

Abstract
Background
Group 2 innate lymphoid cells (ILC2s) expand in the lungs of mice during type 2 inflammation induced by the fungal allergen Alternaria alternata. The increase in ILC2 numbers in the lung has been largely attributed to local proliferation and whether ILC2s migrate from the circulation to the lung following Alternaria exposure is unknown.
Objective
We examined whether human (lung, lymph node, blood) and mouse lung ILC2s express β1 and β2 integrin adhesion molecules, and whether these integrins are required for trafficking of ILC2 into the lungs of mice
Methods
Human and mouse ILC2s were assessed for surface expression of β1 and β2 integrins adhesion molecules by flow cytometry. The role of β1 and β2 integrins in ILC2 trafficking to the lungs was assessed by in vivo blocking of these integrins prior to airway exposure to Alternaria in mice.
Results
Both human and mouse lung ILC2s express high levels of β1 and β2 integrin adhesion receptors. Intranasal administration of Alternaria challenge reduced ILC2s in the bone marrow and concurrently increased blood and lung ILC2 levels. In vivo blocking of β2 integrins (CD18) significantly reduced ILC2 levels in the lungs, but did not alter ILC2 proliferation, apoptosis, and function. In contrast, in vivo blocking of β1 integrins or α4 integrins did not affect lung ILC2 levels.
Conclusion
ILC2s increase in number in the mouse lung not only through local proliferation, but also through trafficking from the circulation into the lung using β2 rather than β1 or α4 integrins.
Keywords: ILC2, Integrin, Adhesion, Alternaria alternata
INTRODUCTION
As ILC2s express high levels of Th2 cytokines (IL-4, IL-5, and IL-13), studies have focused on understanding their role in the pathogenesis of human diseases including asthma, allergic rhinitis, and atopic dermatitis.1–5 While ILC2s and Th2 lymphocytes are both prominent sources of Th2 cytokines, ILC2s do not express antigen receptors and are instead activated by cytokines (i.e. IL-33, TSLP, IL-25) and lipid mediators (i.e. cysteinyl leukotrienes and prostaglandin D2).6, 7 Prostaglandin D2 induces ILC2 Th2 cytokine production and is also a chemotactic factor for ILC2s through the binding to CRTH2, which is highly expressed by ILC2s.8 Mediators that activate ILC2s are known to be elevated in different Th2 disease states, including asthma and allergic rhinitis. The number of ILC2s is increased in the peripheral blood, BAL, and sputum of allergic and/or asthmatic individuals, and several studies demonstrate increased ILC2s in the lung of mice during Th2 inflammation.9–13
While it is well accepted that T lymphocytes utilize adhesion molecules to traffic from the circulation to the lung following inhalation of allergen14, studies have not yet examined whether ILC2s migrate from the circulation to the lung following allergen challenge, and if so which adhesion receptors expressed by ILC2s may mediate this function. Several reports have demonstrated that local proliferation of ILC2s occurs in the tissues, including the lung.15–17 However, ILC2s are bone marrow-derived cells and mechanisms that regulate trafficking from blood to tissues could be important during inflammatory responses.18 Mice deficient in bone marrow CRTH2-expressing ILC2s had diminished ILC2 levels in the lungs following N. brasiliensis infection19, suggesting that a trafficking defect may be involved, but whether ILC2-expressed adhesion receptors may account for this reduction in ILC2 levels in the lung was not examined. We hypothesized that bone marrow derived ILC2s migrating through the blood express adhesion molecules that firmly bind to counter-receptors expressed by lung endothelium during allergic inflammation. This firm adhesion of ILC2s to pulmonary endothelium then allows for their subsequent migration into the tissues through chemotactic mediators, such as prostaglandin D2.
The most common method by which leukocytes (other than ILC2s) are known to migrate from the blood into tissues and specifically the airways is mediated by leukocyte-expressed selectins and integrins.20 Leukocyte-expressed selectins, such as L-selectin (CD62L), mediate the first step of leukocyte tethering to endothelium in vivo, whereas leukocyte-expressed β1 and β2 integrins mediate leukocyte firm adhesion to endothelium. Integrins are heterodimers comprised of α and β subunits. β1 (CD29) integrins, such as VLA-4 (α4/β1 or CD49d/CD29), are highly expressed on eosinophils, T lymphocytes, and basophils, but not neutrophils.21, 22 VLA-4 expressed by leukocytes binds to vascular cell adhesion molecule (VCAM-1 or CD106) expressed by endothelium. In addition to β1 integrins, β2 and β7 integrins expressed by leukocytes also mediate firm adhesion. For example, lymphocytes utilize β2 (CD18) integrins, such as LFA-1 (αL/β2 or CD11a/CD18), to firmly adhere to intercellular adhesion molecule 1 (ICAM-1 or CD54) expressed by endothelium.23 Although lymphocytes also utilize β7 integrins to firmly adhere to endothelium, this interaction of lymphocyte expressed β7 integrins (i.e. α4/β7) with endothelium is more prominent in migration of lymphocytes to the gut24, as the counter-receptor for α4/β7 (i.e. MAdCAM-1) is most highly expressed in the gastro-intestinal tract rather than the lung, which is the focus of our ILC2 studies.
Overall, studies of β integrins and leukocyte trafficking to sites of allergic inflammation have demonstrated an important role for both β1 and β2 integrins in eosinophil adhesion and trafficking to tissues.25 In contrast, for neutrophils β2 integrins, rather than β1 integrins, predominantly mediate neutrophil trafficking to tissue sites of inflammation.26 We therefore profiled expression of integrin adhesion molecules on human and mouse ILC2s to determine which adhesion receptors were highly expressed by ILC2s, and used neutralizing antibodies to demonstrate that ILC2s traffic from the circulation using β2, but not β1, integrins during Alternaria induced lung inflammation.
METHODS
Human ILC2
Human ILC2s were identified by flow cytometry as CD45+Lin−CRTH2+ lymphoid cells8, 27 in single cell suspensions of either post mortem human lungs and lymph nodes, or human peripheral blood, obtained as described in the online supplement.
Mouse ILC2: Airway administration of Alternaria and in vivo blocking of integrins
Mice were challenged intranasally with Alternaria Alternata three times (days 0, 3, and 6) and were sacrificed one day after the final challenge (day 7) as previously described10 and detailed in the online supplement. In experiments involving in vivo blocking of integrins, mice were administered intraperitoneally an integrin specific blocking or control antibody (β1, β2, α4, or αL antibody) (see Supplement Table EII) prior to each Alternaria challenge (days 0, 3, and 6) as detailed in the online supplement.
