

Abstract
Primary adrenal non-Hodgkin’s lymphoma is rarely encountered. Clinical presentation is non-specific with fatigue, abdominal pain and B-symptoms being more commonly reported. We report a case of primary bilateral adrenal lymphoma who initially presented with clinical features suspicious for pheochromocytoma. The patient was recently diagnosed with hypertension and had a family history of early ischaemic heart disease. Plasma free metanephrines were also elevated. Pheochromocytoma was deemed unlikely following multiple investigations and the diagnosis of lymphoma was made following adrenal biopsy. Partial response was noted on positron emission tomography CT scan following four cycles of chemotherapy but failed to remit after six cycles of chemotherapy. Subsequently, he received 20 fractions of radiation to his adrenal glands and is now awaiting further imaging. The presentation of primary adrenal lymphoma can be variable and given that the diagnosis is obtained through tissue sampling, pheochromocytoma should be excluded due to risk of a potentially fatal adrenergic crisis.
Keywords: endocrine system, adrenal disorders, haematology (incl blood transfusion), medical education
Background
Primary adrenal lymphoma (PAL) remains a rare entity although the frequency of diagnosis has increased in the last decade attributed to the availability of more advanced imaging modalities and the increasing awareness of this condition. Within the published literature, descriptions are mostly in the form of case reports and a few case series. Most patients present initially with B-symptoms, abdominal pain and fatigue, with only 1% of cases presenting as an adrenal incidentaloma.1 2
Given that the diagnosis of PAL is confirmed histologically, it is pertinent that pheochromocytoma be excluded as to avoid a potentially fatal adrenergic crisis during biopsy or surgical excision.
We describe a case of bilateral adrenal lymphoma that was referred to the endocrine outpatient clinic with an initial clinical suspicion of bilateral adrenal pheochromocytomas. Given the rarity of this condition, there are no clear treatment protocols for the management of PAL and hence treatments used are as per treatments used in non-PALs. We report the use of venetoclax, a selective B-cell lymphoma 2 (BCL-2) inhibitor more commonly used in patients with chronic lymphocytic leukaemia, with the usual rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone (RCHOP) chemotherapy regimen. This was not curative, thereby resulting in the patient proceeding to radiation therapy to his adrenal glands.
This case report generates further awareness of the variable presentations of this condition, the difficulties in reaching a diagnosis and the need for further research to understand PAL and how best to treat this condition.
Case presentation
A 59-year-old man was referred to endocrine outpatients at the end of April 2016 with bilateral adrenal masses on CT abdomen and elevated plasma metanephrines. He presented to his general practitioner in March 2016 following an episode of severe epigastric pain with associated sweating that resolved after 40 min. He described suffering from abdominal pain and losing 28 kg in weight over the past 6 months. He did not report any night sweats or fevers. Of note, he was commenced on perindopril 5 mg approximately 6 months earlier for a new diagnosis of hypertension. His medical history included an excision of a melanoma skin lesion in 2008 and a diagnosis of gastro-oesophageal reflux disease for which he takes esomeprazole 40 mg once daily.
His family history includes a brother who was diagnosed with coronary artery disease in his 30s and his father who passed away at 62 years old from ischaemic heart disease. At the time of his outpatient review, the patient had quit his job as a fork lift driver in a lettuce factory due to ill health. He is an ex-smoker with a 12 pack-year history of smoking.
The examination revealed a cachectic looking man with no signs of any skin changes and he had a normal cardiovascular and respiratory exam. His blood pressure was 127/80 mm Hg. Postural blood pressure was not checked. Examination of his abdomen revealed palpable masses over the left and right upper quadrants.
Investigations
The CT scan of his abdomen (see figure 1) showed large bilateral adrenal masses measuring 10.9×8×11.9 cm with a density of 67HU on the left adrenal gland; and the right adrenal gland mass measured at 5.6×5.7×7.2 cm with a density of 60HU precontrast. There was minor enhancement on contrast administration with no substantial contrast washout on delayed imaging. The left adrenal mass was found to surround the left renal vessels. There were no other intra-abdominal lesions noted and the CT thorax was reported as normal. Plasma metanephrines were elevated (see table 1).
