

Abstract
Pathological variants in genes encoding calmodulin are associated with severe clinical presentations, including recurrent ventricular fibrillation and sudden death. Beta-receptor antagonists (beta-blockers) and sodium-channel antagonists have been reported as pharmacotherapies in these disorders; however, recent data have demonstrated the importance of derangements in calcium channel inactivation. We report a sustained attempt to use calcium-channel antagonists to treat calmodulinopathy and review the treatment strategies reported in the literature to date.
Keywords: arrhythmias, cardiovascular medicine
Background
Pathological variants in genes encoding calmodulin (CALM1, CALM 2 and CALM3) cause phenotypes similar to long QT syndrome (LQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT).1–3 The first described calmodulin variants were associated with severe clinical presentations, including recurrent ventricular fibrillation and sudden death. Beta-receptor antagonists (beta-blockers) and sodium-channel antagonists have been the most frequently reported pharmacotherapies in these disorders; however, recent data have demonstrated the importance of derangements in calcium channel inactivation.4 We revisit a case that we had previously included in a case series to report a sustained attempt to use calcium-channel antagonists to treat calmodulinopathy and review the treatment strategies reported in the literature to date.5
Case presentation
A 3-year-old boy collapsed suddenly while walking up a flight of stairs in the park with his family. He received immediate bystander CPR and the paramedics delivered a successful automated external defibrillator shock for ventricular fibrillation. Immediately after resuscitation, the corrected QT interval (QTc, Bazett's formula) on his surface ECG was >500 ms and the QTc remained prolonged (460–490 ms; figure 1) during follow-up. Genetic testing was performed, with the results shown below (see the Investigations section).
Figure 1.
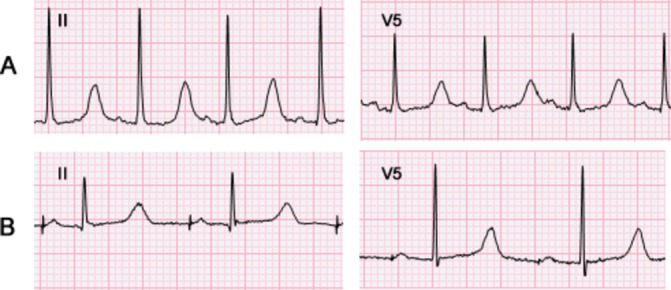
Surface ECG of reported case. Surface ECGs are shown from standard lead II and V5 positions. Panel A was obtained at the time of initial diagnosis, after cardiac arrest, prior to starting medications (QTc 509 ms). Panel B was obtained after the second cardiac arrest, while on atenolol 1.5 mg/kg/day (QTc 471 ms).
The patient received a dual chamber epicardial implantable cardioverter-defibrillator (ICD). The device was programmed with a lower rate limit of 70 beats per minute with rate responsiveness to maintain his heart rate and avoid pause-mediated arrhythmias. He was initially treated with atenolol 1.5 mg/kg/day, divided two times per day, with dose adjustments as needed for growth. Four 24-hour ECG (Holter) recordings were performed during the ensuring 2 years. Mean and peak heart rates were blunted, consistent with beta-blocker compliance (peak heart rate 68%–75% predicted for age) and no significant atrial or ventricular ectopy was observed.
Two years after his initial arrhythmic event, while compliant on medication, he had syncope associated with ventricular fibrillation arrest. He had been ‘active’ at the time of the event, but he was not at peak exertion. His ICD detected ventricular fibrillation and discharged appropriately (figure 2). He awakened immediately with normal neurological status. Atenolol was discontinued; nadolol and mexiletine were initiated (1.2 and 5.2 mg/kg/day respectively). He developed neurological side effects from mexiletine including tremor and recurrent falls. He was admitted to the hospital, mexiletine was stopped and left cardiac sympathetic denervation (LCSD) was discussed, but declined.
Figure 2.

Ventricular fibrillation during the second cardiac arrest, showing beat-to-beat initiation of ventricular fibrillation, followed by a successful ICD discharge. A ventricular escape rhythm follows the successful shock. Four channels are shown. The first channel is a bipolar atrial EGM. The second channel is a bipolar ventricular. The third and fourth channels are atrial and ventricular marker channels respectively, flanked by numbers that show the interval in milliseconds between each marker. The absence of atrial and ventricular EGMs at the beginning of panel A is a normal feature of this manufacturer's ICD system. Ab, atrial blanked event; AP/VP, atrial or ventricular paced event; AR, atrial refractory event; AS/VS, atrial or ventricular sensed event; FS, ventricular sensed event qualifying for the fibrillation counter; 36 J, delivery of a 36 Joule shock.