ILC2 proliferation, apoptosis, and Th2 cytokines
ILC2 proliferation (Ki-67 staining), apoptosis (annexin V staining) and Th2 cytokines (IL-5, IL-13 staining) were quantitated by flow cytometry as described in the online supplement.
Statistical analysis
Statistical analysis was performed using Prism software (Graphpad, La Jolla, CA) as described in the online supplement. P < 0.05 was considered statistically significant.
RESULTS
Human ILC2s express β1 and β2 integrins
To determine whether human ILC2s express adhesion molecules commonly associated with migration of other leukocytes, human ILC2s derived from peripheral blood, lung tissue, and intrathoracic lymph nodes were analyzed for expression of adhesion molecules by flow cytometry. Human ILC2s were gated as CD45+Lin−CRTH2+ lymphoid cells (Figure 1A) and were detected at a mean frequency of 0.15% of CD45+ cells in the blood, 0.04% in the lungs and 0.06% in lung lymph nodes (Figure 1B). Human ILC2s highly express both the integrin chains for β2 integrins (CD18 and CD11a) and moderately express both the integrin chains for β1 integrins (CD29 and CD49d) (Figures 1C–E and Supplement Figures E1–E2). Compared with human CD4 cells, human ILC2s express similar levels of β1 and β2 integrin adhesion molecules (Figure 1F–H).
Figure 1. Human ILC2s express CD18/CD11a (β2/αL) and CD29/CD49d (β1/α4) integrins.
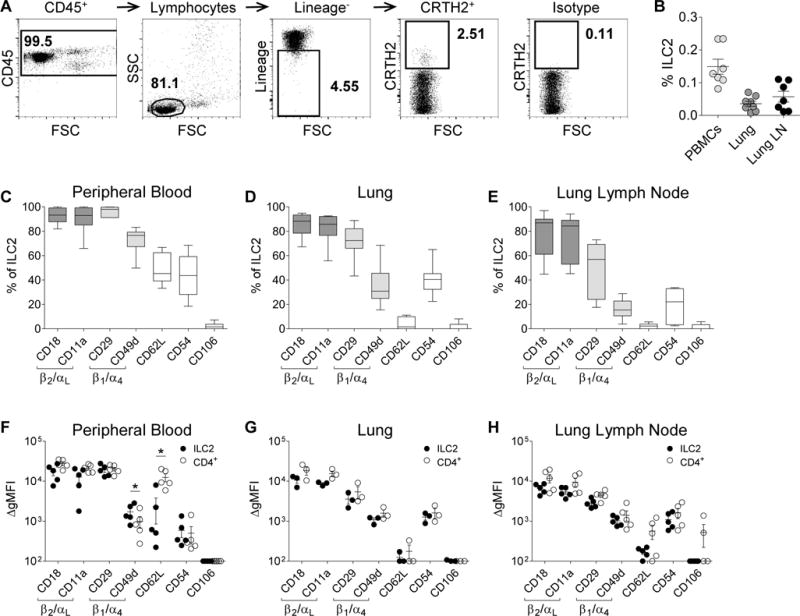
(A) Representative ILC2 gating strategy from human lymph nodes. (B) The frequency of ILC2s from CD45+ human PBMCs, lungs, and lung lymph nodes. Summary of ILC2 adhesion molecule expression from (C) PBMCs (n=7), (D) lung tissue (n=8), and (E) lymph nodes (n=7). Comparison of ILC2 and CD4+CRTH2+ cell adhesion molecule ΔgMFI from (F) PBMCs (n=5), (G) lungs (n=3), and (H) lymph nodes (n=5) of the same donor. Solid circles represent ILC2 populations and open circles represent CD4+CRTH2+ populations. Data depicted as boxplots (whiskers: 10th–90th percentile) or individual points; mean represented as a line ± SEM. *P<0.05 by Student’s paired t test.
Mouse ILC2s express β1 and β2 integrins
To study the functional importance of ILC2 expression of adhesion molecules to recruitment of ILC2 to the lung, we used a well validated model of Alternaria allergen challenge in non-sensitized wildtype mice which induces a rapid innate mediated increase in the number of ILC2s.13 To validate that mouse ILC2s expressed a similar profile of adhesion molecules as human ILC2s, the expression of the same adhesion molecules were assessed by flow cytometry on ILC2s from mouse lungs. Mouse ILC2s were identified as Lin−Thy1.2+ from the CD45+ lymphoid cell population (Figure 2A). We have previously used this gating strategy to show that the Lin−Thy1.2+ are ILC2s in naïve and Alternaria-challenged mice.28 To determine relative levels of adhesion molecule expression, ILC2s and CD4+Thy1.2+ cells from the same naïve mice lungs were analyzed (Figure 2B–C and Supplement Figure E3).
Figure 2. Adhesion molecule expression on mouse lung ILC2s.
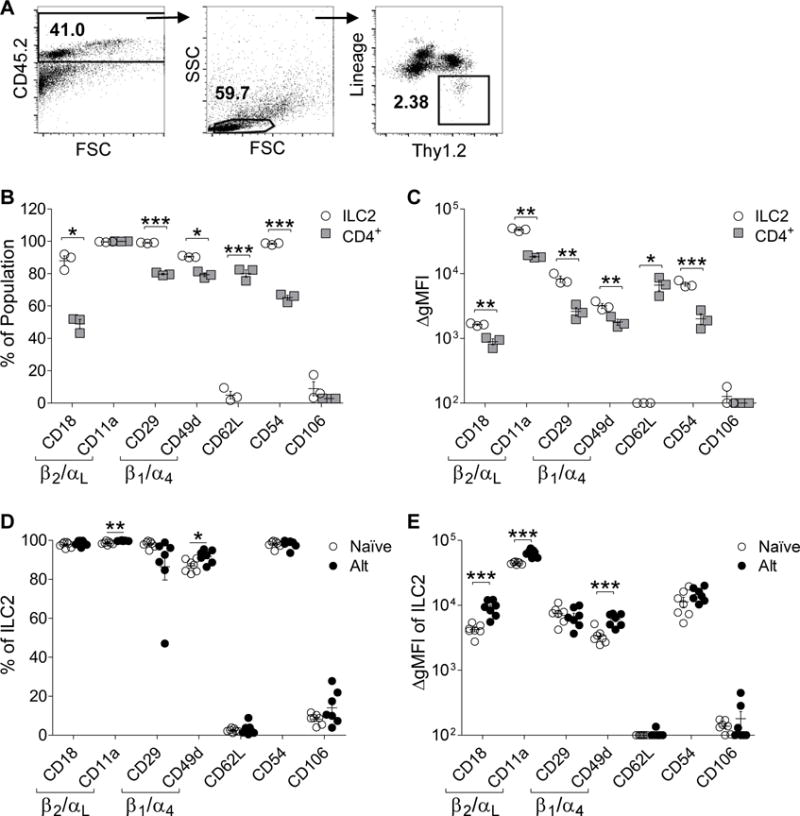
(A) Representative gating strategy of mouse lung ILC2s. Mouse lung ILC2s (open circles) compared to lung CD4+Thy1.2+ cells (gray squares) (B) percent adhesion molecule expression and (C) adhesion molecule ΔgMFI (n=3).Comparison of mouse lung ILC2 (D) percent expression and (E) ΔgMFI of adhesion molecules between naïve mice (open circles) and Alternaria challenged mice (solid circles; n=7). Independent experiments depicted as individual points; mean represented as a line ± SEM. *P<0.05, **P<0.01, ***P<0.001 by Student’s t test.