Figure 1.
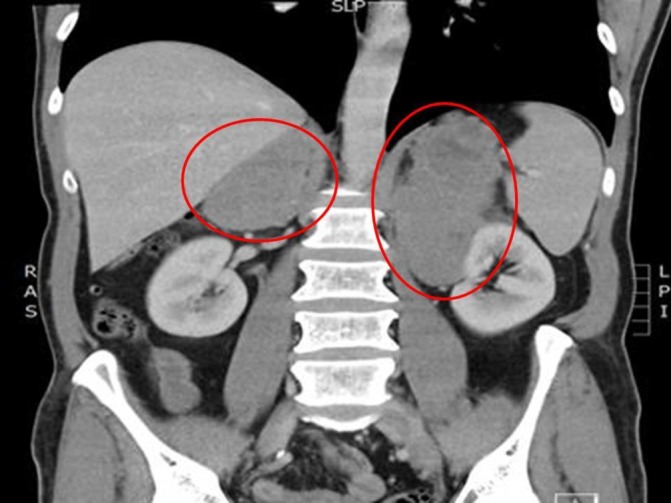
CT abdomen showing bilateral large adrenal masses (circled in red).
Table 1.
Results of plasma free metanephrines
| Plasma metaneprines | Results | Normal range |
| Normetadrenaline | 1110 pmol/L | <970 pmol/L |
| Metadrenaline | 240 pmol/L | <447 pmol/L |
| 3-Methoxytyramine | 220 pmol/L | <181 pmol/L |
Routine blood tests showed mildly elevated alkaline phosphatase 120 (35–110 U/L), alanine aminotransferase 137 (5–40 U/L), gamma-glutamyl transferase 192 (5–50 U/L) and lactate dehydrogenase (LDH) 367 (120–250 U/L). Full blood count results were normal. Both Hepatitis and HIV screen were negative. A 24 hours urine collection for urine vanillylmandelic acid excretion and urinary metanephrine excretion was done which were normal, other than a borderline result for the urinary normetadrenaline level of 2.3 (<2.3 umol/day). Although congenital adrenal hyperplasia was thought to be unlikely to be presenting at this age, a serum 17-hydroxyprogesterone was checked to exclude this and the results were normal at 2.7 (1.3–8.5 nmol/L). A morning cortisol level checked was within normal range at 289 (100–535 nmol/L), but the adrenocorticotropin hormone (ACTH) levels were elevated at 157 (9–51 ng/L). The patient went on to have a Short Synacthen Test (SST) as we wanted to exclude adrenal insufficiency should he require an adrenal biopsy or surgical excision. The SST cortisol response was inadequate (0 min–359 nmol/L, 30 min–395 nmol/L and 60 min346 nmol/L). Serum renin–aldosterone levels were also impaired with results showing renin erect 177 (3.3–4.1 mU/L), aldosterone erect 94 (100–950 pmol/L) and renin upright 156 (3.3–4.1mU/L) and aldosterone upright 79 (100–950 pmol/L) and an aldosterone to renin ratio of <1 (<70). As such, the patient was diagnosed with primary adrenal insufficiency because of his bilateral adrenal masses and was commenced on hydrocortisone and fludrocortisone replacement therapy.
Differential diagnosis
At this point, the differential diagnosis was that of adrenal cortical carcinoma or metastatic disease of unknown primary. Pheochromocytoma was felt to be less likely given the 24 hours urine catecholamine excretion results.
An adrenal biopsy was sought locally however; this could not be performed and the patient was referred to a tertiary centre. The patient was reviewed by the endocrinology team at the tertiary centre who repeated his plasma free metanephrines. The plasma normetadrenaline was elevated at 975 (<900 pmol/L) and the patient underwent an I123 metaiodobenzylguanidine (MIBG) scan to assess for functional status of the adrenal tumour as well as to exclude the likelihood of extra-adrenal pheochromocytomas given the size of the adrenal masses. The I123 MIBG scan results were negative. A positron emission tomography (PET) CT scan (see figure 2) was then requested to rule out a possible primary neoplasm. PET CT scan showed intensely fluorodeoxyglucose (FDG)-avid (SUVmax 19–19.5) lobulated marked enlargement of adrenal glands, left adrenal measuring 15.5x×7.7 cm and right adrenal gland measuring 8.5×1.6 cm with internal necrotic areas.