Investigations
Clinical whole exome sequencing demonstrated a heterozygous missense mutation in CALM1 (c.395 A>T; p.Asp132Val). Two additional variants of unknown significance were noted (GJA5 p.Ala96Ser and DSP p.Pro2777His). No variants were observed in CALM2, CALM3, KCNQ1, KCNH2 or SCN5A. Trio analysis demonstrated that the CALM1 mutation was de novo and that the GJA5 and DSP variants were inherited from the asymptomatic mother (normal echo; QTc 438 ms). His father had none of the observed variants and had a normal ECG.
Treatment
While hospitalised, his nadolol dose was adjusted for weight (1.3 mg/kg/day) and verapamil was initiated (7 mg/kg/day). Impairments of calcium-dependent inactivation have been implicated in the ventricular arrhythmias that affect patients with calmodulinopathy.5–9 A recent evaluation of a pathogenic variant in calmodulin-1 using a human induced pluripotent stem cell model demonstrated prolonged repolarisation as the main functional derangement. In addition to noting sensitivity to β-adrenergic stimulation, impairments of calcium-dependent inactivation of L-type calcium channels (ICaL) were demonstrated and rescued by pharmacological ICaL blockade.4 We predicted that calcium channel blockade would modify this patient's risk of recurrent arrhythmia.
Outcome and follow-up
He has remained arrhythmia-free during the year following the medication changes. Several ECGs performed on each of these medication regimens demonstrated a mean QTc=472–488 ms. The QTc measured by Holter recordings before and after initiation of verapamil was similar (mean QTc 492 vs 495 ms before and after verapamil respectively).
Discussion
This case illustrates two critical points relevant to caring for patients with calmodulinopathy. First, our case reinforces early reports of a high rate of recurrent ventricular fibrillation events in patients with calmodulin variants. Second, this case is an example of beta-blocker therapy failure in calmodulinopathy and offers the first report of safe, sustained use of calcium channel blockade in a patient with a calmodulinopathy.
Case studies and small series have suggested that recurrent VF and failure of monotherapy occur at an unusually high rate in calmodulinopathy, compared with congenital LQTS. Table 1 tabulates data from published cases of cardiac calmodulinopathy with both genotype and phenotype information. In 27 of 29 cases who survived the initial event, beta-blockers were the initial treatment. In 14 of 29 cases, there was a clinical recurrence of sudden death (three cases) or aborted sudden death (11 cases). Recurrent arrest occurred on atenolol in the current case; recurrent arrest has also been published when patients were on nadolol, propranolol and pindolol.1 9
Table 1.
Published cases of calmodulin-associated arrhythmia disorders. Phenotype is as reported in each original manuscript
| Publication | Sex | Age at initial event | Phenotype | Variant | Treatment | Subsequent event | Follow-up treatment |
| Marsman et al 13 | M | 16 years | VF arrest | CALM1-F90L | ICD | No subsequent events | – |
| Marsman et al 13 | F | 10 years | VF arrest | CALM1-F90L | BB, ICD | Recurrent VF with successful ICD discharge | Not reported |
| Marsman et al 13 | F | N/A | Genotype+, phenotype – | CALM1-F90L | No medications, + ICD | None | – |
| Crotti et al 1 | F | Birth | LQTS+2:1 AV block | CALM2-D96V | Propranolol | VF with cardiac arrest | Propranolol, mexiletine, (with subsequent ICD shocks) |
| Chaix et al 14 | F | Neonatal | LQTS | CALM3-D96H | PM, propranolol | No events in 14 years of follow-up | – |