ILC2s showed slightly higher levels of expression compared to CD4 cells of the β1 integrin CD29 (mean 99.1% compared to 79.8%) and CD49d (mean 90.5% compared to 79.4%), as well as the β2 integrin CD18 (mean 88.0% compared to 49.0%). In addition, ICAM-1 was more highly expressed on ILC2 compared to CD4 cells (mean 98.5% compared to 65.2%), while both populations showed equally high percentages of CD11a expression (mean > 99%; Figure 2B). Interestingly, the percent of lung CD4 cells expressing L-selectin (CD62L; mean = 80.1%) was markedly higher compared to lung ILC2 cells expressing L-selectin (mean = 4.9%; p < 0.0001) (Figure 2B).
These differences were further demonstrated by the comparison of ΔgMFI between the ILC2s and CD4+Thy1.2+ populations (Figure 2C). ILC2s showed higher ΔgMFI for the β2 integrins CD18 (ΔΔgMFI = 744) and CD11a (ΔΔgMFI = 29,590), as well as the β1 integrins CD29 (ΔΔgMFI = 5,658) and CD49d (ΔΔgMFI = 1,399), and ICAM-1 (ΔΔgMFI = 4,896). However, ILC2s had decreased ΔgMFI of CD62L compared to the CD4+Thy1.2+ population (ΔΔgMFI = 6,615; Figure 2C). As expected, ILC2 and CD4+Thy1.2+ populations had minimal expression or ΔgMFI for the endothelial adhesion molecule VCAM-1 (CD106). Thus, compared with human ILC2s, mouse lung ILC2s express high levels of both β2 (CD18/CD11a) and β1 (CD29/CD49d) integrins, which are at slightly higher levels compared to CD4 cells.
Effect of Alternaria challenge on levels of ILC2 adhesion molecule expression
Non-sensitized naïve mice challenged with the fungal allergen Alternaria alternata over three days develop innate type 2 inflammation and ILC2 activation.27 Therefore, we assessed lung ILC2 adhesion molecule expression after activation in vivo by Alternaria challenge (Figure 2D–E). Similar to human lung ILC2s, the majority of naïve non-Alternaria challenged mouse lung ILC2s expressed high levels of the β2 integrins CD18 (mean = 97.7%) and CD11a (mean = 98.6%) as well as the β1 integrins CD29 (mean = 98.0%) and CD49d (mean = 86.9%). Levels of ICAM-1 were highly expressed in mouse lung ILC2 (mean = 98.0%). In contrast, CD62L (mean = 2.5%) and VCAM-1 (mean = 8.2%) were expressed at low levels (Figure 2B). After Alternaria challenge, levels of lung ILC2 expression of the β2 integrin CD18 and the β1 integrin CD29, as well as CD62L, ICAM-1, and VCAM-1 did not change. However, there was a minimal but statistically significant increase in the percentage of ILC2s expressing the β2 integrin CD11a and the β1 integrin CD49d (Figure 2D). There was a statistically significant increase in the ΔgMFI of mouse lung ILC2 β2 integrin subunits CD18 (ΔΔgMFI = 4,954) and CD11a (ΔΔgMFI = 19,114), as well as in the β1 integrin subunit CD49d (ΔΔgMFI = 2,723) after Alternaria challenge (Figure 2E). In addition, after Alternaria challenge, levels of lung ILC2 ΔgMFI of CD29, CD62L, ICAM-1, and VCAM-1 did not change (Figure 2E). Thus, Alternaria challenge in mice led to modest increases in β1 and β2 integrin ΔgMFI adhesion molecule expression, but did not alter the overall expression profile.
Alternaria challenge reduced ILC2s in the bone marrow and increased ILC2s in the blood and lungs
To determine whether ILC2s circulate from the bone marrow to the lungs following Alternaria challenge, we assessed ILC2 levels in the bone marrow, blood, and lungs of mice challenged every three days with Alternaria allergen. Mice were sacrificed 24 hours after either the second challenge on day 4, or the third challenge on day 7 (Figure 3). Mouse bone marrow and blood ILC2s were characterized as Lin−Thy1.2+CD127+ST2+ from the CD45+ lymphoid cell population (Supplement Figure E4). Intranasal administration of Alternaria significantly diminished bone marrow ILC2 levels on day 7 compared to both naïve and day 4 mice (Figure 3A). There were little to no detectable blood ILC2s in the naïve and day 4 mice. However, Alternaria challenge significantly increased blood ILC2 levels on day 7 compared to naïve and day 4 mice (Figure 3B). In addition, ILC2 levels in the lung lymph nodes and spleen were found to be increased on day 7 (Supplemental Figures E4 C–D and E5 A–B). As expected, mice sacrificed on both day 4 and day 7 had increased levels of lung ILC2s compared to naïve mice (Figure 3C). Summary of the changes in overall CD45+ cell levels are depicted in Supplement Figure E5 C–G. Together, these studies demonstrate that intranasal administration of Alternaria reduced ILC2s in the bone marrow and concurrently increased blood and lung ILC2 levels on day 7.
Figure 3. ILC2 levels change in bone marrow, blood, and lungs following Alternaria challenge in wt and BMT mice.
