cullum CM. Neuroimaging of cognitive dysfunction and depression in aging retired National Football League players: a cross-sectional study. JAMA Neurol. 2013;70:326–335. doi: 10.1001/2013.jamaneurol.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hashimoto M, Rockenstein E, Crews L, Masliah E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med. 2003;4:21–36. doi: 10.1385/NMM:4:1-2:21. [DOI] [PubMed] [Google Scholar]
- 60.Hashimoto M, Yoshimoto M, Sisk A, Hsu LJ, Sundsmo M, Kittel A, Saitoh T, Miller A, Masliah E. NACP, a synaptic protein involved in Alzheimer’s disease, is differentially regulated during megakaryocyte differentiation. Biochemical and biophysical research communications. 1997;237:611–616. doi: 10.1006/bbrc.1997.6978. [DOI] [PubMed] [Google Scholar]
- 61.Hatcher JM, Richardson JR, Guillot TS, McCormack AL, Di Monte DA, Jones DP, Pennell KD, Miller GW. Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system. Exp Neurol. 2007;204:619–630. doi: 10.1016/j.expneurol.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He Q, Song N, Jia F, Xu H, Yu X, Xie J, Jiang H. Role of alpha-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int J Biochem Cell Biol. 2013;45:1019–1030. doi: 10.1016/j.biocel.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 63.Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M. Manganese Is Essential for Neuronal Health. Annu Rev Nutr. 2015;35:71–108. doi: 10.1146/annurev-nutr-071714-034419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry. 2004;43:16233–16242. doi: 10.1021/bi048453u. [DOI] [PubMed] [Google Scholar]
- 65.Ibanez P, Lesage S, Janin S, Lohmann E, Durif F, Destee A, Bonnet AM, Brefel-Courbon C, Heath S, Zelenika D, Agid Y, Durr A, Brice A, French G, Parkinson’s Disease Genetics Study Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol. 2009;66:102–108. doi: 10.1001/archneurol.2008.555. [DOI] [PubMed] [Google Scholar]
- 66.Jackson MS, Lee JC. Identification of the Minimal Copper(II)-Binding alpha-Synuclein Sequence. Inorganic Chemistry. 2009;48:9303–9307. doi: 10.1021/ic901157w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS letters. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 68.Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 70.Ji H, Liu YE, Jia T, Wang M, Liu J, Xiao G, Joseph BK, Rosen C, Shi YE. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 1997;57:759–764. [PubMed] [Google Scholar]
- 71.Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jorgenson JL. Aldrin and dieldrin: a review of research on their production, environmental deposition and fate, bioaccumulation, toxicology, and epidemiology in the United States. Environ Health Perspect. 2001;109(Suppl 1):113–139. doi: 10.1289/ehp.01109s1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kanthasamy AG, Kitazawa M, Yang Y, Anantharam V, Kanthasamy A. Environmental neurotoxin dieldrin induces apoptosis via caspase-3-dependent proteolytic activation of protein kinase C delta (PKCdelta): Implications for neurodegeneration in Parkinson’s disease. Mol Brain. 2008;1:12. doi: 10.1186/1756-6606-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173:692–702. doi: 10.1111/bph.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, Mbefo MK, Vercruysse F, Dietler G, Lee SJ, Eliezer D, Lashuel HA. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J Biol Chem. 2014;289:21856–21876. doi: 10.1074/jbc.M114.553297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience. 2003;119:945–964. doi: 10.1016/s0306-4522(03)00226-4. [DOI] [PubMed] [Google Scholar]
- 77.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 78.Kostka M, Hogen T, Danzer KM, Levin J, Habeck M, Wirth A, Wagner R, Glabe CG, Finger S, Heinzelmann U, Garidel P, Duan W, Ross CA, Kretzschmar H, Giese A. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J Biol Chem. 2008;283:10992–11003. doi: 10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 79.Kumar RG, Boles JA, Wagner AK. Chronic Inflammation After Severe Traumatic Brain Injury: Characterization and Associations With Outcome at 6 and 12 Months Postinjury. J Head Trauma Rehabil. 2015;30:369–381. doi: 10.1097/HTR.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 80.Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M. Manganese-Induced Parkinsonism and Parkinson’s Disease: Shared and Distinguishable Features. Int J Environ Res Public Health. 2015;12:7519–7540. doi: 10.3390/ijerph120707519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kwik-Uribe C, Smith DR. Temporal responses in the disruption of iron regulation by manganese. J Neurosci Res. 2006;83:1601–1610. doi: 10.1002/jnr.20836. [DOI] [PubMed] [Google Scholar]