| Nyegaard et al 2 | M | 12 years | CPVT | CALM1-N98Sφ | BB | No subsequent events (>20 years f/u) | – |
| Nyegaard et al 2 | M | Unknown (<23 years) |
CPVT | CALM1-N98Sφ | BB | No subsequent events | – |
| Nyegaard et al 2 | F | Unknown | CPVT | CALM1-N98Sφ | BB | No subsequent events | – |
| Nyegaard et al 2 | F | 6 years | CPVT | CALM1-N98Sφ | BB | No subsequent events | – |
| Nyegaard et al 2 | F | 4 years | CPVT | CALM1-N98Sφ | BB | Cardiac arrest 12 years later | BB, ICD |
| Nyegaard et al 2 | F | 4 years | VF arrest; CPVT | CALM1-N98Sφ | BB | Cardiac arrest 11 years later | BB, ICD |
| Makita et al 9 | M | 5 years | LQTS | CALM2-N98S | Propranolol‡ | Syncope and dizziness while running | Changed to metoprolol |
| Jiménez-Jáimez et al 15 | M | 7 years | Sudden death, CPVT |
CALM2-N98S (post-mortem) | N/A | N/A | N/A |
| Jiménez-Jáimez et al 15 | F | 4 years | LQTS | CALM2-N98S | Atenolol, ICD | No subsequent events | – |
| Makita et al 9 | M | 17 months | LQTS | CALM2-N98I | BB, ICD | No subsequent events | – |
| Gomez-Hurtado et al 16 | F | 10 years | CPVT, syncope | CALM3-A103V | BB | No subsequent events | – |
| Crotti et al 1 | F | 6 months | LQTS+2:1 AV block | CALM1-D130G | Propranolol, ICD | Multiple VF episodes | Propranolol, LCSD, RCSD, additional medications† |
| Crotti et al 1 | M | 1 month | LQTS | CALM1-D130G | BB, mexiletine, ICD | Not reported | – |
| Boczek et al 6 | F | 1 day | LQTS+2:1 AV block | CALM2-D130G | BB, ICD at 6 years of age | VF with successful ICD discharge at 11 and 14 years of age | Not reported |
| Boczek et al 6 | M | Birth | LQTS+2:1 AV block, eventually DCM | CALM2-D130V | BB | Torsades de pointesx2 | Beta-blocker, sodium channel blocker, ICD, LCSD |
| Reed et al 3 | M | Birth | LQTS+2:1 AV block | CALM3-D130G | Propranolol, pacemaker | No subsequent events | – |
| Pipilas et al 5 * | M | 3 years | LQTS | CALM1-D132V | Atenolol, ICD | VF with successful ICD discharge | Nadolol, mexiletine, ICD |
| Pipilas et al 5 | M | 1 day | LQTS+2:1 AV block | CALM2-D132H | Propranolol, mexiletine, ICD | Not reported | – |
| Makita et al 9 | F | Neonatal | LQTS, eventually LVNC | CALM2-D132E | Pindolol | Syncope while swimming and non-compliant | Pindolol, propranolol, atenolol all tried |
| Makita et al 9 | M | 6 years | LQTS | CALM2-D134H | Mexiletine | Cardiac arrest while non-compliant | BB, mexiletine, ICD (with one subsequent shock) |
| Makita et al 9 | F | 8 years | LQTS/CPVT | CALM2-Q136P | Nadolol | Sudden death while dancing at age 11 | N/A |
| Boczek et al 6 | M | 3 years | LQTS | CALM1-E141G | BB, sodium channel blocker | No events in 11 years of follow-up | – |
| Boczek et al 6 | F | Birth | LQTS+2:1 AV block | CALM1-F142L | BB, pacemaker | VF with sudden death | N/A |
| Boczek et al 6 | M | Birth | LQTS | CALM1-F142L | BB, pacemaker | VF with sudden death | N/A |
| Crotti et al 1 | M | ? Neonatal | LQTS | CALM1-F142L | BB, mexiletine, LCSD, ICD | Not reported | – |
| Chaix et al 14 | F | Prenatal | LQTS+2:1 AV block | CALM3-F142L | PM, propranolol | No events in 14 years of follow-up | – |
*Case described in this report.
†After VF arrest, a loading dose of mexiletine was attempted (not continued), a trial of verapamil was attempted (not continued) and the patient was maintained on propranolol and flecainide, plus the ICD.
‡A trial dose of mexiletine was given, but was not continued as an outpatient. φ The amino acid number is based on NCBI Reference Sequence NP_008819.
BB, beta-adrenergic blockade without a particular agent specified; LQTS,long QT syndrome;L/RCSD,left/right cardiac sympathetic denervation; LVNC,left ventricular non-compaction.