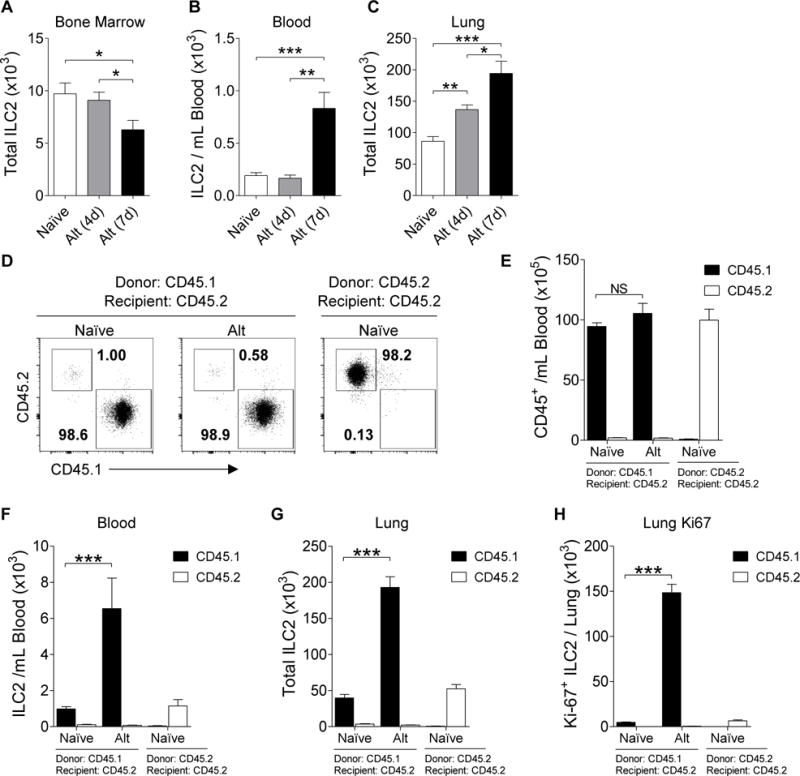
Total ILC2s from (A) bone marrow, (B) blood, and (C) whole lung from naïve (white; n≥23), 4-day Alternaria challenged (gray; n=8), and 7-day Alternaria challenged mice (black; n≥14). CD45.1 or CD45.2 donor bone marrow was transplanted into CD45.2 irradiated recipient mice (D) Representative plots of BMT mouse blood ILC2 expression of CD45.1 and CD45.2. (E) Total CD45.1 (black) and CD45.2 (white) blood cells from BMT mice. Total ILC2s from (F) blood and (G) whole lung from naïve (n=8) or Alternaria challenged (n=4) BMT mice. (H) Total lung Ki-67+ ILC2s from BMT mice. Data summarized as bar graphs representing mean ± SEM. *P< 0.05, **P< 0.01, ***P< 0.001 by Student’s t test.
ILC2s in the lungs following Alternaria challenge originate in bone marrow in mice
In order to determine whether ILC2s migrate from the bone marrow to the airways, we generated bone marrow transplanted (BMT) mice in which either CD45.1 or CD45.2 donor bone marrow was transplanted into irradiated recipient CD45.2 mice, and challenged them with Alternaria (as described in the Methods in the Online Supplement). In both the CD45.1 and the CD45.2 BMT mice, over 95% of all the hematopoietic cells in the blood were of donor origin (Figure 3D–E). Alternaria challenge significantly increased CD45.1 ILC2 levels in the blood and lungs of the CD45.1 donor BMT mice, but did not increase CD45.2 ILC2 levels (Figure 3F–G and Supplemental figure E6 A–C). In addition, there was a significant increase in the total number of proliferating (Ki-67 expressing) CD45.1 ILC2s in the Alternaria challenged CD45.1 BMT mice compared to the naïve CD45.1 BMT mice (Figure 3H). There was little Ki-67 expressing CD45.2 ILC2s in both the naïve and Alternaria challenged CD45.1 BMT lungs (Supplemental Figure E6 D). These results suggest that ILC2s migrate from the bone marrow to the lungs.
In vivo blocking of β2 integrins reduced ILC2 recruitment to the lung
Due to the high levels of β2 integrins we noted to be expressed on both human and mouse ILC2s we examined whether administering neutralizing antibodies to β2 integrins would inhibit the accumulation of ILC2s in the lungs of Alternaria challenged mice (Figure 4A). Alternaria challenged mice treated with control antibody had significantly higher total numbers of ILC2s in the lungs compared to naïve mice (Figure 4B). In contrast, Alternaria-challenged mice pretreated with an αCD18 β2 integrin blocking antibody had significantly reduced absolute ILC2s per lung compared to Alternaria challenged mice (61.3% reduction of Alternaria-induced ILC2 increase in lung) (Figure 4B).
Figure 4. In vivo blocking of CD18 diminishes Alternaria-induced ILC2 recruitment to the airways.