Figure 2.
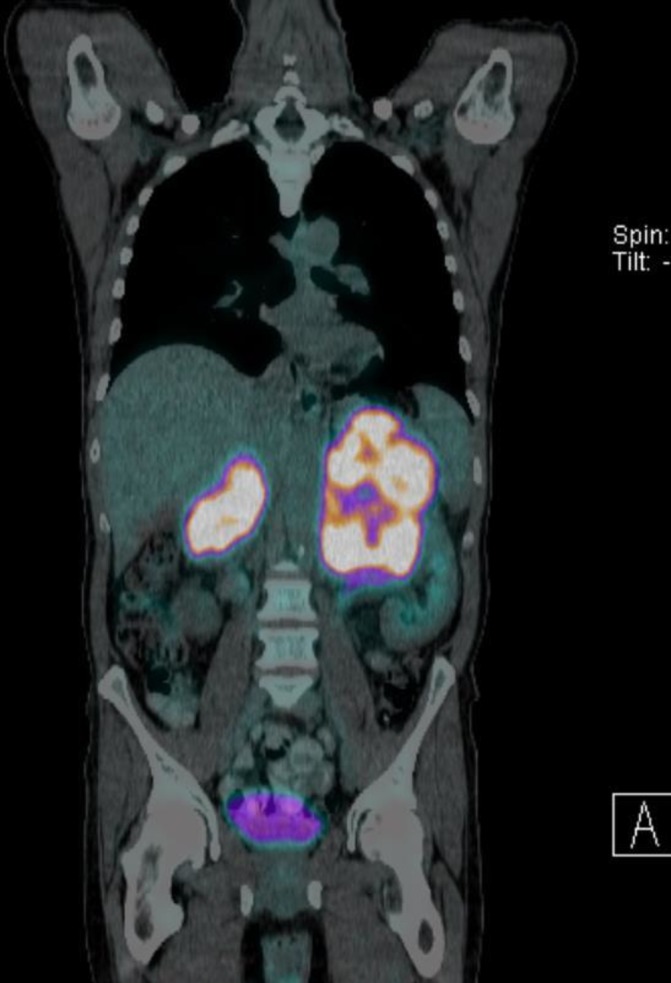
Positron emission tomography CT showing enlarged, fluorodeoxyglucose-avid adrenal glands with internal necrotic areas.
The patient went on to have a CT-guided biopsy and histological results revealed diffuse large B-cell lymphoma (DLBCL) positive for CD20, Bcl2, Bcl6 and multiple myeloma oncogene 1 placing the diagnosis in a non-germinal centre B cell subgroup. The Ki 67 stain, demonstrated a proliferative index of >80%. Epstein Barr Virus encoded RNA and CD10 were negative. Fluorescent in situ hybridisation analysis testing for MYC was negative. Bone marrow aspirate and biopsy results did not show any evidence of marrow involvement.
Treatment
The patient was enrolled into a phase 1B/11 chemotherapy trial (Cavalli trial for DLBCL) under the care of the haematology team and has now recently completed six cycles of RCHOP chemotherapy with Venetoclax. He had a repeat positron emission tomography (PET-CT) scan done post four cycles of chemotherapy and the results were encouraging, showing a decrease in size of both adrenal masses (left 6.2×5.3 cm and right 3.4×1.6 cm) with decreased FDG-avidity implying partial response to treatment (see figures 3 and 4). Following completion of six cycles of chemotherapy, however, a repeat PET CT scan did not show resolution of the disease.
Figure 3.