- 82.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 83.Lazaro DF, Rodrigues EF, Langohr R, Shahpasandzadeh H, Ribeiro T, Guerreiro P, Gerhardt E, Krohnert K, Klucken J, Pereira MD, Popova B, Kruse N, Mollenhauer B, Rizzoli SO, Braus GH, Danzer KM, Outeiro TF. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014;10:e1004741. doi: 10.1371/journal.pgen.1004741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM., Jr Pathology of inherited manganese transporter deficiency. Ann Neurol. 2014;75:608–612. doi: 10.1002/ana.24131. [DOI] [PubMed] [Google Scholar]
- 85.Lee HJ, Lee SJ. Characterization of cytoplasmic alpha-synuclein aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J Biol Chem. 2002;277:48976–48983. doi: 10.1074/jbc.M208192200. [DOI] [PubMed] [Google Scholar]
- 86.Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. The international journal of biochemistry & cell biology. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 87.Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry. 2001;40:11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 88.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nature medicine. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 89.Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Y, Sun L, Cai T, Zhang Y, Lv S, Wang Y, Ye L. alpha-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain Res Bull. 2010;81:428–433. doi: 10.1016/j.brainresbull.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 91.Liang G, Qin H, Zhang L, Ma S, Huang X, Lv Y, Qing L, Li Q, Xiong Y, Huang Y, Chen K, Huang Y, Shen Y, Nong J, Yang X, Zou Y. Effects of chronic manganese exposure on the learning and memory of rats by observing the changes in the hippocampal cAMP signaling pathway. Food Chem Toxicol. 2015;83:261–267. doi: 10.1016/j.fct.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 92.Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 93.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 94.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE. Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med. 2013;62:65–75. doi: 10.1016/j.freeradbiomed.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain: a journal of neurology. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michalke B, Fernsebner K. New insights into manganese toxicity and speciation. J Trace Elem Med Biol. 2014;28:106–116. doi: 10.1016/j.jtemb.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 98.Mondello S, Buki A, Italiano D, Jeromin A. alpha-Synuclein in CSF of patients with severe traumatic brain injury. Neurology. 2013;80:1662–1668. doi: 10.1212/WNL.0b013e3182904d43. [DOI] [PubMed] [Google Scholar]
- 99.More SV, Kumar H, Kim IS, Song SY, Choi DK. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediators Inflamm. 2013;2013:952375. doi: 10.1155/2013/952375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Navarro-Otano J, Gelpi E, Mestres CA, Quintana E, Rauek S, Ribalta T, Santiago V, Tolosa E. Alpha-synuclein aggregates in epicardial fat tissue in living subjects without parkinsonism. Parkinsonism Relat Disord. 2013;19:27–31. doi: 10.1016/j.parkreldis.2012.07.005. discussion 27. [DOI] [PubMed] [Google Scholar]
- 101.Negro A, Brunati AM, Donella-Deana A, Massimino ML, Pinna LA. Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2002;16:210–212. doi: 10.1096/fj.01-0517fje. [DOI] [PubMed] [Google Scholar]
- 102.Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, Hyman BT. Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J Neuropathol Exp Neurol. 1999;58:1263–1268. doi: 10.1097/00005072-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 103.Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- 104.Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, Said J, Marsico G, Verbavatz JM, Rodrigo-Angulo M, Gille G, Funk RH, Reichmann H. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Scientific reports. 2012;2:898. doi: 10.1038/srep00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petty MA, Lo EH. Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002;68:311–323. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- 106.Povlishock JT. The classification of traumatic brain injury (TBI) for targeted therapies. J Neurotrauma. 2008;25:717–718. doi: 10.1089/neu.2008.9964. [DOI] [PubMed] [Google Scholar]