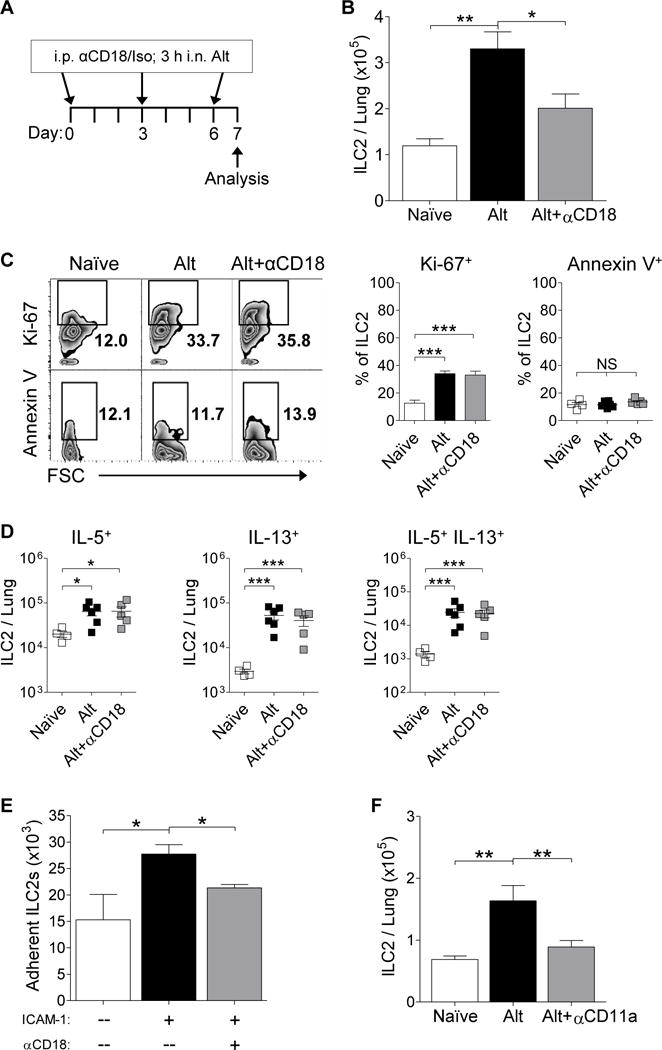
(A) Mice treated with anti-CD18 antibody (αCD18) or the isotype control and challenged with Alternaria (Alt) (B) Total ILC2s per mouse whole lung (n≥10). (C) Representative plots of ILC2 Ki-67 and Annexin V expression and summary of the percentage of Ki-67+ ILC2s (n≥10) and live Annexin V+ ILC2s (n≥4) per mouse lung. (D) Summary of total IL-5, IL-13, and IL-5/IL-13 producing ILC2s in the lung (n≥4; data was log transformed for normalcy). (E) Total number of sorted ILC2s adherent to ICAM-1 coated wells compared to control coated wells ± preincubation of ILC2s with αCD18 (two independent experiments). (F) Total ILC2s per lung of mice treated ± Alt and ± anti-CD11a antibody (n≥6). Data summarized as bar graphs or individual points; mean represented as a line ± SEM. (NS) not significant, *P< 0.05, **P< 0.01, ***P< 0.001 by Student’s t test.
In vivo blocking of β2 integrins does not inhibit ILC2 proliferation, or induce ILC2 apoptosis
To confirm that the effect of the αCD18 antibody on the levels of lung ILC2 following Alternaria challenge was due to inhibition of ILC2 migration rather than inhibition of ILC2 proliferation or induction of ILC2 apoptosis, ILC2 levels of Ki-67 and Annexin V were determined (Figure 4C). Alternaria challenge induced increased lung ILC2 proliferation, as previously described.29 Administration of the αCD18 antibody to Alternaria challenged mice did not inhibit lung ILC2 proliferation (Ki-67+ ILC2s) or ILC2 apoptosis (Annexin V+ ILC2s) (Figure 4C). Thus, β2 integrins are required for the accumulation of lung ILC2s from the blood without affecting proliferation or apoptosis.
Effect of in vivo blocking of β2 integrins on ILC2 Th2 cytokine expression
To determine whether pretreatment with the αCD18 antibody had an effect on the activation of ILC2s, we assessed IL-5 and IL-13 production. Alternaria allergen challenge induced increased numbers of IL-5+ ILC2, IL-13+ ILC2, as well as ILC2s that were double positive for IL-5 and IL-13 (Figure 4D). The ILC2s in the lung following Alternaria allergen challenge include ILC2 expressing Th2 cytokines IL-5 and IL-13 (approximately 23% and 17% of total lung ILC2 respectively), as well as a population of ILC2 not expressing Th2 cytokines. Administration of the αCD18 antibody did not reduce cytokine-producing ILC2 numbers (Figure 4D), despite reducing the total number of ILC2 (Figure 4B) suggesting a reduction in the number of ILC2s that do not express Th2 cytokines after administration of the αCD18 antibody (Supplement Figure E7).
In vitro adhesion of ILC2s to ICAM-1 is dependent on β2 integrins
In order to determine direct interaction between ILC2s and ICAM-1, sorted mouse lung ILC2s were plated on ICAM-1 coated plates and allowed to adhere for 90 minutes (as described in the Methods in the Online Supplement). There was a significant increase in the number of adherent ILC2s in the ICAM-1 coated wells compared to control coated wells (Figure 4E). To determine the role of β2 in the adherence of ILC2s to ICAM-1, sorted ILC2s were preincubated in the presence or absence of the αCD18 antibody prior to plating on ICAM-1 coated plates. There was a significant reduction in the number of ICAM-1 adherent ILC2s in wells containing ILC2s preincubated with αCD18 compared to control ILC2s (Figure 4E). This demonstrates the direct interaction between ILC2s and ICAM-1 in vitro, which is dependent on ILC2 expression of CD18.
In vivo blocking of the β2 binding integrin, αL, inhibits ILC2 recruitment to the airways
Both human and mouse ILC2s had high expression of αL, which is one of the α integrins that interacts with β2. Therefore, we tested whether blocking the integrin αLβ2 (LFA-1), would also inhibit ILC2 recruitment to the airways. Alternaria challenged mice pretreated with a blocking anti-αL integrin (αCD11a) antibody30 had a significant reduction in lung ILC2 levels compared to the lung ILC2 levels of Alternaria challenged mice that were pretreated with the isotype control (Figure 4F).
In vivo blocking of either β1 or α4 integrins does not inhibit ILC2 recruitment to the airways
As ILC2s also express high levels of β1 integrins, we investigated whether blocking this pathway would inhibit ILC2 numbers in the lung following Alternaria challenge. Alternaria challenged mice pretreated with a blocking anti-β1 integrin (αCD29) antibody31 had no reduction in total lung ILC2 (Figure 5A). Administration of the αCD29 antibody to Alternaria challenged mice did not inhibit lung ILC2 proliferation (Figure 5B). Similarly, administration of the αCD29 antibody to Alternaria challenged mice did not induce lung ILC2 apoptosis (Figure 5C).
Figure 5. In vivo blocking of CD29 and CD49d does not alter Alternaria–induced ILC2 recruitment.