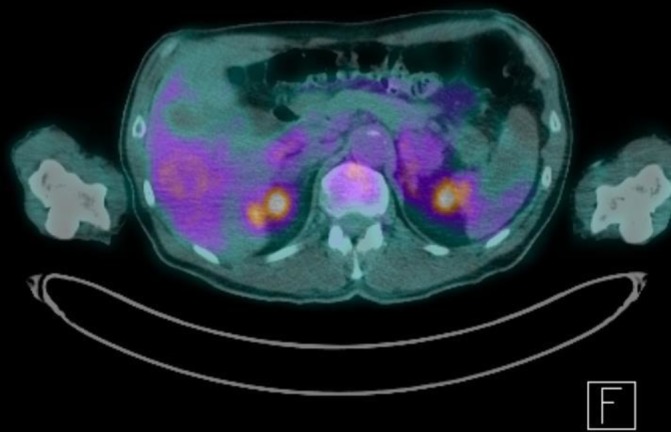
Positron emission tomography CT (axial view) post four cycles of chemotherapy showing partial response.
Figure 4.
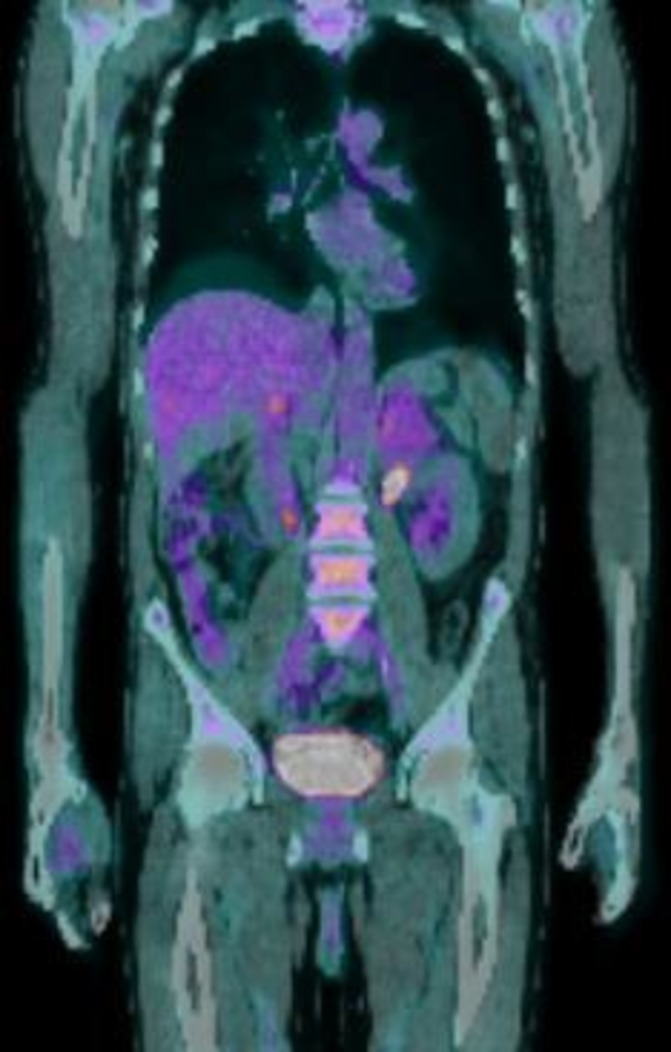
Positron emission tomography (PET) CT (coronal view) post four cycles of chemotherapy showing partial response.
Outcome and follow-up
The patient went on to have 20 fractions of radiotherapy to his adrenal glands and is currently awaiting a repeat PET scan.
Discussion
Although still rare, cases of PAL are increasingly being reported, likely due to improved imaging modalities and awareness of this condition. About 70% of cases of PAL involve both adrenal glands.3 Other causes of large bilateral adrenal masses include metastatic disease (lung cancer, lymphoma, melanoma, renal and ovarian cancer), adrenocortical carcinoma, pheochromocytoma, myelolipoma, macronodular adrenal hyperplasia, congenital adrenal hyperplasia, functioning adenomas, infiltrative causes (sarcoidosis and amyloidosis) and infections (tuberculosis, cryptococcosis and histoplasmosis). Currently, there are fewer than 200 cases of PAL within the published literature.