- 107.Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson’s disease: a metaanalysis. Environ Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- 108.Pyatigorskaya N, Sharman M, Corvol JC, Valabregue R, Yahia-Cherif L, Poupon F, Cormier-Dequaire F, Siebner H, Klebe S, Vidailhet M, Brice A, Lehericy S. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov Disord. 2015;30:1077–1084. doi: 10.1002/mds.26218. [DOI] [PubMed] [Google Scholar]
- 109.Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO. Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc Natl Acad Sci U S A. 2005;102:4294–4299. doi: 10.1073/pnas.0407881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rochet JC, Conway KA, Lansbury PT., Jr Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39:10619–10626. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- 111.Roth JA. Correlation between the biochemical pathways altered by mutated parkinson-related genes and chronic exposure to manganese. Neurotoxicology. 2014;44:314–325. doi: 10.1016/j.neuro.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 112.Rybicki BA, Johnson CC, Uman J, Gorell JM. Parkinsons-Disease mortality and the industrial use of heavy-metals in Michigan. Movement Disorders. 1993;8:87–92. doi: 10.1002/mds.870080116. [DOI] [PubMed] [Google Scholar]
- 113.Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT, T. Workshop Scientific. M. Advisory Panel Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saberzadeh J, Arabsolghar R, Takhshid MA. Alpha synuclein protein is involved in Aluminum-induced cell death and oxidative stress in PC12 cells. Brain Res. 2016;1635:153–160. doi: 10.1016/j.brainres.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 115.Sanchez-Ramos J, Facca A, Basit A, Song S. Toxicity of dieldrin for dopaminergic neurons in mesencephalic cultures. Exp Neurol. 1998;150:263–271. doi: 10.1006/exnr.1997.6770. [DOI] [PubMed] [Google Scholar]
- 116.Schildknecht S, Bachschmid M, Ullrich V. Peroxynitrite provides the peroxide tone for PGHS-2-dependent prostacyclin synthesis in vascular smooth muscle cells. FASEB J. 2005;19:1169–1171. doi: 10.1096/fj.04-3465fje. [DOI] [PubMed] [Google Scholar]
- 117.Serbest G, Burkhardt MF, Siman R, Raghupathi R, Saatman KE. Temporal profiles of cytoskeletal protein loss following traumatic axonal injury in mice. Neurochem Res. 2007;32:2006–2014. doi: 10.1007/s11064-007-9318-9. [DOI] [PubMed] [Google Scholar]
- 118.Sheftel AD, Mason AB, Ponka P. The long history of iron in the Universe and in health and disease. Biochim Biophys Acta. 2012;1820:161–187. doi: 10.1016/j.bbagen.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- 120.Shin EC, Cho SE, Lee DK, Hur MW, Paik SR, Park JH, Kim J. Expression patterns of alpha-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cells. 2000;10:65–70. doi: 10.1007/s10059-000-0065-x. [DOI] [PubMed] [Google Scholar]
- 121.Spillantini MG, Divane A, Goedert M. Assignment of human alpha-synuclein (SNCA) and beta-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics. 1995;27:379–381. doi: 10.1006/geno.1995.1063. [DOI] [PubMed] [Google Scholar]
- 122.Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- 123.Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiology of aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun F, Anantharam V, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Dieldrin induces ubiquitin-proteasome dysfunction in alpha-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. The Journal of pharmacology and experimental therapeutics. 2005;315:69–79. doi: 10.1124/jpet.105.084632. [DOI] [PubMed] [Google Scholar]
- 125.Surguchov A, Surgucheva I, Solessio E, Baehr W. Synoretin–A new protein belonging to the synuclein family. Mol Cell Neurosci. 1999;13:95–103. doi: 10.1006/mcne.1999.0735. [DOI] [PubMed] [Google Scholar]
- 126.Tang-Schomer MD, Patel AR, Baas PW, Smith DH. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:1401–1410. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tavassoly O, Nokhrin S, Dmitriev OY, Lee JS. Cu(II) and dopamine bind to alpha-synuclein and cause large conformational changes. Febs Journal. 2014;281:2738–2753. doi: 10.1111/febs.12817. [DOI] [PubMed] [Google Scholar]
- 128.Tobe T, Nakajo S, Tanaka A, Mitoya A, Omata K, Nakaya K, Tomita M, Nakamura Y. Cloning and characterization of the cDNA encoding a novel brain-specific 14-kDa protein. J Neurochem. 1992;59:1624–1629. doi: 10.1111/j.1471-4159.1992.tb10991.x. [DOI] [PubMed] [Google Scholar]