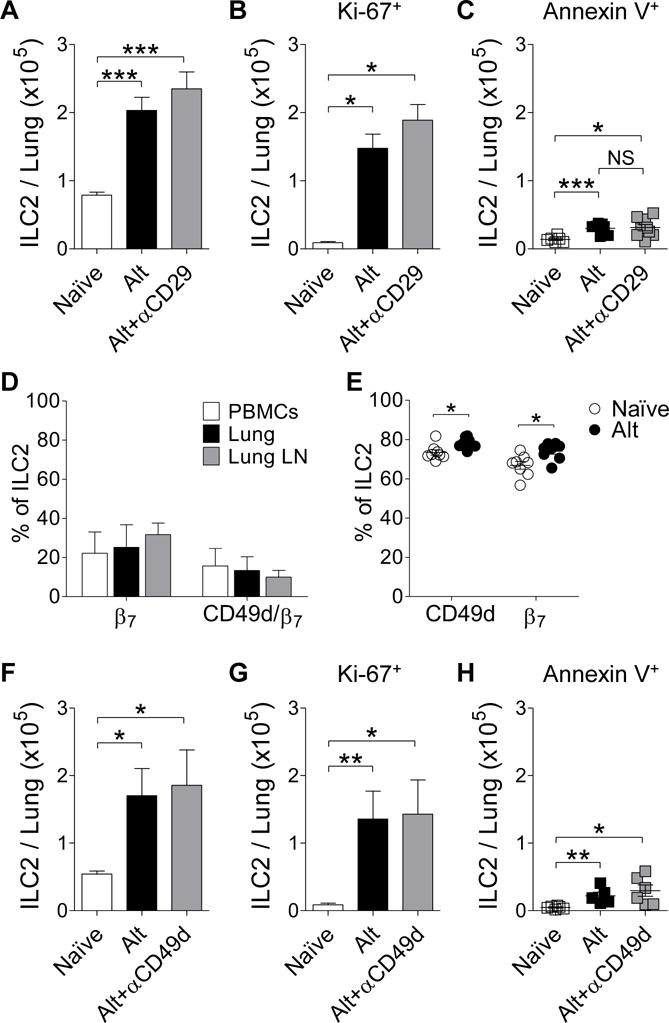
(A) Total number of lung ILC2s, (B) total Ki-67+ ILC2s, and (C) live Annexin V+ ILC2s per mouse treated ± anti-CD29 antibody (n≥6). (D) Percentage of human ILC2s expressing integrin β7 and both CD49d and β7 from PBMCs (white; n=4), lung tissue (black; n=3), and lymph nodes (gray; n=4). (E) Summary of the percentage of mouse lung ILC2s expressing CD49d and integrin β7 from both naïve and Alt challenged mice (n=8). (F) Total number of lung ILC2s, (G) total Ki-67+ ILC2s, and (H) live Annexin V+ ILC2s per mouse treated ± anti-CD49d antibody (n≥5). Mean represented as a line ± SEM. (NS) not significant, *P< 0.05, **P< 0.01, ***P< 0.001 by Student’s t test.
As the α4 integrin (CD49d), may associate with either a β1 integrin (α4β1) or a β7 integrin (α4β7), we also examined whether they expressed β7 integrins. Human ILC2s from PBMCs, lymph nodes, and lungs were assessed for the surface expression of integrin β7 alone and for dual expression of CD49d and β7 (Figure 5D). Human ILC2s, regardless of tissue source, showed low percentages of ILC2s expressing β7 and even lower levels of ILC2s expressing both CD49d and β7 (Figure 5D). In contrast to human ILC2s, mouse ILC2s expressed high levels of β7 integrin in both naïve mice (mean = 67.8%) and Alternaria challenged mice (mean = 74.0%; Figure 5E). Due to the high levels of α4β1 and α4β7 expression on mouse lung ILC2s, the role of blocking α4 integrins in Alternaria-induced ILC2 recruitment was assessed.
Alternaria challenged mice pretreated with a blocking anti-α4 integrin (αCD49d) antibody32 also had no reduction in total lung ILC2 (Figure 5F). Administration of the αCD49d antibody to Alternaria challenged mice did not inhibit lung ILC2 proliferation (Figure 5G). Similarly, administration of the αCD49d antibody to Alternaria challenged mice did not induce lung ILC2 apoptosis (Figure 5H). These studies suggest that neither α4β1 nor α4β7 integrins play a significant role in ILC2 recruitment to the lung.
DISCUSSION
ILC2s expand in the lungs of mice during type 2 inflammation induced by Alternaria challenge.13, 27 The increase in ILC2 numbers in the lung has previously been attributed to local proliferation alone17 as studies have not demonstrated that ILC2 migrate from the circulation to the lung following allergen challenge. We report that both human and mouse ILC2s express high levels of β1 and β2 integrin adhesion receptors, known to be used by other circulating leukocytes to adhere to endothelium and traffic to the lung. In addition, we show that after the 7 day Alternaria challenge ILC2 levels in the bone marrow decreased, while they increased in the circulation and lungs of mice, suggesting that ILC2s are released from the bone marrow to traffic to the lung following Alternaria challenge. In addition, BMT studies demonstrated that ILC2s derived from the bone marrow traffic to the lung following Alternaria challenge. In vivo blocking of β2 integrins (CD18) prior to Alternaria allergen challenge in mice significantly diminished ILC2 levels in the lungs. The effect of the αCD18 blocking antibody was specific to trafficking, as αCD18 treatment did not affect Alternaria-induced ILC2 proliferation, apoptosis, or Th2 cytokine expression. Moreover, in vitro we demonstrated that ILC2s adhere to ICAM-1 and that the adherence was inhibited by blocking antibodies to the β2 integrin. This was further confirmed by in vivo blocking of the αL integrin, a subunit of integrin αLβ2 (LFA-1), which showed a reduction in lung ILC2 levels when αCD11a was administered. Importantly, we also demonstrate that in vivo blocking of β1 or α4 integrins did not affect Alternaria-induced ILC2 levels in the airways. Therefore, we demonstrate an additional novel mechanism by which Alternaria allergen challenge increases airway ILC2 levels by recruiting ILC2s from the bone marrow to the circulation through integrin mediated trafficking that is dependent on β2 integrins, but not β1 integrins.