The highest reported cases of PAL are in Asia (54%).1 Worldwide, males tend to be affected more commonly than females with a male to female ratio of 1.8:1.1 The mean age of onset is around 68 years.3 Of note 70% of PAL cases involve both adrenal glands and in 85%–91% cases of PAL, the diagnosis was DLBCL.1 4–6 A large proportion of PAL case reports showed evidence of complete or partial adrenal insufficiency.1 5 7 Notably, the size of the underlying adrenal lymphoma did not necessarily correlate with the presence of adrenal insufficiency.6 Further research is required to investigate the underlying mechanisms and risk factors for developing adrenal insufficiency in these groups of patients.
The pathophysiology remains debatable. It is widely proposed that like thyroid lymphoma where the risk is that of prior autoimmune thyroiditis, adrenal lymphoma may arise in the context of prior autoimmune adrenalitis. This theory is questionable given a recent meta-analysis published did not identify a single case where a prior history of autoimmune adrenalitis was reported.1 Another proposed theory is that lymphoma cells may arise from haematopoietic rest cells within the adrenal gland as these rest cells have been found in cases of myelolipomas. The other possible aetiologies linked to PAL include mutations in the p53 and c-kit genes, Epstein-Barr virus and history of immune dysfunction.6 8
Cases of PAL have been encountered and reported by general physicians, endocrinologists, urologists and haematologists. The path to reaching a diagnosis of PAL poses many challenges. We note some case reports within the published literature that did not report results of plasma or urine catecholamines or metanephrines. It is important that any patient with bilateral adrenal masses suspicious for malignancy be checked for possible pheochromocytomas even when there is no evidence of hypertension, given the risk of a potentially fatal adrenergic crisis in the setting of an adrenal biopsy or surgical excision. A recently published case report described a normotensive patient confirmed to have both PAL and pheochromocytoma within the same adrenal gland following adrenalectomy.9 This patient only had mildly elevated serum and urine catecholamine levels.9
The use of imaging plays a significant role in distinguishing benign lesions from malignant lesions. However, confirming the diagnosis based on imaging alone among lesions of concern for malignancy is difficult. In our patient, the initial non-contrast CT scan showed masses with densities more than 10HU and mass size of more than 4 cm, which was suspicious for malignancy.10 11
Following delayed images, there was minimal contrast washout further implying malignant lesions. He had multiple features to suggest the possibility of bilateral pheochromocytomas including raised plasma metanephrines, recent diagnosis of hypertension, symptoms of epigastric pain with sweating and a family history of early onset heart disease. Consequently, he had an I123 MIBG scan performed to assess for adrenal catecholamine functionality and to rule out the possibility of multiple tumours (paragangliomas). As this was negative, it was reassuring that this was unlikely to be a pheochromocytoma but still concerning for some other underlying malignancy. An 18F FDG-PET CT scan was performed to rule out other possible primary sources of malignancy which showed abnormal uptake within the adrenal glands only. In hindsight, the only findings that hinted towards the possibility of a lymphoma was the raised serum LDH level. Ultimately, the diagnosis of PAL is achieved only through the means of obtaining a tissue sample.1
Currently, there are no clear treatment protocols for primary DLBCL of the adrenal gland. Treatment regimens used are as per usual treatment regimens used in non-primary adrenal DLBCL cases. Adrenalectomy is controversial given the risk of disease recurrence later, the associated high morbidity and mortality risk and no effect on overall survival.12 The use of RCHOP chemotherapy has improved survival rates (51%—2-year progression free survival and 68.3% survive at 2 years).12 Our patient also received Venetoclax, a selective inhibitor of BCL-2 more commonly used in patients with chronic lymphocytic leukaemia, in addition to the standard RCHOP therapy. This combination therapy appeared encouraging during the first half of the treatment but failed to result in a curative outcome. At this stage, he has completed 20 fractions of radiation therapy and will be having a follow-up restaging scan soon.
We believe this case provides additional value to the current literature regarding the variable clinical presentation of PAL and the challenges faced in distinguishing the underlying cause of large bilateral adrenal masses. This case also stresses the need to exclude the presence of pheochromocytoma given that the diagnosis of PAL is made from tissue sampling. The patient has now completed six cycles of RCHOP chemotherapy in combination with Venetoclax and thereafter required 20 fractions of radiotherapy. The use of Venetoclax is novel in this condition. The optimal management of PAL is yet to be determined and requires further studies.
Learning points.
Primary adrenal lymphoma (PAL) remains relatively rare and frequently involves both adrenal glands.
Clinical presentation is variable and may be suggestive of other causes of bilateral adrenal gland masses including pheochromocytoma, adrenocortical cancer and metastatic disease of unknown primary.
Given that the diagnosis of PAL is made histologically and such presentations may be encountered by multiple medical and surgical specialties, it is important to assess for functionality of these masses. The presence of an underlying pheochromocytoma needs to be excluded due to risk of a potentially fatal adrenergic crisis.
Optimal management of PAL is not known and is based on results of small studies implying the need for further research into this condition.
Footnotes
Contributors: FGJ recruited patient for case report, data collection, literature review and write up of case report. SC and DG have contributed by reviewing the available literature.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Rashidi A, Fisher SI. Primary adrenal lymphoma: a systematic review. Ann Hematol 2013;92:1583–93. 10.1007/s00277-013-1812-3 [DOI] [PubMed] [Google Scholar]
- 2.Ellis RD, Read D. Bilateral adrenal non-Hodgkin's lymphoma with adrenal insufficiency. Postgrad Med J 2000;76:508–9. 10.1136/pmj.76.898.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong A, Cui Y, Wang Y, et al. (18)F-FDG PET/CT of adrenal lesions. AJR Am J Roentgenol 2014;203:245–52. 10.2214/AJR.13.11793 [DOI] [PubMed] [Google Scholar]
- 4.Laurent C, Casasnovas O, Martin L, et al. Adrenal lymphoma: presentation, management and prognosis. QJM 2017;110:103–9. 10.1093/qjmed/hcw174 [DOI] [PubMed] [Google Scholar]
- 5.Kasaliwal R, Goroshi M, Khadilkar K, et al. Primary adrenal lymphoma: a single-center experience. Endocr Pract 2015;21:719–24. 10.4158/EP14471.OR [DOI] [PubMed] [Google Scholar]
- 6.Grigg AP, Connors JM. Primary adrenal lymphoma. Clin Lymphoma 2003;4:154–60. 10.3816/CLM.2003.n.024 [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Sun NC, Renslo R, et al. Clinically silent primary adrenal lymphoma: a case report and review of the literature. Am J Hematol 1998;58:130–6. [DOI] [PubMed] [Google Scholar]
- 8.Padhi S, Sahoo J. Primary adrenal non Hodgkin lymphoma: changing trends. Turk J Gastroenterol 2015;26:85–6. 10.5152/tjg.2015.4882 [DOI] [PubMed] [Google Scholar]
- 9.Babinska A, Peksa R, Sworczak K. Primary malignant lymphoma combined with clinically ‘silent’ pheochromocytoma in the same adrenal gland. World J Surg Oncol 2015;13:289 10.1186/s12957-015-0711-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angeli A, Osella G, Alì A, et al. Adrenal incidentaloma: an overview of clinical and epidemiological data from the National Italian Study Group. Horm Res 1997;47:279–83. 10.1159/000185477 [DOI] [PubMed] [Google Scholar]
- 11.Hamrahian AH, Ioachimescu AG, Remer EM, et al. Clinical utility of noncontrast computed tomography attenuation value (hounsfield units) to differentiate adrenal adenomas/hyperplasias from nonadenomas: Cleveland Clinic experience. J Clin Endocrinol Metab 2005;90:871–7. 10.1210/jc.2004-1627 [DOI] [PubMed] [Google Scholar]
- 12.Kim YR, Kim JS, Min YH, et al. Prognostic factors in primary diffuse large B-cell lymphoma of adrenal gland treated with rituximab-CHOP chemotherapy from the Consortium for Improving Survival of Lymphoma (CISL). J Hematol Oncol 2012;5:49–57. 10.1186/1756-8722-5-49 [DOI] [PMC free article] [PubMed] [Google Scholar]