Several features are remarkable in the published cohort of calmodulinopathy cases with a prolonged QT interval. The age at presentation is younger than is typical for congenital LQTS. For neonates with calmodulinopathy, 2:1 AV block secondary to QTc prolongation is more common than in the usual congenital LQTS population. Even in patients who were first identified outside the neonatal period, an initial presentation of sudden death or aborted sudden death was common.
In LQT1, LQT2, and LQT3, beta-blockers have a track record of success in preventing recurrent arrhythmias, especially among compliant patients.10 11 The high rate of beta-blocker failure in patients with pathological calmodulin variants is unusual and suggests that calmodulinopathy should not be considered simply a form of congenital LQTS. Even knowing that perfect compliance is rarely obtainable in clinical practice, the nearly 50% recurrence rate of VF and cardiac arrest during beta-blocker therapy in the literature suggests that additional protection may be required in some patients with symptomatic calmodulinopathy.
The literature currently describes nine patients who were treated with monotherapy and who were successfully resuscitated from a subsequent cardiac event. In the four cases where multiple classes of medications were tried, the long-term management strategy was a beta-blocker combined with a sodium channel blocker. However, isolated cell preparations have failed to show markedly abnormal sodium channel function associated with calmodulin variants.6 7 There is strong in vitro evidence that calcium handling is abnormal in the setting of pathological calmodulin variants.1 3–6 8 9 12 The one published attempt to use verapamil clinically in calmodulin-positive LQTS was aborted when no change in the surface electrogram was noted after the administration of a single dose.1 To the best of our knowledge, the current publication is the first sustained clinical attempt to use calcium channel blockade to treat calmodulinopathy. Because the mechanism underlying calmodulinopathy with prolonged QTc involves impaired calcium-dependent inactivation leading to enhanced L-type calcium current, calcium channel blockade may potentially modify the risk of arrhythmia.
Importantly, we observed no change in QTc interval duration in our patient after steady-state administration of verapamil at 7 mg/kg/day. We did not determine if a dose-dependent change in the QTc might occur with this drug. Our findings are in contrast to a recent in vitro study performed with human induced pluripotent stem cell derived cardiomyocytes with the CALM1-F142L mutation.4 This work reported prolongation of the rate-corrected field potential duration in CALM1-F142L cardiomyocytes and this was observed to be partially rescued by verapamil. The actions of the drug on isolated cardiomyocytes may not adequately predict its clinical effects. Further, verapamil primarily affects peak calcium current rather than altering calcium channel inactivation. Drugs capable of modulating inactivation may be better suited to treating calmodulinopathy. Roscovitine, an investigational cyclin-dependent kinase inhibitor, enhances voltage-dependent inactivation of cardiac calcium channels and was shown to rescue in vitro electrophysiological abnormalities associated with Timothy syndrome4.
In summary, we present a case of recurrent life-threatening arrhythmia despite compliance with beta-blocker therapy in a patient with a pathological calmodulin mutation. In the highly selected group of patients published thus far with calmodulinopathy, the rate of recurrent ventricular fibrillation and sudden death during treatment is high, nearly 50%. Genotype testing that includes CALM1-CALM3 should be considered in any young patient with a severe ventricular arrhythmia phenotype and especially considered in infants and toddlers with a severely prolonged QT interval coupled with 2:1 AV block or a presentation of aborted sudden death. Patients with calmodulin mutations may require more comprehensive pharmacological and device-based treatment early in the clinical course and further studies of calcium antagonists are warranted in this disease. Advances in understanding the basic physiological mechanism of arrhythmia in patients with calmodulinopathy may lead to more effective treatment strategies.
Patient's perspective.
In April of 2014, our son passed out for unknown reasons while going up a flight of steps at an outdoor park. The ambulance arrived on the scene and assisted with the first aid that was already being done. He was transported to the hospital. He was stabilized and at the time it was determined he needed an ICD. He was also put on medicines to help prevent the same thing from happening again, but he had another incident and his ICD went off. After that, we tried another beta-blocker, nadolol, and a medicine called mexiletine, but he had side effects from the mexiletine. Those side effects were disturbing. About an hour after taking the medicine, his hands would shake so badly he couldn't hold a cup, he became clumsier and had difficulties walking and climbing stairs. He was then switched to his current medicine regimen of verapamil and nadolol and the side effects disappeared. Since switching over to the current regimen, his "beep beep" (his word for his ICD) has been quiet.