We have previously demonstrated that human subjects with allergic rhinitis due to cat allergen exposure, when challenged with cat allergen in the laboratory develop a significant increase in numbers of peripheral blood ILC2s, which is not noted on a separate diluent nasal challenge visit.9 In addition, ILC2s levels in grass pollen allergic individuals increase in the blood during the grass pollen season.33 These studies demonstrate that exposure to inhaled allergens in allergic subjects can significantly increase peripheral blood ILC2 levels. At present it is not known in humans whether the increase in peripheral blood ILC2s following exposure to inhaled allergens is due to a signal to the bone marrow to release ILC2 from the bone marrow which then traffic to tissue sites such as the upper or lower airway, where they can express Th2 cytokines and contribute to allergic inflammation. In contrast, studies in mice have provided evidence that ILC2s have tissue residency in the airways and do not recirculate.16, 17 However, these studies have not examined whether the expansion of ILC2 numbers in the lung following Alternaria challenge may be mediated by trafficking of ILC2 from the bone marrow to the lung. Our study provides evidence that ILC2 numbers increase in the mouse lung following Alternaria allergen challenge through mechanisms that include β2 integrin, but not β1 integrin, mediated trafficking from the circulation to the lung, as well as due to local proliferation of ILC2s in response to local inflammatory mediators. These results are in agreement with a prior study by our group, showing Alternaria challenge increases the percentage of Ki-67 proliferating ILC2s in the lungs.15 When considered together, it is likely that one subset of ILC2s that are increased in the lung following Alternaria challenge are a result of migration from the blood, while a second subset of ILC2s are the pre-existing lung ILC2s that proliferate and increase ILC2 lung numbers independent of migration from the circulation.
As ILC2 express high levels of the β2 integrin which is known to mediate firm adhesion of other leukocytes to ICAM-1 expressed by endothelium, the reduction in lung ILC2 we have noted in Alternaria challenged mice treated with an αCD18 antibody can best be explained by reduced ILC2 β2 integrin adhesion to ICAM-1 expressed by endothelium in the lung. Our in vitro studies demonstrated that ILC2s adhere to ICAM-1 in a β2 integrin dependent manner. As ILC2s and endothelium both express ICAM-1, administration of an anti-ICAM-1 antibody to Alternaria challenged mice would unfortunately not only block ICAM-1 function on endothelium, but also ICAM-1 function on ILC2 making interpretation of this in vivo experiment problematic. One of the weaknesses of this study is the lack of the blocking of CD18 specifically on ILC2s, which would demonstrate direct CD18 expression on ILC2 involvement in ILC2 migration. In addition, our study demonstrates that the αCD18 antibody does not inhibit the numbers of ILC2 in the subset of ILC2 expressing Th2 cytokines. This lack of inhibition of ILC2 Th2 cytokine expression would be anticipated as there is currently no evidence that inhibition of β2 integrins inhibits Th2 cytokine responses.
Although we demonstrated that both human and mouse ILC2s expressed significant levels of β1 integrins (α4β1 or VLA-4), blocking β1 integrin did not reduce the number of ILC2 in the lungs of Alternaria challenged mice. Thus, β1 integrins, are not as central as β2 integrins in mediating the increase in number of lung ILC2 following Alternaria challenge. In addition to β1 and β2 integrins, β7 integrins (in particular α4β7) can mediate adhesion to MAdCAM-1 expressed by endothelium. In this study we found that β7 integrin is expressed at low levels on human ILC2s isolated from PBMCs, lung tissue, and lung lymph nodes. Prior studies have also not detected significant expression of β7 integrin on mouse lung ILC2s and human peripheral blood ILC2s.34, 35 Thus, β7 integrins are unlikely to play a major role in human ILC2 adhesion. In contrast to human lung ILC2s, we did note high levels of expression of β7 integrin on mouse lung ILC2s. Prior studies have reported the expression of CD49d/β7 integrin on the surface of mouse ILC precursors in the bone marrow18, but minimal expression on mouse lung ILC2s.34, 35 Despite the varying levels of mouse ILC2 CD49d/β7 integrin expression reported in different studies, we demonstrated that the inhibition of CD49d did not alter ILC2 levels in the airways and therefore CD49d/β7 is unlikely to play a significant role in ILC2 migration to the lungs following Alternaria challenge.
β1 and β2 integrins, expressed by ILC2s, bind to known counter-receptors such as VCAM-1 and ICAM-1 expressed by vascular endothelium. Alternaria inhalation challenge is known to induce high levels of lung IL-33 within hours of challenge.13 In addition, IL-33 has been shown to induce VCAM-1 and ICAM-1 expression by endothelial cells.36 As this study demonstrated that blocking β2 integrins (i.e. the ICAM-1 counter-receptor pathway) reduced numbers of ILC2s in the lung following Alternaria challenge, whereas blocking the β1 integrin pathway (i.e. the VCAM-1 counter-receptor pathway) did not reduce lung ILC2 numbers, it suggests that either the VLA-4/VCAM pathway only mediates minor firm adhesion of ILC2s to endothelium pathway that can be bypassed by the β2 integrin/ICAM-1 pathway, or that VLA-4 expressed on ILC2 is in a low affinity state37 which does not permit firm adhesion to endothelium. We also demonstrated that mouse lung ILC2s did not express VCAM-1, but did express ICAM-1, which is in agreement with another study.34
In summary, this study makes the novel observation that the expansion of ILC2s in the lung following Alternaria challenge is not only mediated by the well described local lung proliferation of ILC2s17, but is also mediated by recruitment of circulating ILC2s from the blood to the lungs. Interestingly, ILC2 expression of β2 integrins plays an important role in the recruitment of ILC2s to the lung, as demonstrated in in vivo studies blocking β2 integrins prior to Alternaria allergen challenge. In contrast, in vivo blocking of β1 integrins, or α4 integrins, did not affect ILC2 levels in the airways. In vivo blocking of β2 integrins did not inhibit ILC2 proliferation, induce ILC2 apoptosis, or inhibit Th2 cytokine responses. Thus, ILC2 numbers increase in the lung following Alternaria allergen challenge through mechanisms that include β2 integrin, but not β1 integrin, mediated trafficking of ILC2s from the circulation to the lung, as well as due to local proliferation of ILC2s in response to local inflammatory mediators.
Supplementary Material
KEY MESSAGES.
Both human and mouse lung ILC2s express high levels of β1 and β2 integrin adhesion receptors.
ILC2s increase in number in the mouse lung not only through local proliferation, but also through trafficking from the circulation into the lung using β2 rather than β1 or α4 integrins.
CAPSULE SUMMARY.
ILC2s, which express high levels of Th2 cytokines, are not only resident in the lung, but also traffic from the bone marrow to the lung upon exposure to Alternaria, an allergen associated with severe asthma.
Acknowledgments
Funding: This work was supported by NIH grants AI 107779, AI 38425, AI 70535, AI 242236, and AI 72115 to DB, NIH T32 AI 007469 to MK, and NIH AI 114585 to TD.
ABBREVIATIONS
- BM
Bone marrow
- BMT
Bone marrow transplanted
- CRTH2
Chemoattractant receptor-homologous molecule expressed on Th2 cells
- FSC
Forward scatter
- gMFI
Geometric mean of fluorescent intensity
- ICAM-1
Intercellular adhesion molecule-1
- ILC2
Group 2 innate lymphoid cells
- LFA-1
Lymphocyte function-associated antigen-1
- Lin
Lineage
- LN
Lymph nodes
- MAdCAM-1
Mucosal vascular addressin cell adhesion molecule-1
- PBMCs
Peripheral blood mononuclear cells
- SSC
Side scatter
- VCAM-1
Vascular cell adhesion molecule-1
- VLA-4
Very late antigen-4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Doherty TA, Broide DH. Group 2 innate lymphoid cells: new players in human allergic diseases. J Investig Allergol Clin Immunol. 2015;25:1–11. [PMC free article] [PubMed] [Google Scholar]
- 2.Kim BS, Artis D. Group 2 innate lymphoid cells in health and disease. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernink JH, Germar K, Spits H. The role of ILC2 in pathology of type 2 inflammatory diseases. Curr Opin Immunol. 2014;31:115–20. doi: 10.1016/j.coi.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, Hulse KE, et al. Thymic stromal lymphopoietin activity is increased in nasal polyps of patients with chronic rhinosinusitis. J Allergy Clin Immunol. 2013;132:593–600. doi: 10.1016/j.jaci.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT, Jr, Rollins DR, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136:59–68. doi: 10.1016/j.jaci.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karta MR, Broide DH, Doherty TA. Insights into Group 2 Innate Lymphoid Cells in Human Airway Disease. Curr Allergy Asthma Rep. 2016;16:8. doi: 10.1007/s11882-015-0581-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlow JL, McKenzie AN. Type-2 innate lymphoid cells in human allergic disease. Curr Opin Allergy Clin Immunol. 2014;14:397–403. doi: 10.1097/ACI.0000000000000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang JE, Doherty TA, Baum R, Broide D. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J Allergy Clin Immunol. 2014;133:899–901. doi: 10.1016/j.jaci.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doherty TA, Scott D, Walford HH, Khorram N, Lund S, Baum R, et al. Allergen challenge in allergic rhinitis rapidly induces increased peripheral blood type 2 innate lymphoid cells that express CD84. J Allergy Clin Immunol. 2014;133:1203–5. doi: 10.1016/j.jaci.2013.12.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134:671–8. doi: 10.1016/j.jaci.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O’Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137:75–86. doi: 10.1016/j.jaci.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 12.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–8. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 13.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–13. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. 2005;26:360–6. doi: 10.1016/j.it.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Kim HK, Lund S, Baum R, Rosenthal P, Khorram N, Doherty TA. Innate type 2 response to Alternaria extract enhances ryegrass-induced lung inflammation. Int Arch Allergy Immunol. 2014;163:92–105. doi: 10.1159/000356341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. 2016;17:76–86. doi: 10.1038/ni.3309. [DOI] [PubMed] [Google Scholar]
- 17.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–5. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508:397–401. doi: 10.1038/nature13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8:1313–23. doi: 10.1038/mi.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692–704. doi: 10.1038/nri3908. [DOI] [PubMed] [Google Scholar]
- 21.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–59. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 22.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 23.Hogg N, Patzak I, Willenbrock F. The insider’s guide to leukocyte integrin signalling and function. Nat Rev Immunol. 2011;11:416–26. doi: 10.1038/nri2986. [DOI] [PubMed] [Google Scholar]
- 24.Sakai Y, Kobayashi M. Lymphocyte ‘homing’ and chronic inflammation. Pathol Int. 2015;65:344–54. doi: 10.1111/pin.12294. [DOI] [PubMed] [Google Scholar]
- 25.Johansson MW, Mosher DF. Integrin activation States and eosinophil recruitment in asthma. Front Pharmacol. 2013;4:33. doi: 10.3389/fphar.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zarbock A, Ley K. Mechanisms and consequences of neutrophil interaction with the endothelium. Am J Pathol. 2008;172:1–7. doi: 10.2353/ajpath.2008.070502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol. 2012;303:L577–88. doi: 10.1152/ajplung.00174.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132:205–13. doi: 10.1016/j.jaci.2013.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walford HH, Lund SJ, Baum RE, White AA, Bergeron CM, Husseman J, et al. Increased ILC2s in the eosinophilic nasal polyp endotype are associated with corticosteroid responsiveness. Clin Immunol. 2014;155:126–35. doi: 10.1016/j.clim.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Madrid F, Davignon D, Martz E, Springer TA. Antigens involved in mouse cytolytic T-lymphocyte (CTL)-mediated killing: functional screening and topographic relationship. Cell Immunol. 1982;73:1–11. doi: 10.1016/0008-8749(82)90431-2. [DOI] [PubMed] [Google Scholar]
- 31.Ridger VC, Wagner BE, Wallace WA, Hellewell PG. Differential effects of CD18, CD29, and CD49 integrin subunit inhibition on neutrophil migration in pulmonary inflammation. J Immunol. 2001;166:3484–90. doi: 10.4049/jimmunol.166.5.3484. [DOI] [PubMed] [Google Scholar]
- 32.Brocke S, Piercy C, Steinman L, Weissman IL, Veromaa T. Antibodies to CD44 and integrin alpha4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc Natl Acad Sci U S A. 1999;96:6896–901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lao-Araya M, Steveling E, Scadding GW, Durham SR, Shamji MH. Seasonal increases in peripheral innate lymphoid type 2 cells are inhibited by subcutaneous grass pollen immunotherapy. J Allergy Clin Immunol. 2014;134:1193–5. doi: 10.1016/j.jaci.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 34.Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity. 2015;42:538–51. doi: 10.1016/j.immuni.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiter B, Patil SU, Shreffler WG. Vitamins A and D have antagonistic effects on expression of effector cytokines and gut-homing integrin in human innate lymphoid cells. Clin Exp Allergy. 2015;45:1214–25. doi: 10.1111/cea.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demyanets S, Konya V, Kastl SP, Kaun C, Rauscher S, Niessner A, et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011;31:2080–9. doi: 10.1161/ATVBAHA.111.231431. [DOI] [PubMed] [Google Scholar]
- 37.Sung KL, Li Y, Elices M, Gang J, Sriramarao P, Broide DH. Granulocyte-macrophage colony-stimulating factor regulates the functional adhesive state of very late antigen-4 expressed by eosinophils. J Immunol. 1997;158:919–27. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.