- 129.Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003;278:44405–44411. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- 130.Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner GG. Re-examination of exboxers’ brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol. 1991;82:280–285. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- 131.Tsunemi T, Krainc D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23:2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Uversky VN, Li J, Bower K, Fink AL. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson’s disease. Neurotoxicology. 2002;23:527–536. doi: 10.1016/s0161-813x(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 135.Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem. 2001;276:44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 136.Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett. 2001;500:105–108. doi: 10.1016/s0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- 137.Valensin D, Dell’Acqua S, Kozlowski H, Casella L. Coordination and redox properties of copper interaction with alpha-synuclein. J Inorg Biochem. 2016 doi: 10.1016/j.jinorgbio.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 138.Verina T, Schneider JS, Guilarte TR. Manganese exposure induces alpha-synuclein aggregation in the frontal cortex of non-human primates. Toxicol Lett. 2013;217:177–183. doi: 10.1016/j.toxlet.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Arguelles S, Delgado-Cortes MJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, Cano J, Machado A. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson’s disease. J Neurochem. 2010;114:1687–1700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
- 140.Visanji NP, Brooks PL, Hazrati LN, Lang AE. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta neuropathologica communications. 2013;1:2. doi: 10.1186/2051-5960-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang X, Moualla D, Wright JA, Brown DR. Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J Neurochem. 2010;113:704–714. doi: 10.1111/j.1471-4159.2010.06638.x. [DOI] [PubMed] [Google Scholar]
- 142.White AR, Kanninen KM, Crouch PJ. Editorial: Metals and neurodegeneration: restoring the balance. Front Aging Neurosci. 2015;7:127. doi: 10.3389/fnagi.2015.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wills J, Credle J, Oaks AW, Duka V, Lee JH, Jones J, Sidhu A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One. 2012;7:e30745. doi: 10.1371/journal.pone.0030745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xiang W, Schlachetzki JC, Helling S, Bussmann JC, Berlinghof M, Schaffer TE, Marcus K, Winkler J, Klucken J, Becker CM. Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci. 2013;54:71–83. doi: 10.1016/j.mcn.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 145.Xu B, Wu SW, Lu CW, Deng Y, Liu W, Wei YG, Yang TY, Xu ZF. Oxidative stress involvement in manganese-induced alpha-synuclein oligomerization in organotypic brain slice cultures. Toxicology. 2013;305:71–78. doi: 10.1016/j.tox.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 146.Yousefi Babadi V, Sadeghi L, Shirani K, Malekirad AA, Rezaei M. The toxic effect of manganese on the acetylcholinesterase activity in rat brains. J Toxicol. 2014;2014:946372. doi: 10.1155/2014/946372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yuan YH, Yan WF, Sun JD, Huang JY, Mu Z, Chen NH. The molecular mechanism of rotenone-induced alpha-synuclein aggregation: emphasizing the role of the calcium/GSK3beta pathway. Toxicol Lett. 2015;233:163–171. doi: 10.1016/j.toxlet.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 148.Zarbiv Y, Simhi-Haham D, Israeli E, Elhadi SA, Grigoletto J, Sharon R. Lysine residues at the first and second KTKEGV repeats mediate alpha-Synuclein binding to membrane phospholipids. Neurobiol Dis. 2014;70:90–98. doi: 10.1016/j.nbd.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 149.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of neurology. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 150.Zhang H, Griggs A, Rochet JC, Stanciu LA. In vitro study of alpha-synuclein protofibrils by cryo-EM suggests a Cu(2+)-dependent aggregation pathway. Biophys J. 2013;104:2706–2713. doi: 10.1016/j.bpj.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]