

Abstract
The DNA base guanine (G) can be oxidatively modified to 8-oxo-7,8-dihydroguanine (OG). Extraction of genomic DNA followed by nuclease digestion and mass spectrometry analysis have found OG is present at background levels of ~1 out of 106 Gs; however, this approach cannot determine the locations for the OGs in the genome. Thus, in this methods report, we outline three different methods (A, B, and C) for sequencing OG in DNA. Method A sequences OG by utilizing the base excision repair pathway to delete the OG nucleotide from the DNA that is then detected by Sanger sequencing as a deletion signature. Method B sequences OG by harnessing the base excision repair pathway to convert OG to an unnatural DNA base pair followed by Sanger sequencing to locate the unnatural base pair indicating where OG was located. Method C (i.e., OG-Seq) takes genomic DNA sheared to ~150 bps followed by selectively biotinylating the OG-containing fragments for affinity purification and enrichment of the OG-modified strands. The OG-modified fragments are sequenced on a next-generation sequencing platform to locate OG on the genomic scale with a resolution of ~150 bps. The methods outlined are then compared and contrasted allowing researchers to select the one that best suits their experimental goals.
1. INTRODUCTION
In DNA, the heterocyclic bases can be oxidatively modified by reactive oxygen species to yield many products (Cadet et al., 2014; Delaney et al., 2012; Fleming and Burrows, 2016). A prominent oxidative compound observed is 8-oxo-7,8-dihydroguanine (OG), a primary two-electron oxidation product of guanine (G). Cell-based experiments suggest that OG causes mutations suspected in disease initiation and progression (Al-Tassan et al., 2002); additionally, OG can modulate gene expression (Fleming et al., 2016; Pan et al., 2016; Pastukh et al., 2015). In vitro polymerase bypass and cell studies that knocked out proteins responsible for OG removal from the genome have identified that OG causes G→T transversion mutations due to OG Hoogsteen base pairing with adenine (Al-Tassan et al., 2002; Shibutani et al., 1991). Confirmation of the cellular mutation results was achieved by synthesizing OG site specifically into a plasmid and allowing replication of the plasmid to occur in prokaryotic cells followed by sequence analysis of the replicons (Wood et al., 1992). This method verified that OG induces G→T transversion mutations. Recently, the modified base OG has been suspected in modulating gene expression on the basis of in vitro and in vivo studies (Fleming et al., 2016; Hailer-Morrison et al., 2003; Moore et al., 2013; Pan et al., 2016; Pastukh et al., 2015; Ramon et al., 1999; Tornaletti et al., 2004). For instance, in vitro studies with OG located in transcription factor binding sequences identified a position-dependent impact on protein binding that could possibly alter gene expression (Hailer-Morrison et al., 2003; Moore et al., 2013; Ramon et al., 1999). When OG was synthesized in a plasmid in the template strand of the coding region for a reporter protein, the presence of OG negatively impacted protein expression in mammalian cells (Allgayer et al., 2016). In contrast, when OG was located in the TNFα or VEGF promoters in mammalian cells, gene activation was observed that was suspected to result from coordination of DNA repair with gene regulatory factors (Fleming et al., 2016; Pan et al., 2016; Pastukh et al., 2015). These studies indicate that OG has many roles in biological processes; therefore, to better understand these roles, sequencing DNA for OG is essential.
The presence of OG in the genome is traditionally determined by extraction of genomic DNA followed by nuclease digestion and mass spectrometry analysis that has found a background level of ~1 OG out of every 106 Gs (Gedik and Collins, 2005); however, this approach cannot determine the locations of the OGs in the genome. Thus, sequencing the genome for the thousands of OGs present will allow the identification of their locations to better understand the mutagenesis and gene regulatory effect of OG. Implementation of OG sequencing on the genomic scale requires a high-throughput method or sequencing for specific genomic loci. Additionally, synthetic incorporation of OG into plasmids requires OG sequencing to confirm the synthesis was successful; this can be achieved with targeted OG sequencing. Direct sequencing for OG is achievable by SMRT or nanopore sequencing methods on native DNA (Clark et al., 2011; Schibel et al., 2010); however, a drawback to these methods is that the DNA cannot be PCR amplified and retain the OG location during this step. This methods paper outlines three approaches developed in our laboratory for sequencing OG in DNA (Ding et al., 2017; Riedl et al., 2015a; Riedl et al., 2015b). The three methods described allow PCR amplification of the OG-containing DNA. In two of the three methods, the location of OG is marked prior to PCR amplification allowing single-nucleotide resolution of the OG location (Methods A and B). In the third method, fragments of genomic DNA containing OG are purified away from the other fragments and then sequenced (Method C). The three methods are compared and contrasted to inform researchers for selecting the method that best suits the goals of their experiments. Other research groups have sequenced OG from sheared genomic DNA via enrichment with an OG-binding antibody with a resolution ranging from ~102–106 bps (Ohno et al., 2006; Pastukh et al., 2015; Yoshihara et al., 2014); the antibody-based methods will not be described in these protocols.
2. METHOD A: SEQUENCING OG BY CONVERSION TO A DELETION SIGNATURE
2.1 THEORY
Sequencing OG by conversion to a deletion signature harnesses enzymes of the base excision repair pathway to delete OG from the DNA strand leading to a deletion signature at the OG site by Sanger sequencing (Riedl et al., 2015b). The DNA glycosylase formamidopyrimidine DNA glycosylase (Fpg) removes OG from duplex DNA by its N-glycosylase and AP-lyase activities (Fig. 1 Step I) (David and Williams, 1998). The action of Fpg yields a gapped site with 5′ and 3′ phosphates. Next, endonuclease IV (endo IV) catalyzes removal of the 5′ phosphate to furnish a ligatable gap (Fig. 1 Step II) (David and Williams, 1998). The processed gapped site is a substrate for T4 DNA ligase that furnishes a ligated product strand one nucleotide shorter resulting from removal of the OG (Fig. 1 Step III). After exponential PCR amplification of the duplex (Fig. 1 Step IV), the amplicons are subjected to Sanger sequencing to produce a chromatogram that has two peaks out of register by 1 nucleotide starting at the OG site (Fig. 1 Step V) (Riedl et al., 2015b). The two peaks are observed in the chromatogram because the OG-containing strand is one nucleotide shorter after processing than the complementary strand, and both are amplified during PCR prior to Sanger sequencing. This approach is capable of sequencing one OG or two OGs in the same strand separated by at least five nucleotides (Riedl et al., 2015b), as this is the closest Fpg can recognize and operate efficiently on two substrates (Cunniffe et al., 2007). It is conceivable that more than two OGs could be sequenced via this method, although this concept has yet to be verified.
Fig 1.

Outline for sequencing OG via generation of a characteristic deletion signature in a Sanger sequencing chromatogram.
2.2 EQUIPMENT
PCR thermal cycler
Sanger sequencing facility
2.3 MATERIALS
Fpg (8,000 units/mL; New England Biolabs)
Endo IV (10,000 units/mL; New England Biolabs)
T4 DNA ligase (400,000 units/mL; New England Biolabs)
Reaction buffer (25 mM HEPES pH 7.5, 10 mM MgCl2, 5 mM KCl, 1 mM DTT, and 1 mM EDTA)
ATP (2 mM stock in ddH2O; New England Biolabs)
Phusion® High-fidelity DNA polymerase (2,000 units/mL; New England Biolabs)
dNTP solution mix (10 mM of each dNTP; New England Biolabs)
PCR primers (forward and reverse in 8 μM stock solutions)
ddH2O (autoclaved)
DMSO
Agarose
TAE buffer (1× = 40 mM Tris, 20 mM acetic acid, and 1 mM EDTA)
Zymoclean™ Gel DNA Recovery kit
Ethidium bromide (10 mg/mL)
2.4 NOTES
This procedure can be implemented on linear DNA duplexes or plasmid DNA. The method outlined in this report is for plasmid DNA.
The example provided had OG synthetically incorporated in the pBR322 plasmid by a protocol outlined in the original publication of this method (Riedl et al., 2015b).
The PCR primers are designed to start polymerase extension at least 70 nucleotides away from the OG site in the plasmid. This distance is necessary for the Sanger sequencing chromatogram to provide reliable sequencing reads in the region of interest.
2.5 PROCEDURE
Suspend 5 ng of OG-containing plasmid DNA in 8.4 μL of reaction buffer in a 0.2-mL PCR tube.
Thermally equilibrate the sample at 37 °C in a PCR thermal cycler for 10 min prior to initiation of the reaction.
After thermal equilibration at 37 °C, add 5 units of Fpg (0.6 μL) and 10 units of endo IV (1.0 μL) to achieve a final volume of 10 μL. Allow the reaction to proceed for 30 min to generate a ligatable gap at the OG site (Fig. 1 Steps I and II).
Following the 30 min reaction, change the thermal cycler temperature to 25 °C and allow the sample to thermally equilibrate for 10 min at the new temperature.
Once the sample is thermally equilibrated at 25 °C, add ATP to a concentration of 83 μM (0.5 μL) and 600 units of T4 DNA ligase (1.5 μL) to achieve a final volume of 12 μL. Next, allow the reaction to incubate for 2 h at 25 °C (Fig. 1 Step III).
-
After ligation of the gap, conduct exponential PCR to produce two product duplexes that differ in length by one nucleotide, in which the missing nucleotide in the shorter duplexes was the original site of OG (Fig. 1 Step IV).
-
(F.1)
PCR reaction conditions
2.0-μL processed plasmid reaction mixture
4.0-μL Phusion® GC buffer
0.4-μL dNTP solution mixture
0.6-μL DMSO
2.5-μL forward primer
2.5-μL reverse primer
1.0-μL Phusion® High-Fidelity DNA polymerase
7.0-μL ddH2O
Total volume = 20 μL
-
(F.2)
PCR thermal cycler method
Initiate the PCR with a single denaturing step for 2 min at 95 °C.
-
Next, conduct 20 cycles of PCR with the following settings.
Denaturation = 95 °C for 45 sec
Annealing = 55 °C for 30 sec
Extension = 72 °C for 1.5 min
After 20 cycles of PCR, conduct a final extension for 5 min at 72 °C.
-
(F.1)
-
Once the PCR is complete, the product duplexes are separated from the plasmid template by agarose gel electrophoresis followed by removal of the PCR product strands from the gel with a Zymoclean™ Gel DNA Recovery kit.
-
(G.1)
Agarose gel electrophoresis
Prepare an agarose gel with ethidium bromide (0.5 μg/mL) following your laboratory’s established protocol for the plasmid that is to be sequenced.
After loading the samples into the agarose gel, conduct electrophoresis in 1× TAE buffer using power setting appropriate for the plasmid analyzed.
-
(G.2)
PCR product purification from the agarose gel
Utilize a UV light box to locate and cut the PCR-product band from the gel
Extract the product DNA using a Zymoclean™ Gel DNA Recovery kit following the manufacturer’s protocol. Additional information: Gel DNA extraction can be performed using other kits or established protocols in your laboratory.
-
(G.1)
The purified PCR product is then submitted for standard Sanger sequencing. Because Sanger sequencing is not routinely conducted in most laboratories, refer to your sequencing facility for their instructions on how to prepare and submit the samples to be analyzed.
After Sanger sequencing, the data are visually inspected to locate the OG site(s). In the Sanger sequencing chromatogram, the start of two peaks appearing out of register by one nucleotide reveals the location of the OG (Fig. 1 Step V). If more than one OG nucleotides are in the strand analyzed, refer to the original publication for analysis to identify the position of each OG (Riedl et al., 2015b).
3. METHOD B: SEQUENCING OG BY CONVERSION TO AN UNNATURAL DNA BASE PAIR
3.1 THEORY
This approach for sequencing OG harnesses the base excision repair pathway to convert a duplex with an OG-containing base pair into a PCR-amplifiable unnatural DNA base pair (e.g., dNaM:d5SICS. Fig. 2A) (Riedl et al., 2015a). Post PCR amplification, the amplicons are sequenced to reveal the location of the unnatural base pair, and hence OG. This method utilizes the N-glycosylase and AP-lyase activities of Fpg for removal of the OG nucleoside to yield a gapped site with phosphates on the 5′ and 3′ sides (Fig. 2B Step I) (David and Williams, 1998). Removal of the 5′ phosphate is catalyzed by endo IV to generate a competent gap for introduction of an unnatural nucleotide (Fig. 2B Step II). Next, the gapped site is filled with an unnatural nucleotide by Klenow fragment of DNA polymerase I deficient in exonuclease activity (Kf exo−; Fig. 2B Step III). The unnatural nucleotide introduced at the gap site includes dNaMTP, dMMO2TP, or d5SICSTP that were developed in the Romesberg laboratory (Fig. 2A) (Malyshev et al., 2012; Malyshev et al., 2009; Seo et al., 2011). Polymerase insertion of the unnatural nucleotide into the gap yields a nick on the 3′ side of the incorporated unnatural nucleotide base paired with a canonical nucleotide (i.e., A or C). The nick is then sealed with T4 DNA ligase to furnish an intact duplex (Fig. 2B Step IV). Upon exponential PCR amplification of the duplex labeled with an unnatural nucleotide in the presence of unnatural complementary nucleotides, new amplicons are generated bearing an unnatural base pair (Fig. 2A) at the site of OG (Fig. 2B Step V). The unnatural DNA nucleotides developed by Romesberg and coworkers are PCR amplifiable with high efficiency and retention in the presence of natural dNTPs (Malyshev et al., 2009). Prior to sequencing, the unnatural base pair containing duplex must be purified from the other duplex, which is outlined below. Sequencing for the dNaM:d5SICS or dMMO2:d5SICS base pair is traditionally achieved by Sanger sequencing to locate a strong stop in the sequencing chromatogram (Fig. 2B Step VI). The strong stop is observed because the unnatural base pair that occurs where OG was originally positioned in the DNA cannot be sequenced past by Sanger sequencing that only looks for canonical nucleotides.
Figure 2.

Method outline to replace an OG-containing base pair with a dNaM:d5SICS or dMMO2:d5SICS unnatural DNA base pairs. (A) The dNaM:d5SICS and dMMO2:d5SICS structures. (B) Steps in the method to replace an OG-containing base pair with the dNaM:d5SICS unnatural DNA base pair. aFor clarity only dNaMTP is shown in the figure; however, this step can also be achieved by allowing the polymerase to insert d5SICSTP. bFor the sake of brevity, amplicons resulting from the unnatural nucleotide containing strand are shown and sequenced. In practice, both strands from Step IV will amplify yielding a mixture of two duplexes, one with the unnatural base pair and the other without. A method to purify the unnatural base pair-containing duplex away from the other duplex is presented in a note below.
3.2 EQUIPMENT
PCR thermal cycler
Sanger sequencing facility
3.3 MATERIALS
Fpg (8,000 units/mL; New England Biolabs)
Endo IV (10,000 units/mL; New England Biolabs)
T4 DNA ligase (400,000 units/mL; New England Biolabs)
Reaction buffer (25 mM HEPES pH 7.5, 10 mM MgCl2, 5 mM KCl, and 1 mM EDTA)
ATP (2 mM stock in ddH2O; New England Biolabs)
dNaMTP (500 μM stock in ddH2O)
d5SICSTP (500 μM stock in ddH2O)
dMMO2SSBIOTP (500 μM stock in ddH2O)
One Taq® DNA Polymerase (5,000 units/mL; New England Biolabs)
Klenow fragment deficient in exonucease activity (5,000 units/mL; New England Biolabs)
dNTP solution mix (10 mM of each dNTP; New England Biolabs)
PCR primers (forward and reverse in 8 μM solutions)
ddH2O (autoclaved)
DMSO
DTT (300 mM stock in ddH2O)
TAE buffer (1× = 40 mM Tris, 20 mM acetic acid, and 1 mM EDTA)
Agarose
Zymoclean™ Gel DNA Recovery kit
Ethidium bromide (10 mg/mL)
Streptavidin-coated magnetic beads
3.4 NOTES
This procedure can be implemented on linear DNA duplexes or plasmid DNA. The method outlined in this report is for plasmid DNA.
The example provided had OG synthetically incorporated site specifically in the pBR322 plasmid by a protocol outlined in the original publication of this method (Riedl et al., 2015a).
The PCR primers are designed to start polymerase extension at least 70 nucleotides away from the OG site in the plasmid. This distance is necessary for the Sanger sequencing chromatogram to provide reliable sequencing reads in the region of interest.
The dNaM and d5SICS nucleosides can be purchased from Berry & Associates and need to be converted to the nucleotide triphosphates by literature protocols to utilize this OG-sequencing protocol (Malyshev et al., 2014; Malyshev et al., 2009).
During PCR amplification, each member strand of the duplex will amplify (Fig. 2 Step IV), one has the unnatural DNA nucleotide and the other does not. For Sanger sequencing, the amplicon with the unnatural DNA base pair must be purified away from the amplicon with only canonical base pairs. This can be achieved by adding an unnatural nucleotide triphosphate functionalized with a biotin and a linker containing a disulfide during PCR amplification (e.g., dMMO2SSBIOTP. Fig. 3A) (Seo et al., 2011). The functionalized unnatural nucleotide triphosphate allows affinity purification of the duplex with the unnatural base pair via streptavidin (STP) coated magnetic beads, followed by release from the beads by reduction of the disulfide bond in the linker with DTT (Fig. 3B). At present, the dMMO2SSBIOTP nucleotide required in this step must be synthesized by literature methods (Seo et al., 2011).
Figure 3.

Functionalized unnatural nucleotide utilized for selective purification of duplex DNA with an unnatural base pair at the site OG originally occupied. (A) Structure of dMMO2SSBIOTP. (B) Scheme for affinity purification of duplexes with the dMMO2SSBIO nucleotide. STP = streptavidin BTN = biotin
3.5 PROCEDURE
Suspend 50 ng of OG-containing plasmid in 8.4 μL of reaction buffer in a 0.2-mL PCR tube.
Next, thermally equilibrate the PCR tube in a thermal cycler at 37 °C for 10 min.
After thermal equilibration, add 5 units of Fpg (0.6 μL) and 10 units of endo IV (1.0 μL) and allow the reaction to incubate for 30 min at 37 °C. This enzyme combination will remove the OG nucleotide to furnish a gapped site with a 3′ OH on the 5′ side of the gap and a 5′ phosphate on the 3′ side of the gap (Fig. 2B Steps I and II).
After conversion of OG to a competent gapped site add d5SICSTP to a concentration of 150 μM (3 μL) and 7 units of Kf exo− (1.4 μL). Allow the reaction to progress at 37 °C for 1 h (Fig. 3B Step III). Additional information: This reaction can also be conducted with dNaMTP using identical reaction conditions as reported with the d5SICSTP.
After polymerase insertion of the unnatural nucleotide into the gapped site, the reaction is quenched by heating at 90 °C for 10 min in a preheated water bath.
After quenching Kf exo−, change the thermal cycler temperature to 25 °C, and allow the mixture to thermally equilibrate for 10 min.
Once the sample is thermally equilibrated at 25 °C, add ATP to achieve a concentration of 63 μM (0.5 μL) and 600 units of T4 DNA ligase (1.5 μL) with a final volume of 16.5 μL. Next, allow the reaction to incubate for 16 h during which the nicked site with an unnatural nucleotide replacing OG is sealed (Fig. 2B Step IV).
-
After ligation, exponential PCR is conducted to yield two product duplexes, one of which has an unnatural DNA base pair and the other does not. To allow purification of the unnatural-base pair containing duplex, the PCR reaction is conducted in the presence of d5SICSTP and dMMO2SSBIOTP to yield the d5SICS:dMMO2SSBIO base pair (Figs. 2A and 3).
-
(H.1)
Conditions for the PCR amplification reaction.
2.0-μL processed plasmid reaction mixture
4.0-μL OneTaq® reaction buffer
0.2-μL dNTP solution mixture
4.0-μL d5SICSTP
4.0-μL dMMO2SSBIOTP
2.5-μL forward primer
2.5-μL reverse primer
0.5-μL OneTaq® DNA polymerase
0.3-μL ddH2O (total volume = 20 μL)
-
(H.2)
Settings for the PCR thermal cycler.
Conduct an initial denaturation step for 2 min at 95 °C.
-
Next, conduct 20 cycles of PCR using the following method.
Denaturation = 95 °C for 45 sec
Annealing = 55 °C for 30 sec
Extension = 72 °C for 4 min
After 20 cycles of PCR, conduct a final extension for 5 min at 72 °C.
-
(H.1)
The presence of dMMO2SSBIOTP allows affinity purification of the PCR-generated duplex with an unnatural base pair via STP-coated magnetic beads (Fig. 3). Follow the manufacturer’s protocol for proper use of the STP-coated magnetic beads during the purification step. After affinity purification of the duplexes is completed, release the DNA from the beads by reduction of the disulfide bond in the linker between the biotin and DNA with 30 mM DTT for 30 min at room temperature (Fig. 3).
After completion of the previous step, submit the sample for standard Sanger sequencing. Because Sanger sequencing is not routinely conducted in most laboratories, refer to your sequencing facility for their instructions on how they want the sample prepared.
After Sanger sequencing, the data are visually inspected to reveal the location of the unnatural DNA base pair by observation of an abrupt stop in the sequencing chromatogram (Fig. 2 Step VI). The stop is observed because Sanger sequencing requires an additional PCR step conducted without the unnatural nucleotides. Consequently, sequencing OG by conversion to an unnatural base pair allows detection of one OG per strand by Sanger sequencing.
4. METHOD C: SEQUENCING OG BY OG-SEQ
4.1 THEORY
The OG-Seq protocol was developed and implemented on mouse genomic DNA after enriching OG-containing fragments by OG-selective biotinylation (Fig. 4A) and affinity purification (Ding et al., 2017). The enriched fragments were then sequenced on a next generation sequencing platform (NGS, Figs. 4B and 4C) (Ding et al., 2017). The fragment size will determine the resolution, which OG is sequenced in the genome; in our case, the fragments were ~150 bps. Selective biotinylation of OG is possible because the reduction potential of OG is sufficiently low such that only OG is oxidized with the mild one-electron oxidant K2IrBr6 (Ding et al., 2017). Upon two-electron oxidation of OG, a reactive electrophilic intermediate is formed that reacts with a nucleophilic primary amine analog of biotin (BTN-NH2) to yield a stable covalent adduct (Fig. 4A) (Hosford et al., 2004; Xue and Greenberg, 2007). The duplex fragments containing the biotin-adducted OG are then enriched and purified by STP-coated magnetic beads. In this version of OG-Seq, the complementary strands to the biotinylated OG are released from the beads by NaOH denaturation and submitted for Illumina NGS. The NGS data are analyzed by aligning the reads to a reference genome to identify mapped reads that form regions of enrichment when compared to an input control experiment without OG enrichment (Fig. 4C). Further analysis of the sequencing reads can be tailored using any bioinformatic pipeline that is suitable for the analysis of these NGS data types.
Figure 4.
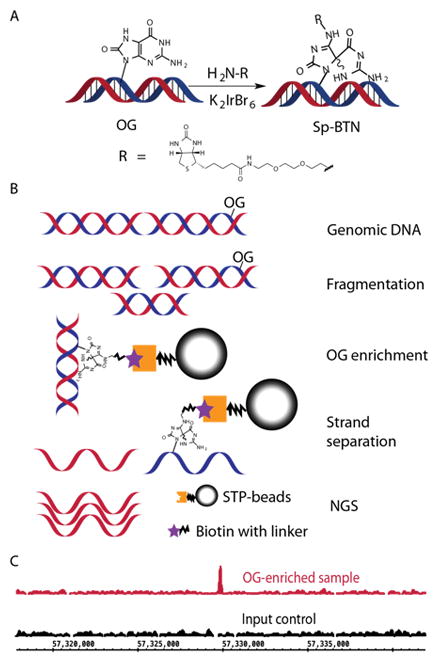
Method overview for implementation of OG-Seq. (A) Mechanism for selectively labeling OG with biotin (BTN). (B) Sequence of steps to prepare ~150-mer DNA strands enriched in OG for submission to NGS. (C) Example data showing a region in the mouse genome where OG was found fivefold enriched.
4.2 EQUIPMENT
DynaMag™- Spin Magnet (ThermoFisher)
S2 Focused-ultrasonicator (Covaris)
Qubit Fluorimeter (ThermoFisher)
NanoDrop 1000 (ThermoFisher)
Rotisserie mixer
NGS facility
4.3 MATERIALS
Genomic DNA (~30 μg)
ddH2O (autoclaved)
Desferal (100 mM solution in ddH2O, Sigma Aldrich)
Butylated hydroxytoluene (BHT, 100 mM solution in DMSO, Sigma Aldrich)
Centricon-10 Concentrators (Sigma Aldrich)
Micro Bio Spin P-6 gel columns (Bio-Rad)
Qubit ssDNA assay kit (ThermoFisher)
ssDNA/RNA clean & concentrator kit (Zymo Research)
Dneasy blood and tissue kit (Qiagen)
PCR clean-up kit (Mo Bio)
Amine-PEG2-Biotin (BTN-NH2, 500 mM solution in ddH2O, ThermoFisher)
K2IrBr6 (100 mM solution in ddH2O, Alfa Aesar)
NaPi buffer (100 mM solution at pH 8.0)
Dynabeads MyOne Streptavidin C1 (Invitrogen)
B&W buffer A (2x, 10 mM Tris pH 7.5, 1 mM EDTA, 2 M NaCl, 0.1% Tween-20)
B&W buffer B (2x, 10 mM Tris pH 7.5, 1 mM EDTA, 2 M NaCl)
Buffer 1 (150 mM NaCl, 0.01 M Tris pH7.0)
TE buffer (10 mM Tris pH 8.0, 1 mM EDTA)
NaOH (150 mM)
Tris buffer (10 mM solution at pH 8.0)
Accel-NGS® 1S Plus DNA Library Kit (Swift Biosciences).
4.4 NOTES
OG-Seq was developed to sequence OG in genomic DNA. In the method outlined, the genomic DNA was derived from mouse embryonic fibroblast cells. The details of how these cells were grown can be found in the original publication (Ding et al., 2017); however, this method can be applied to any genomic DNA sample of interest, as long as you have enough DNA for implementation of this method (~30 μg for samples containing ~1 OG per 106 Gs).
The OG-enriched strands obtained by the method below were sequenced on an Illumina Hi-Seq platform, although any NGS platform would be suitable for data collection.
Bioinformatic analysis requires an input control that is comprised of genomic DNA that was sheared to the desired length and sequenced but not treated with the enrichment steps.
4.5 PROCEDURE
If mammalian cells are the source of OG for application of OG-Seq, obtain ~107 cells (~30 μg of genomic DNA) grown under conditions of interest to your research. Pellet the cells by centrifugation via standard laboratory methods. Additional information: If studies are conducted on other cells harvest enough cells to obtain ~30 μg of genomic DNA if OG is present at ~1 OG per 106 Gs.
Utilize a genomic DNA extraction kit, such as a DNeasy blood and tissue kit, to obtain pure genomic DNA following the manufacturer’s protocol. Before commencing the extraction, add BHT and desferal to the solutions of the DNA extraction kit to a final concentration of 100 μM for each compound. Additional information: The desferal binds iron ions and BHT is a reductant to prevent additional OG formation in the DNA during genomic DNA harvesting (Taghizadeh et al., 2008).
Depending on the genomic DNA extraction kit used, the volume obtained will vary. Measure the genomic DNA concentration by either NanoDrop or Quibit™ analysis following the protocols for the instrument selected.
The genomic DNA is then sheared by sonication to fragments of ~150 bps. In our studies, a Covaris S2 ultrasonicator was used that requires sample sizes of 5 μg per 130 μL of Tris buffer with 100 μM of BHT and desferal present. Use the concentration obtained in the previous step to adjust the concentrations accordingly prior to sonication. Sonicate the samples with the settings set to the following values, duty cycle = 10%; intensity = 5; cycles/burst = 200; and time = 430 s. Additional information: If using a different ultrasonicator or shearing method, consult the manufacturer’s protocol to obtain sheared DNA fragments of the desired length. In our published studies, ~150 bp lengths were the shortest that could be reliably obtained. If a different shearing method is selected, the resolution of the OG-Seq will be the average length of the sheared DNA.
After sonication, exchange the residual Tris buffer from the previous steps with NaPi buffer (100 mM pH 8.0) using a Micro Bio Spin P-6 gel column following the manufacturer’s protocol. This step will also concentrate the sample to a volume of 20 μL. Additional information: It is important to exchange the Tris buffer with NaPi buffer prior to the next step. During oxidation of OG an electrophile is formed that should react with the BTN-NH2 nucleophile; however, Tris is a primary amine, and if it is present, it will compete with BTN-NH2 for reacting with OG that would cause a loss in the enrichment of OG-containing fragments upon STP affinity purification.
Take the concentrated sample and add 100 μL of 100 mM NaPi (pH 8.0) buffer with BTN-NH2 (5 μL or 20 mM) followed by heat the sample to 75 °C for 10 min. After thermal equilibration, add K2IrBr6 (6.3 μL or 5 mM) and allow the react to progress for 1 h.
After the biotinylation of OG, the excess BTN-NH2 and K2IrBr6 are removed with a PCR clean-up kit following the manufacturer’s protocol. Adjust the volume to 125 μL with Tris buffer.
-
Use STP-coated magnetic beads (Dynabeads) to extract the DNA fragments biotinylated at OG. The steps for extraction are as follows.
-
(H.1)
Take 100 μL of the Dynabeads for each sample to be analyzed and wash them with 1 mL of 1× B&W buffer A four times. Between each wash step, use the DynaMag™-Spin magnet to retain the Dynabeads.
-
(H.2)
Using the retained Dynabeads from the previous step, wash them with 1 mL of 1× B&W buffer B using a DynaMag™-Spin magnet to retain the Dynabeads after the wash.
-
(H.3)
Using the retained Dynabeads from the previous step, wash them with 1 mL of buffer 1 using a DynaMag™-Spin magnet to retain the Dynabeads after the wash.
-
(H.4)
Resuspend the washed Dynabeads in 125 μL of B&W buffer B.
-
(H.5)
Add the 125 μL of OG-biotinylated sample from Step G to the washed beads from Step H.4 and allow them to incubate overnight at 4 °C on a rotisserie shaker.
-
(H.6)
After the overnight incubation, the Dynabeads are washed twice with 1 mL of 1× B&W buffer B using a DynaMag™-Spin magnet to retain the Dynabeads after the wash.
-
(H.7)
The sample is then washed a final time with 50 μL of TE buffer.
-
(H.8)
To release the complementary strands to the bound OG-biotinylated strands, the beads are incubated in 150 mM NaOH at 20 °C for 30 min.
-
(H.9)
Separate the supernatant with the complementary strands from the beads.
-
(H.10)
Use a single-strand DNA/RNA concentrator kit to elute the supernatant containing the complementary strands into 10 μL of ddH2O following the manufacturer’s protocol.
-
(H.11)
Determine the sample concentration by Qubit analysis prior to submission of the sample to NGS to ensure that you have enough material for sequencing. The amount of DNA required is dependent on the NGS platform and you should consult your sequencing facility to determine the amounts required.
-
(H.1)
Most laboratories do not possess an NGS instrument; therefore, refer to your NGS facility for further sample preparation prior to sequencing. If using an Illumina platform, the NGS library is constructed with an Accel-NGS® 1S Plus DNA Library Kit.
The data are initially analyzed by aligning the reads to a reference genome using NovoAlgin. The enriched peaks from the alignment are called from the mapped reads by MACS 2.0 using the input sample as a control (Zhang et al., 2008). Additional information: The bioinformatic analysis of the data can be further tailored using other pipelines that fit your experimental needs.
5. COMPARISON AND CONTRAST OF THE THREE OG SEQUENCING METHODS
5.1 SEQUENCING PLATFORMS
The three OG sequencing methods outlined (A, B, and C) can be applied using different sequencing platforms. In Method A, OG is sequenced by deletion that is best observed in a Sanger sequencer because this platform will generate the two peaks out of register required for OG determining the location of OG (Fig. 1 Step V and Table 1). Utilization of Method A on an NGS platform would be problematic because deletion signatures are common sequencing errors that would require very high sequencing depth to reliably identify if they were real (Goodwin et al., 2016). At present, Method B that sequences OG by conversion to an unnatural DNA base pair (e.g., d5SICS:dNaM; Fig. 2A) would be conducted on a Sanger sequencer (Fig. 2B Step VI and Table 1); however, d5SCIS and dNaM were recently sequenced using the MspA protein nanopore (Craig et al., 2015). This opens the possibility of sequencing these unnatural nucleotides by Oxford Nanopore’s MinION high-throughput NGS platform that uses a protein nanopore (Jain et al., 2015); however, this has yet to be achieved. Lastly, sequencing OG by OG-Seq (i.e., Method C) utilizes affinity purification for enrichment of randomized OG-containing, genomic-DNA fragments by selective biotinylation of the OG. This approach yields a large mixture of sequences to be analyzed that is best suited for high-throughput NGS platforms (Fig. 4B and Table 1).
Table 1.
Comparisons of three OG sequencing methods.
| METHOD A Sequencing OG by formation of a deletion signature |
METHOD B Sequencing OG by conversion to an unnatural base pair |
METHOD C Sequencing OG by OG-Seq |
|
|---|---|---|---|
| Suitable Sequencing Platforms | Sanger | Sanger & NGSa | NGS |
| DNA Contexts | Targeted sequencing of plasmids or genomic loci | Targeted sequencing of plasmids or genomic loci & the genomic scalea | Genomic scale |
| Resolution of sequenced OG | Single nucleotide | Single nucleotide | ~150 bps |
| Ability to detect more than one OG per strand | Yes | No & Yesa | No |
| Custom chemical synthesis required | No | Yes | No |
The ability to sequence OG by conversion to an unnatural DNA base pair on an NGS platform, the genomic scale, and more than one OG per strand analyzed is achievable as soon as the d5SICS, dNaM, and dMMO2 unnatural DNA bases can be sequenced on a commercial NGS platform. The concept of sequencing d5SICS and dNaM with the MspA protein nanopore has been demonstrated (Craig et al., 2015).
5.2 DNA CONTEXTS FOR SEQUENCING OG
The three sequencing methods outlined can be applied to different DNA sources. Method A requires PCR workup followed by Sanger sequencing, and therefore, this approach would best be applied to targeted sequences of established regions (Riedl et al., 2015b). Method A would best confirm synthesis of plasmids with a site-specific OG or to interrogate single, specific loci in a genome (Table 1). On the same hand, Method B that finds OG by conversion to an unnatural DNA base pair followed by PCR amplification (Riedl et al., 2015a), at present, would be applied to confirm synthesis of plasmids with a site-specific OG or for interrogation of single, specific loci in a genome. However, when sequencing d5SCIS or dNaM by commercial nanopore NGS technology is realized, OG-sequencing by this approach could be applied on the genomic scale (Table 1). Method C was developed and is applicable for sequencing OG on the genomic scale (Table 1) (Ding et al., 2017).
5.3 RESOLUTION FOR SEQUENCING OG
Methods A and B achieve OG sequencing at the single-nucleotide resolution (Table 1) (Riedl et al., 2015a; Riedl et al., 2015b). In contrast, Method C (i.e., OG-Seq) has a resolving power equal to the shearing size and the published version of this method had a resolution of ~150 bps (Table 1) (Ding et al., 2017). We anticipate new iterations of OG-Seq to emerge in the future with the power to sequence OG at singe-nucleotide resolution.
5.4 ABILITY TO SEQUENCE MORE THAN ONE OG PER STRAND
Method A detects OG by conversion to a deletion signature that is initiated with the DNA glycosylase Fpg and completed by the next steps of the base excision repair process; therefore, more than one OG can be detected per strand, as long as the OGs are spaced >5 nucleotides apart for Fpg to find and operate on both substrates (Table 1). On the other hand, Method B, at present, can only be used to detect a single OG per strand analyzed (Table 1); however, this limitation will be lifted once NGS sequencing for unnatural DNA nucleotides are achieve on a commercial instrument such as the Pacific Biosciences or Oxford Nanopore platforms. In the present version of Method C (i.e., OG-Seq) only one OG per 150-mer strand can be sequenced (Table 1).
5.5 CUSTOM CHEMICAL SYNTHESIS REQUIREMENTS
All materials needed to implement Methods A or C can be obtained from commercial suppliers. Also, the sequencing facilities required to implement Methods A or C for OG sequencing can be found at most research institutions (Table 1). In contrast, Method B requires custom synthesis of the unnatural dNTPs d5SICSTP, dNaMTP, and/or dMMO2SSBIOTP, while the sequencing facility to conduct this method can be found at most research institutions (Table 1).
Acknowledgments
Financial support for development of these methods was obtained from the National Institutes of Health (R01 GM093099), the National Cancer Institute (R01 CA090689), and a seed grant from the Nuclear Control Program at Huntsman Cancer Institute at the University of Utah (P30 CA042014). We thank the DNA/Peptide core facility at the University of Utah for synthesizing the oligomers and the Genomics core facility at the University of Utah for conducting the sequencing experiments; both are supported in part by a National Cancer Institute Cancer Center Support Grant (P30 CA042014). We are grateful to Dr. Jan Riedl for his hard work in developing Methods A and B.
References
- Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- Allgayer J, Kitsera N, Bartelt S, Epe B, Khobta A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016;44:7267–7280. doi: 10.1093/nar/gkw473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J, Wagner JR, Shafirovich V, Geacintov NE. One-electron oxidation reactions of purine and pyrimidine bases in cellular DNA. Int J Radiat Biol. 2014;90:423–432. doi: 10.3109/09553002.2013.877176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Spittle KE, Turner SW, Korlach J. Direct detection and sequencing of damaged DNA bases. Genome Integr. 2011;2:2–10. doi: 10.1186/2041-9414-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig JM, Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Nova IC, Doering K, Tickman BI, Svet MT, Gundlach JH. