

Abstract
Intracellular delivery of mRNA holds great potential for vaccine1–3 and therapeutic4 discovery and development. Despite increasing recognition of the utility of lipid-based nanoparticles (LNPs) for intracellular delivery of mRNA, particle engineering is hindered by insufficient understanding of endosomal escape, which is believed to be a main limiter of cytosolic availability and activity of the nucleic acid inside the cell. Using a series of CRISPR-based genetic perturbations of the lysosomal pathway, we have identified that late endosome/lysosome (LE/Ly) formation is essential for functional delivery of exogenously presented mRNA. Lysosomes provide a spatio-temporal hub to orchestrate mTOR signaling and are known to control cell proliferation, nutrient sensing, ribosomal biogenesis, and mRNA translation. Through modulation of the mTOR pathway we were able to enhance or inhibit LNP-mediated mRNA delivery. To further boost intracellular delivery of mRNA we screened 212 bioactive lipid-like molecules that are either enriched in vesicular compartments or modulate cell signaling. Surprisingly, we have discovered that leukotriene-antagonists, clinically approved for treatment of asthma and other lung diseases, enhance intracellular mRNA delivery in vitro (over 3-fold, p<0.005) and in vivo (over 2-fold, p<0.005). Understanding LNP-mediated intracellular delivery will inspire the next generation of RNA therapeutics that have high potency and. limited toxicity.
Keywords: Intracellular Delivery, mRNA, Lipid Nanoparticles, Endosome Escape, Bioactive Lipids, mTOR
Graphical abstract
RNA therapeutics constitute a class of powerful new drug candidates with the potential to transform modern medicine2,5–7. Recent technological advances have improved mRNA design for reduced innate immune response8–11, leading to a surge in mRNA-based therapeutics that are now entering clinical trials1,2. Intracellular delivery of mRNA boasts rapid genetic transfer in recalcitrant cell populations and averts challenges traditionally associated with DNA-based gene therapy including undesirable genomic integration and the requirement for nuclear localization11–14. The most advanced systems for non-viral delivery of nucleic acids are LNPs, which are formulated using precise molar ratios of phospholipids, cationic/ionizable amino lipids, poly-(ethylene) glycol (PEG)-lipid, and cholesterol15–18. With proper engineering, LNPs effectively encapsulate negatively-charged mRNA19,20, aid in protection against serum nucleases, bypass the mononuclear phagocyte system, and facilitate cellular uptake for gene delivery21,22.
LNPs gain entry into cells by exploiting membrane-derived endocytic pathways. Once endocytosed encapsulated genetic material must egress to the cytosol for translation and subsequent protein expression23,24. Studies suggest that endosomal escape of nucleic acids is spatio-temporally limited, occurring only during a brief stage in endo-lysosomal maturation. Net egress of LNP-delivered nucleic acids is a mere 1–2%, dooming the remainder for lysosomal degradation24. Our understanding of the mechanisms of endosomal escape remains limited25. It is thought that the buffering capacity of a nanoparticle can attenuate the decline in endosomal pH leading to increased osmotic pressure and eventual endosomal rupture26. Another hypothesis suggests that cationic LNPs interact with anionic lipids on the endosomal membrane causing the formation of a hexagonal phase (from a disordered or bilayer phase) which enables cytosolic bioavailability of nucleic acids by facilitating escape27. Furthermore, there is a growing body of evidence that transporters located on endocytic compartments can efflux nucleic acids across endosomes28,29. For example, Niemann-Pick Type C-1 (NPC1), a transmembrane cholesterol transporter located on late LE/Ly, can efflux LNPs containing siRNA to the extracellular millieu29. Antisense oligonucleotides are also known to interact with proteins within the LE/Ly to facilitate delivery across endosomal barriers30. Despite gains in understanding from these studies, insight into endosomal escape pathways remains insufficient for the rational design of nanocarriers for gene delivery25,31.
Because nanocarrier-delivered mRNA rarely reaches the cytosol, traditional microscopy techniques are suboptimal for revealing productive endosomal escape sites due to sensitivity limitations. Even with recent mechanistic insights into escape pathways, engineering LNP formulations for increased functional delivery remains challenging32. Therefore, we employed a two-pronged strategy to begin to resolve these issues. First, we tested the ability of clinically relevant nanoparticles to functionally deliver mRNA in cell lines that were made genetically devoid of key LE/Ly trafficking proteins. Second, to enhance transfection, we screened a library of bioactive lipids known to affect signaling and enrich endo/lysosomal compartments. We have identified new cell-signaling pathways that contribute to enhanced gene expression and found that LEs act as a hub for cell signaling effectors capable of potentiating mRNA translation. These studies show that signaling from lysosomal compartments are essential for mRNA translation and identify new bioactive lipid-like molecules that enhance intracellular delivery.
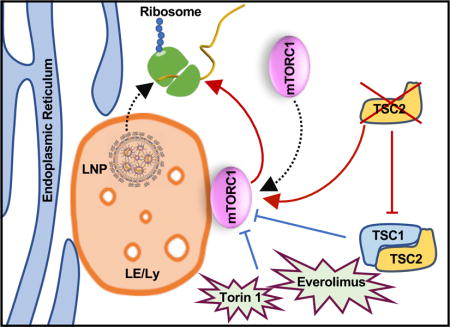
To gain insight into intracellular trafficking and identify important sites of endosome escape of nanoparticle-delivered mRNA, we utilized haploid cells (HAP1) genetically edited to be devoid of Rab5A, Rab4A, or Rab7A to interrupt biogenesis of early, recycling, and late endosomes, respectively (Fig. 1a)33. Dose-response and time course studies were performed to establish the kinetics of mRNA expression using commercially available lipoplex and proprietary ionizable LNPs (~100 nm in size with over 90% encapsulation efficiency) that have been previously published (Fig. S1a, b)34,35. We found that the absence of Rab4A or Rab5A had limited influence on transfection in the HAP1 cells indicating that neither endosomal recycling nor transition from early to late stage was critical in mRNA delivery (Fig. 1b, c). Conversely, Rab7A-deficient cells exhibited a dramatic decrease in gene expression (Fig. 1d, e) while having little effect on mRNA uptake (data not shown) demonstrating that late endosome (LE) formation is required for gene delivery. Electron microscopy showed enlarged endosomes in Rab7A-deficient cells (~540 nm) as compared to wild-type cells (WT) (~90nm), confirming compromised LE/Ly biogenesis (Fig. 1f i–iii). We further tested whether bypassing the endo/lysosomal system restores mRNA delivery. To this end, we electroporated free mRNA into the HAP1-WT and Rab7A knockout (KO) cells and were unable to rescue the transfection, potentially indicating that downstream cell signaling pathways originating from the LE/Ly drive mRNA translation (Fig. 1g). Interestingly, it has been shown that upon activation mechanistic target of rapamycin complex 1 (mTORC1) resides on the lysosomal surface, which serves as a hub for mTORC1 to bind several downstream signaling effectors that control multiple cellular processes including mRNA translation and ribosomal biogenesis. Upon cellular nutrient uptake, mTORC1 is sequestered from the cytosol to LE/Ly membrane where it initiates proliferation events including protein translation36–38. We hypothesized that arresting endosome maturation through the deletion of Rab7A would prevent mTORC1 from triggering translation of delivered mRNA. To confirm that loss of translation was due to the disruption of mTORC1 signaling, we employed selective pharmacological mTORC1 inhibitors including Torin 1 and Everolimus to impair signaling. As expected, this resulted in a decrease in successful lipoplex-delivered mRNA even at nanomolar inhibitory concentrations (Fig. S2a). Constitutive activation of mTORC1 through genetic deletion of an upstream effector, Tuberous Sclerosis Complex 2 (TSC2), enhanced gene delivery of exogenous mRNA transfected through electroporation (Fig. S2b), further implicating the involvement of mTORC1 signaling in LE-mediated mRNA expression39. These studies suggest that lysosomes known to serve as a hub for mTORC1 dependent signaling36, promotes translation of synthetically delivered mRNA (Fig. S2c). Tampering with lysosomal biogenesis to reduce nanoparticle delivery to these degradative compartments can therefore be counterproductive for gene delivery applications.
Figure 1. Late endosomes are essential for mRNA transfection.
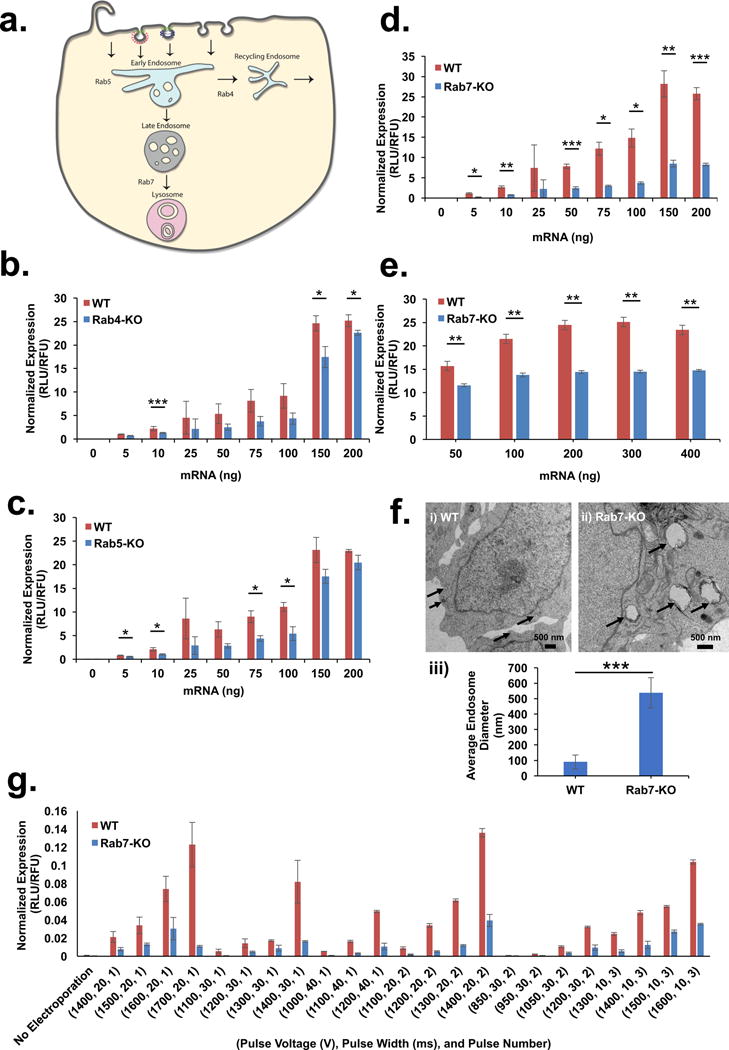
(a) Schematic representation of Rab proteins localizing to various stages of endocytic process. (b–d) HAP1 cells with deletions of (b) Rab4A, (c) Rab5A, and (d) Rab7A were transfected with mRNA packaged inside lipoplexes and luciferase expression was compared to wild-type. (e) HAP1-Rab7-KO cells were transfected with LNPs at a range of mRNA doses and normalized luciferase expression was compared to wild-type (f) Electron micrographs of (i) HAP1-WT (scale bar = 500 nm) and (ii) HAP1-Rab7-KO cells (scale bar = 500 nm) showing enlarged endosomes in Rab7-KO cells indicated by arrows. (iii) Average size of endosomes (indicated by inset arrow) in HAP1-WT and HAP1-Rab7-KO, n = 4, mean ± SD. (g) Luciferase expression in HAP1-WT and HAP1-Rab7-KO cells transfected with free mRNA using electroporation at various pulse voltages, pulse widths and pulse numbers. All experiments were conducted with n = 3; mean ± SD, unless indicated otherwise. Statistical analysis of the data was assessed by Student’s t-test (0.05 ≥ *p > 0.01, 0.01 ≥ **p > 0.005, ***p ≤ 0.005).
Previous reports have shown a significant number of molecules that increase transfection of nucleic acids40–43. We wanted to test whether a small set of molecules (Table S1) had the capability to show similar activity in relation to mRNA delivery. Despite minor condition-dependent activity (time and co-incubation parameters) of these compounds, our studies failed to elicit any dramatic increase in transfection (Fig. S3a–d). To uncover other molecules capable of enhancing intracellular delivery, we screened a bioactive lipid library (212 compounds), which included prostaglandins, isoprostanes, thromboxanes, leukotrienes, lipoxins and several other complex polyunsaturated fatty acids (Supplementary Excel File). It is well known that beyond their role as structural components44, lipids also serve as signaling molecules45, play a crucial role in vesicular trafficking46, and can be easily incorporated into a liposomal formulation. As compared to other known endosomal escape agents (Table S1), exposure of cells to select bioactive lipids can impact mRNA transfection by inducing LE/Ly maturation or modulating cell signaling events. We pre-treated cells with the compound library prior to transfection with lipoplexes and identified several transfection-enhancing compounds (Fig. 2a). Further validation of several compounds revealed that pre-treatment of cells with only one compound, MK-571 (a leukotriene inhibitor) resulted in 200% increase in transfection (Fig. 2b; Fig. S4a i–ii; Table S2). Leukotrienes are inflammatory lipid mediators and metabolites of arachidonic acid. Leukotriene D4 (LTD4), binds the Cysteinyl Leukotriene Receptor 1 (CysLT1), a G-protein coupled receptor (GPCR) that is internalized through the endo/lysosomal system. CysLT1 activation contributes to various allergic reactions including bronchial constriction and asthma47. MK-571 is an orally active, potent and selective LTD4 antagonist (competes for CysLT1), and multidrug resistance protein 1 (MRP1) inhibitor48. While arachidonic acid by itself showed no improvements in transfection (Fig. S4b), we tested three clinically approved leukotriene antagonists (Montelukast, Pranlukast, and Zafirlukast) and identified two compounds, Pranlukast (Brandname – Onon and Azlaire) and Zafirlukast (Brandname - Accolate, Accoleit, and Vanticon), of comparable efficacy (Fig. S4c, d)49. We further found that pre-incubating cell cultures with MK-571 led to a 200% increase LNP-mediated gene expression (Fig. 2c). Furthermore, we developed nanoparticle formulations containing MK-571 for co-delivery with mRNA (LNP-MK571). LNPs formulated with and without MK-571 exhibited no significant differences in size or encapsulation efficiency (Fig. S5a). Gel electrophoresis showed that LNPs or LNP-MK571 remained stable in presence of heparin, a negatively charged polymer, while Triton X-100 disassembled both particles similarly, suggesting a general physical compatibility between MK-571 and components of LNP (Fig. S5b). LNP-MK571 outperformed LNPs in both HeLa and HepG2 cells (3-fold and 5-fold, respectively; Fig. 2d, e). We provide additional data that compares intracellular delivery of LNP-MK571 in Rab5A-, Rab4A-, or Rab7A-depleted cells to WT. LNP-mediated mRNA transfection remained unaltered despite defects in formation of early and recycling endosomes but was significantly diminished when the biogenesis of late endosomes was impaired (Fig. S5c–e), as was observed earlier with nanoparticles alone (Fig. 1b–d). This is to be expected since defects in late endosome formation diminishes mTOR signaling, which in turn reduces mRNA translation (Fig. S2a–c) that cannot be rescued by MK-571. In addition, we compared transfection efficiency of LNPs and LNP-MK571 in Rab7A-deficient cells. We found that LNP-MK571 led to a significantly higher gene delivery as compared to its counterpart (Fig. S5f), further suggesting that MK-571 boosts intracellular delivery of LNP encapsulated mRNA. Finally, we compared the in vivo delivery of LNPs and LNP-MK571 after intravenous (IV) administration in BALB/c mice. Both nanoparticles showed gene expression in the liver and spleen, with LNP-MK571 exhibiting significant increase (over two-fold, p<0.005) after six hours in both organs (Fig. 2f, g; Fig. S5g, h), demonstrating that this bioactive lipid can enhance transfection both in vitro and in vivo. These studies suggest that bioactive lipophilic compounds can be incorporated into LNPs to promote gene expression.
Figure 2. MK-571 a leukotriene antagonist improves intracellular delivery to enhance gene delivery in vitro and in vivo.
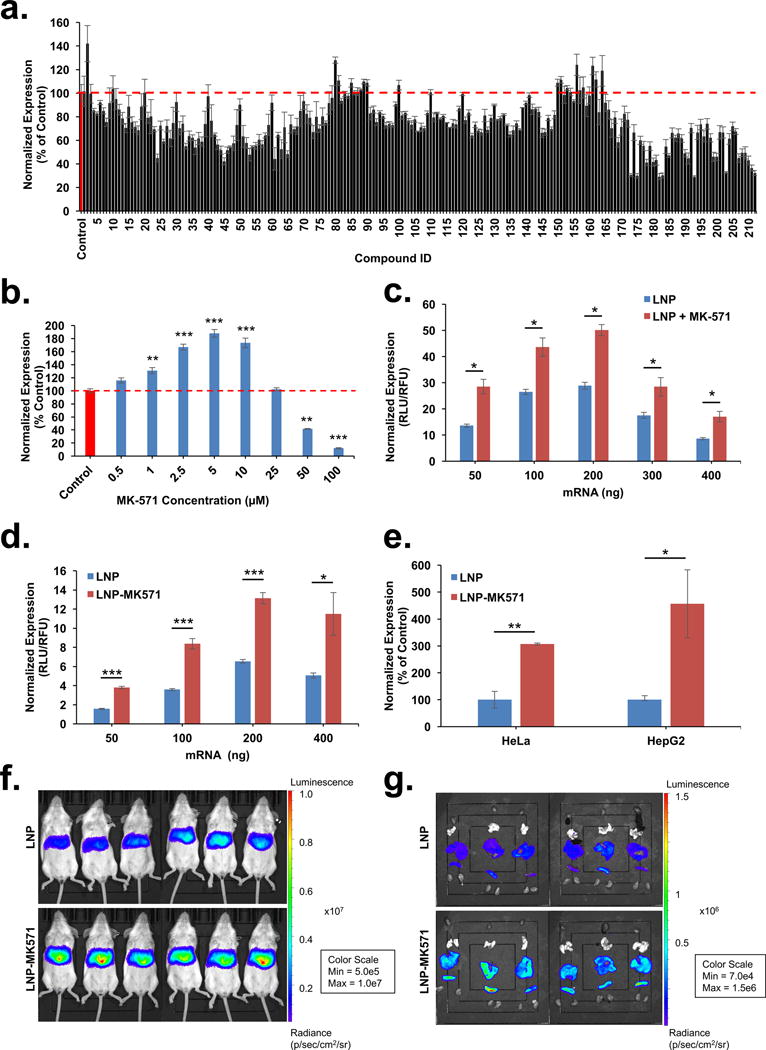
(a) Bioactive lipid library screening showing in vitro transfection with mRNA-lipoplexes (50 ng mRNA in the presence of 1 μM bioactive lipids) normalized to mRNA-lipoplex control (100%). (b) mRNA-lipoplex transfection of 50 ng mRNA in the presence of varying concentrations of MK-571 (0 – 100 μM) relative to mRNA-lipoplex control. Increasing amounts of mRNA were transfected using (c) LNPs with and without 24 hr pre-incubation with MK-571 (5 μM) or (d) with and without MK-571 loaded LNPs (LNP-MK571). (e) Comparison of in vitro transfection of two different cell lines using 400 ng mRNA delivered by LNP-MK571. LNPs served as positive control (100% expression) (f) Transfection in BALB/c mice treated with 0.05 mg/kg mRNA delivered through an IV injection of LNP or LNP-MK571; n=6; mean ± SD. (g) Isolated organs showing localized expression in liver and spleen. Luciferase intensity measured at 6 hrs post injection, scale represented as radiance (p/sec/cm2/sr); n=6; mean ± SD. All experiments were conducted with n = 3; mean ± SD, unless indicated otherwise. Statistical analysis of the data was assessed by student’s t-test (0.05 ≥*p > 0.01, 0.01 ≥ **p > 0.005, ***p ≤ 0.005).
Endosomal processing remains the rate-limiting step in efficient intracellular drug and gene delivery25,30,50–52. Endosomal escape is a transient event, which restricts the delivery of nucleic acids across endosomal membranes53. Powerful imaging methods have been employed to reveal dynamic endosomes that allow for siRNA escape, albeit at extremely low amounts, but sufficient to result in gene silencing50,51. Advances in cell biology have shown that Rab proteins localized on the surface of the vesicles control endosomal biogenesis, transport, and distribution within a cell33. The transient inhibition of Rabs or localization of nanoparticles with endosomes bearing them has demonstrated that siRNA escape may occur during endosomal maturation50,51. In this study, we have utilized CRISPR/Cas9-based gene editing to cause permanent disruption of different stages of trafficking. This method provides a platform to measure functional delivery of cargo in the absence of specific endosomal processes as opposed to other cumbersome and artefact-ridden methods23,54. While siRNA delivery has been limited due to endocytic recycling through the late endosomes and requires escape from the early endosomes, our studies indicate that late endosome formation is essential for mRNA delivery. We have shown using gene-editing tools that late endosomes serve as a host to mTOR-based cell signaling that regulates translation of exogenously delivered mRNA. Lysosomes, long underestimated as static dead catabolic organelles, have been shown to be a hub of enormous activity; they control cellular metabolism, host numerous transporters, promote nutrient and cholesterol sensing, and orchestrate mTOR-dependent intracellular signaling36,36,38,55,56. The lysosomal subpopulation, either in the periphery or near the perinuclear region, has distinct pH57. LNP sensing through a lysosome can trigger a signaling cascade that, depending on spatio-temporal localization of these compartments, may affect mRNA delivery. It is possible that differences in mTOR signaling in various tissues can lead to differential mRNA expression, and in cancers like TSCs where mTOR is constitutively active mRNA therapeutics may be effective at extremely low doses in these cancer cell populations. We further speculate that understanding LNP interactions with lysosomes and the overall lysosomal milieu remains critical to uncover the mechanisms of endosomal escape of nucleic acids, an area that we are currently investigating. However, it is likely that external interference on lysosomal biogenesis will have negative consequences on effective gene delivery. Lysosomes can form transient membrane contact sites with the endoplasmic reticulum (ER) using Rab7, so it is possible that these organelles are required for subcellular targeting to the active site of mRNA translation i.e. the ER58. It is likely that various therapeutic RNAs (mRNA, miRNA, sgRNA, siRNA, etc.) can differ in their itinerary for escape through interactions with distinct proteins within the endo/lysosomal system. We have further used bioactive lipids that modulate cellular signaling, are known to enrich endo/lysosomal compartment, and can be easily formulated with liposomal formulations to enhance gene delivery. Leukotriene inhibitors identified from our screen are currently in clinical use for treatment of asthma. We posit that co-delivery of these molecules with a therapeutic mRNA can be used for synergistic treatment especially of different lung diseases59, including cystic fibrosis60. Unlocking intracellular transport mechanisms for mRNA delivery will lead to engineering of the next generation of drug delivery systems by enabling high potency with limited toxicity.
Materials and Methods
Chemicals
Guanabenz acetate, heparin sodium salt, ethanol, Synperonic PE/P84 (Pluronics® P84) were purchased from Sigma-Aldrich. Bioactive Lipid Library II in addition to compounds L1–L12 was obtained from Cayman Chemicals (Details in Supplementary Excel File). All leukotriene inhibitors were purchased from Cayman Chemicals. Chloroquine diphosphate was purchased from MP Biomedicals. Everolimus and Torin 1 were purchased from Cell Signaling Technologies. Pluronic® F127 and Pluronic® P103 were purchased from BASF. Muramyl dipeptide (MDP) was purchased from InvivoGen. Triton X-100 was purchased from Acros Organics. Quanti-iT RiboGreen RNA reagent and rRNA standards were purchased from Life Technologies. CellTiter Fluor Cell Viability Assay and One-Glo Luciferase Assay was purchased from Promega. Firefly luciferase mRNA (FLuc mRNA, Moderna) and lipid formulation (ionizable lipid: structural lipid: cholesterol: PEG-lipid) were obtained from Moderna Therapeutics. All cell culture reagents were purchased from Corning.
Formulation
All particles contained FLuc mRNA unless otherwise noted.
Lipoplexes: Stemfect lipid reagent (Stemgent) and mRNA was diluted with transfection buffer (Stemgent) in separate tubes according to manufacturer’s protocol. Lipid solution was then added to mRNA solution and homogenized by pipette mixing. The formulation incubated at room temperature for 10 mins and was used to transfect cells at desired concentrations.
Lipid-based nanoparticles (LNPs): LNPs were prepared by combining an aqueous phase (mRNA diluted in 50 mM citrate buffer, pH 4, and supplemented with 5 μM MK-571 when specified) and organic phase (ionizable lipid: structural lipid: cholesterol: PEG-lipid dissolved in ethanol at molar ratios of 50:10:38.5:1.5, respectively) at a 3:1 ratio through syringe mixing or a microfluidic mixer (Precision Nanosystems). Aqueous and organic phases were injected into the microfluidic device at a combined volumetric flow rate of 12 mL/min. Formulations were filtered, washed with Phosphate Buffered Saline (PBS, pH 7.2) and concentrated using Amicon Ultra Centrifugal Filters (EMD Millipore) and stored at 4 °C until use.
Particle characterization
LNPs were characterized using dynamic light scattering (DLS, Malvern Zetasizer). mRNA encapsulation efficiency was evaluated by modified Quanti-iT RiboGreen RNA reagent assay (Life Technologies). LNP sample or PBS (negative control) was diluted into TE buffer down to a mRNA concentration between 2–5 ng/μL. Aliquots of each LNP working solution was further diluted 1:1 in TE buffer (measuring unencapsulated mRNA) or 1:1 in TE buffer with 2% Triton-X100 (measuring total mRNA – both encapsulated within LNPs and unencapsulated, “free” mRNA). Samples were prepared in duplicate. Quanti-iT™ RiboGreen RNA reagent was added to each sample and fluorescent signal was quantified (Tecan i-Control v. 3.8.2.0, Tecan Infinite M200 Pro Multimode Plate Reader). Encapsulation efficiency was determined as follows:
Cell culture
Cells were grown in appropriate growth media (Table 1) supplemented with 10% fetal bovine serum (Corning) and 5% Penicillin/Streptomycin (Corning). All cultures were grown in 37 °C incubators supplemented with 5% CO2 and were cultured according to suppliers’ instructions. The HAP1 cells generated by Horizon Discovery were edited using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 based tools. See table for details.
Table 1.
Cell Lines and Corresponding Culture Conditions
| Cell Line | Medium | Source |
|---|---|---|
| HeLa | DMEM | Langer Lab |
| HepG2 | DMEM | Sun Lab |
| HAP1 WT | IMDM | Horizon Discovery |
| HAP1 Rab4A− | IMDM | Horizon Discovery |
| HAP1 Rab5A− | IMDM | Horizon Discovery |
| HAP1 Rab7A− | IMDM | Horizon Discovery |
| Immortalized MEFs TSC2+/+ and TSC2−/− |
DMEM | Kwiatkowski Lab (Sarkar Lab) |
In vitro transfection
Cells were plated in white, clear-bottom 96-well plates. The number of cells plated per well varied by cell type: all diploid adherent cells (HeLa and MEFs) were plated at 4,000 cells/well, haploid cells were plated at 20,000 cells/well, Stemfect and LNPs: Cells were allowed to adhere and grow for 24 hours prior to LNP or LNP-MK571 transfection. Nanoparticles were added dropwise on top of the media.
General protocol for screening
After a 2-hour settling period, cells were pre-treated with bioactive lipid (concentrations in text), mTOR inhibitors, or leukotriene inhibitors for 24 hours prior to transfection using Stemfect (50 ng/well mRNA).
Electroporation
Cells were transfected with free FLuc mRNA using the Neon transfection system (Thermo Scientific) according to manufacturer’s protocol. Cells recovered in a 24-well plate for 24 hours prior to trypsinization and were transferred to a 96-well plate for analysis.
Analysis
Cell viability (CellTiter Fluor Cell Viability Assay, Promega) and luciferase expression (One-Glo Luciferase Assay, Promega) were measured after 24 hours.
Transmission Electron Microscopy
The cells were fixed in cold Karnovsky fixative (2% paraformaldehyde, 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer pH 7.2), pre-embed in 6% bovine serum albumin (BSA) and prepared for microwave-assisted processing in a Pelco BioWave Microwave. In the BioWave, the samples were rinsed in 0.1 M sodium cacodylate buffer; incubated in reduced osmium tetroxide (1.5% potassium ferrocyanide in 2% OsO4); rinsed in water and en bloc stained with aqueous 0.5% uranyl acetate. Following the uranyl acetate incubation, samples were dehydrated in an aqueous series of 50%, 75% and 95% acetone, followed by two exchanges in 100% acetone. Epon resin infiltration was facilitated by incubation in a 1:1 solution of 100% acetone: freshly-made Epon resin, followed by 4 exchanges in 100% freshly-made Epon. Samples were removed from the BioWave and transferred into embedding capsules (BEEM) filled with freshly-made Epon and cured at 60°C for 36 hours. Thin sections (70 nm) obtained from the block face were imaged at 80 kV on a FEI-Tecnai 12 system interfaced to a digital camera and associated software (Advanced Microscopy Techniques, Danvers, MA).
Gel Retardation Assay
LNP and LNP-MK571 were incubated for 5 mins with heparin (5–20 μg) or Triton X-100 (1–10%). Free mRNA, LNP, LNP-MK571, heparin- or Triton X-treated LNPs, and LNP-MK571 were then run on a 1% agarose gel at 250 μg mRNA per well and visualized by ethidium bromide staining using myECL Imager (Thermo Scientific).
In vivo transfection
All animal studies were conducted at Oregon Health and Sciences University, were approved by the Institutional Animal Care and Use Committee (IACUC), and were compliant with local, state, and federal regulations. Female BALB/c mice (Charles River Laboratories) received tail vein injections of LNP or LNP-MK571 at a mRNA dose of 0.05 mg/kg body weight. Bioluminescent imaging was performed on mice or isolated organs using the IVIS Lumina XRMS imaging system (Perkin Elmer) following intraperitoneal injection of 200 μL of D-luciferin substrate (Perkin Elmer, 150 mg/kg body weight). Image acquisition and analysis was conducted using IVIS Living Image software (Perkin Elmer).
Statistical analysis
Significance was determined using Student’s t-test for all comparisons.
Supplementary Material
Acknowledgments
This project was supported through funding from Moderna Therapeutics (GS), OSU College of Pharmacy startup funding (GS), National Institute of Biomedical Imaging and Bioengineering 1R15EB021581-01 (GS), National Institute of General Medical Sciences (NIGMS) 1R35GM119839-01 (C.S.) and Cystic Fibrosis Foundation (GS). We thank Dr. D.J. Kwiatkowski and Dr. Sovan Sarkar for TSC2−/− MEFs and Dr. Adam Alani for access to Malvern Zetasizer. Electron microscopy was performed at the Multiscale Microscopy Core (MMC) with technical support from the Oregon Health & Science University (OHSU) FEI Living Lab and the OHSU Center for Spatial Systems Biomedicine (OCSSB). We would like to thank Mr. Fabian Wieghardt (Univ Wurzburg) for technical assistance. We thank Dr. John Joyal and Dr. Luis Brito (Moderna Therapeutics) for insightful comments.
Footnotes
CONTRIBUTIONS:
GS conceived the idea and directed research through input from ÖA, CS, KB. GS provided ideas for SP to perform screens with bioactive lipid screen and use of gene editing tools to dissect intracellular trafficking. NA, ER, AD and CS provided support with in vitro and in vivo assays. All authors wrote the paper.
COMPETING FINANCIAL INTERESTS:
KB, CM and OA are Moderna Therapeutics employees; GS received funding from and is a consultant with Moderna Therapeutics.
SUPPLEMENTAL INFORMATION ARE AVAILABLE ONLINE.
References
- 1.Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, Laska ME, Smith M, Almarsson Ö, Thompson J, Ribeiro AM, Watson M, Zaks T, Ciaramella G. Mol Ther J Am Soc Gene Ther. 2017 doi: 10.1016/j.ymthe.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS. Cell. 2017;168(6):1114–1125.e10. doi: 10.1016/j.cell.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rauch S, Lutz J, Kowalczyk A, Schlake T, Heidenreich R. Methods Mol Biol Clifton NJ. 2017;1499:89–107. doi: 10.1007/978-1-4939-6481-9_5. [DOI] [PubMed] [Google Scholar]
- 4.DeRosa F, Guild B, Karve S, Smith L, Love K, Dorkin JR, Kauffman KJ, Zhang J, Yahalom B, Anderson DG, Heartlein MW. Gene Ther. 2016;23(10):699–707. doi: 10.1038/gt.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cullis PR, Hope MJ. Mol Ther J Am Soc Gene Ther. 2017 [Google Scholar]
- 6.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Nat Rev Genet. 2014;15(8):541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 7.Stewart MP, Sharei A, Ding X, Sahay G, Langer R, Jensen KF. Nature. 2016;538(7624):183–192. doi: 10.1038/nature19764. [DOI] [PubMed] [Google Scholar]
- 8.Karikó K, Buckstein M, Ni H, Weissman D. Immunity. 2005;23(2):165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Uchida S, Kataoka K, Itaka K. Pharmaceutics. 2015;7(3):137–151. doi: 10.3390/pharmaceutics7030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D. J Control Release Off J Control Release Soc. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahin U, Kariko K, Tureci O. Nat Rev Drug Discov. 2014;13(10):759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 12.Seiffert M, Stilgenbauer S, Döhner H, Lichter P. Leukemia. 2007;21(9):1977–1983. doi: 10.1038/sj.leu.2404863. [DOI] [PubMed] [Google Scholar]
- 13.Youn H, Chung JK. Expert Opin Biol Ther. 2015;15(9):1337–1348. doi: 10.1517/14712598.2015.1057563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou S, Scarfo K, Nantz MH, Hecker JG. Int J Pharm. 2010;389(1–2):232–243. doi: 10.1016/j.ijpharm.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, Sah DWY, Stebbing D, Crosley EJ, Yaworski E, Hafez IM, Dorkin JR, Qin J, Lam K, Rajeev KG, Wong KF, Jeffs LB, Nechev L, Eisenhardt ML, Jayaraman M, Kazem M, Maier MA, Srinivasulu M, Weinstein MJ, Chen Q, Alvarez R, Barros SA, De S, Klimuk SK, Borland T, Kosovrasti V, Cantley WL, Tam YK, Manoharan M, Ciufolini MA, Tracy MA, de Fougerolles A, MacLachlan I, Cullis PR, Madden TD, Hope MJ. Nat Biotechnol. 2010;28(2):172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 16.Allen TM, Cullis PR. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 17.Dong Y, Love KT, Dorkin JR, Sirirungruang S, Zhang Y, Chen D, Bogorad RL, Yin H, Chen Y, Vegas AJ, Alabi CA, Sahay G, Olejnik KT, Wang W, Schroeder A, Lytton-Jean AKR, Siegwart DJ, Akinc A, Barnes C, Barros SA, Carioto M, Fitzgerald K, Hettinger J, Kumar V, Novobrantseva TI, Qin J, Querbes W, Koteliansky V, Langer R, Anderson DG. Proc Natl Acad Sci U S A. 2014;111(11):3955–3960. doi: 10.1073/pnas.1322937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen D, Love KT, Chen Y, Eltoukhy AA, Kastrup C, Sahay G, Jeon A, Dong Y, Whitehead KA, Anderson DG. J Am Chem Soc. 2012;134(16):6948–6951. doi: 10.1021/ja301621z. [DOI] [PubMed] [Google Scholar]
- 19.Fenton OS, Kauffman KJ, Kaczmarek JC, McClellan RL, Jhunjhunwala S, Tibbitt MW, Zeng MD, Appel EA, Dorkin JR, Mir FF, Yang JH, Oberli MA, Heartlein MW, DeRosa F, Langer R, Anderson DG. Adv Mater Deerfield Beach Fla. 2017 doi: 10.1002/adma.201606944. [DOI] [PubMed] [Google Scholar]
- 20.Kaczmarek JC, Kowalski PS, Anderson DG. Genome Med. 2017;9(1):60. doi: 10.1186/s13073-017-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, Jayaraman M, Rajeev KG, Cantley WL, Dorkin JR, Butler JS, Qin L, Racie T, Sprague A, Fava E, Zeigerer A, Hope MJ, Zerial M, Sah DW, Fitzgerald K, Tracy MA, Manoharan M, Koteliansky V, de Fougerolles A, Maier MA. Mol Ther. 2010;18(7):1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Nat Rev Genet. 2014;15(8):541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 23.Sahay G, Alakhova DY, Kabanov AV. J Control Release Off J Control Release Soc. 2010;145(3):182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stöter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M, Zerial M. Nat Biotechnol. 2013;31(7):638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 25.Dowdy SF. Nat Biotechnol. 2017;35(3):222–229. doi: 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 26.Behr J. Chimia. 1997;(51):34–36. [Google Scholar]
- 27.Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, Sah DWY, Stebbing D, Crosley EJ, Yaworski E, Hafez IM, Dorkin JR, Qin J, Lam K, Rajeev KG, Wong KF, Jeffs LB, Nechev L, Eisenhardt ML, Jayaraman M, Kazem M, Maier MA, Srinivasulu M, Weinstein MJ, Chen Q, Alvarez R, Barros SA, De S, Klimuk SK, Borland T, Kosovrasti V, Cantley WL, Tam YK, Manoharan M, Ciufolini MA, Tracy MA, de Fougerolles A, MacLachlan I, Cullis PR, Madden TD, Hope MJ. Nat Biotechnol. 2010;28(2):172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 28.Kaufmann AM, Krise JP. J Biol Chem. 2008;283(36):24584–24593. doi: 10.1074/jbc.M803715200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, Buganim Y, Schroeder A, Langer R, Anderson DG. Nat Biotechnol. 2013;31(7):653–658. doi: 10.1038/nbt.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crooke ST, Wang S, Vickers TA, Shen W, Liang X. Nat Biotechnol. 2017;35(3):230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 31.Suh J, Dawson M, Hanes J. Adv Drug Deliv Rev. 2005;57(1):63–78. doi: 10.1016/j.addr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Huang L. Nat Biotechnol. 2013;31(7):611–612. doi: 10.1038/nbt.2634. [DOI] [PubMed] [Google Scholar]
- 33.Zerial M, McBride H. Nat Rev Mol Cell Biol. 2001;2(2):107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 34.Turnbull IC, Eltoukhy AA, Fish KM, Nonnenmacher M, Ishikawa K, Chen J, Hajjar RJ, Anderson DG, Costa KD. Mol Ther. 2016;24(1):66–75. doi: 10.1038/mt.2015.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akinc A, Battaglia G. Cold Spring Harb Perspect Biol. 2013;5(11):a016980. doi: 10.1101/cshperspect.a016980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Efeyan A, Zoncu R, Sabatini DM. Trends Mol Med. 2012;18(9):524–533. doi: 10.1016/j.molmed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perera RM, Zoncu R. Annu Rev Cell Dev Biol. 2016;32:223–253. doi: 10.1146/annurev-cellbio-111315-125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim CY, Zoncu R. J Cell Biol. 2016;214(6):653–664. doi: 10.1083/jcb.201607005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM. Mol Biol Cell. 2010;21(5):833–841. doi: 10.1091/mbc.E09-09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varkouhi AK, Scholte M, Storm G, Haisma HJ. J Controlled Release. 2011;151(3):220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Gilleron J, Paramasivam P, Zeigerer A, Querbes W, Marsico G, Andree C, Seifert S, Amaya P, Stöter M, Koteliansky V, Waldmann H, Fitzgerald K, Kalaidzidis Y, Akinc A, Maier MA, Manoharan M, Bickle M, Zerial M. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang B, Ming X, Cao C, Laing B, Yuan A, Porter MA, Hull-Ryde EA, Maddry J, Suto M, Janzen WP, Juliano RL. Nucleic Acids Res. 2015;43(4):1987–1996. doi: 10.1093/nar/gkv060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Batrakova EV, Kabanov AV. J Control Release Off J Control Release Soc. 2008;130(2):98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H, Leal C. ACS Nano. 2015;9(10):10214–10226. doi: 10.1021/acsnano.5b03902. [DOI] [PubMed] [Google Scholar]
- 45.Bissig C, Johnson S, Gruenberg J. Methods Cell Biol. 2012;108:19–46. doi: 10.1016/B978-0-12-386487-1.00002-X. [DOI] [PubMed] [Google Scholar]
- 46.Bissig C, Gruenberg J. Cold Spring Harb Perspect Biol. 2013;5(10):a016816. doi: 10.1101/cshperspect.a016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Funk CD. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 48.Peterson BG, Tan KW, Osa-Andrews B, Iram SH. Pharmacol Res. 2017;119:313–326. doi: 10.1016/j.phrs.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Yang H, Wang Z, Xie X, Nan F. ACS Med Chem Lett. 2016;7(3):335–339. doi: 10.1021/acsmedchemlett.5b00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J. Nat Biotech. 2015;33(8):870–876. doi: 10.1038/nbt.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stöter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M, Zerial M. Nat Biotechnol. 2013;31(7):638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 52.Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, Buganim Y, Schroeder A, Langer R, Anderson DG. Nat Biotechnol. 2013;31(7):653–658. doi: 10.1038/nbt.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart MP, Lorenz A, Dahlman J, Sahay G. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015 doi: 10.1002/wnan.1377. [DOI] [PubMed] [Google Scholar]
- 54.Ivanov AI. Methods Mol Biol Clifton NJ. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- 55.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. EMBO J. 2012;31(5):1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle REN, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS, Zoncu R. Science. 2017;355(6331):1306–1311. doi: 10.1126/science.aag1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson DE, Ostrowski P, Jaumouillé V, Grinstein S. J Cell Biol. 2016;212(6):677–692. doi: 10.1083/jcb.201507112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raiborg C, Wenzel EM, Stenmark H. EMBO J. 2015;34(14):1848–1858. doi: 10.15252/embj.201591481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riccioni G, Bucciarelli T, Mancini B, Di Ilio C, D’Orazio N. Curr Med Chem. 2007;14(18):1966–1977. doi: 10.2174/092986707781368522. [DOI] [PubMed] [Google Scholar]
- 60.Liu YC, Khawaja AM, Rogers DF. Br J Pharmacol. 1998;124(3):563–571. doi: 10.1038/sj.bjp.0701886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.