Learning points.
Early reports of pathogenic calmodulin variants are associated with a young age at presentation and a high rate of recurrent ventricular fibrillation events.
Beta-blocker therapy failure in calmodulinopathy may be more common than in other forms of channelopathy and this is the first report of safe, sustained use of calcium channel blockade in this disease.
The efficacy of calcium channel blockade remains unproven for calmodulinopathy. A larger clinical sample will be required to determine if verapamil is effective and if therapeutic serum levels change physiologic parameters such as QTc interval on the surface ECG.
Footnotes
Contributors: This manuscript was conceived (GW/ALG), drafted (GW/ZJS) and revised (GW/ZJS/ALG) by the authors.
Funding: Research reported in this publication was supported, in part, by the National Institutes of Health's National Center for Advancing Translational Sciences, Grant Number KL2TR001424. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests: None declared.
Patient consent: Obtained from guardian.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Crotti L, Johnson CN, Graf E, et al. . Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013;127:1009–17. 10.1161/CIRCULATIONAHA.112.001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nyegaard M, Overgaard MT, Søndergaard MT, et al. . Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet 2012;91:703–12. 10.1016/j.ajhg.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reed GJ, Boczek NJ, Etheridge SP, et al. . CALM3 mutation associated with long QT syndrome. Heart Rhythm 2015;12:419–22. 10.1016/j.hrthm.2014.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rocchetti M, Sala L, Dreizehnter L, et al. . Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc Res 2017;113:531–41. 10.1093/cvr/cvx006 [DOI] [PubMed] [Google Scholar]
- 5. Pipilas DC, Johnson CN, Webster G, et al. . Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Heart Rhythm 2016;13:2012–9. 10.1016/j.hrthm.2016.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boczek NJ, Gomez-Hurtado N, Ye D, et al. . Spectrum and prevalence of CALM1-, CALM2-, and CALM3-Encoded calmodulin variants in Long QT syndrome and functional characterization of a novel long QT Syndrome-Associated calmodulin missense variant, E141G. Circ Cardiovasc Genet 2016;9:136–46. 10.1161/CIRCGENETICS.115.001323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin G, Hassan F, Haroun AR, et al. . Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc 2014;3:e000996 10.1161/JAHA.114.000996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Limpitikul WB, Dick IE, Joshi-Mukherjee R, et al. . Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol 2014;74:115–24. 10.1016/j.yjmcc.2014.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makita N, Yagihara N, Crotti L, et al. . Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet 2014;7:466–74. 10.1161/CIRCGENETICS.113.000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilde AA, Moss AJ, Kaufman ES, et al. . Clinical aspects of type 3 Long-QT syndrome: an International Multicenter Study. Circulation 2016;134:872–82. 10.1161/CIRCULATIONAHA.116.021823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moss AJ, Zareba W, Hall WJ, et al. . Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000;101:616–23. 10.1161/01.CIR.101.6.616 [DOI] [PubMed] [Google Scholar]
- 12. Søndergaard MT, Tian X, Liu Y, et al. . Arrhythmogenic calmodulin mutations affect the activation and termination of cardiac ryanodine Receptor-mediated Ca2+ release. J Biol Chem 2015;290:26151–62. 10.1074/jbc.M115.676627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yazawa M, Hsueh B, Jia X, et al. . Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011;471:230–4. 10.1038/nature09855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaix MA, Koopmann TT, Goyette P, et al. . Novel CALM3 mutations in pediatric long QT syndrome patients support a CALM3-specific calmodulinopathy. HeartRhythm Case Rep 2016;2:250–4. 10.1016/j.hrcr.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiménez-Jáimez J, Palomino Doza J, Ortega Á, et al. . Calmodulin 2 mutation N98S is associated with unexplained cardiac arrest in infants due to low clinical Penetrance electrical disorders. PLoS One 2016;11:e0153851 10.1371/journal.pone.0153851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gomez-Hurtado N, Boczek NJ, Kryshtal DO, et al. . Novel CPVT-Associated calmodulin mutation in CALM3 (CALM3-A103V) Activates arrhythmogenic Ca waves and sparks. Circ Arrhythm Electrophysiol 2016;9:e004161 10.1161/CIRCEP.116.004161 [DOI] [PMC free article] [PubMed] [Google Scholar]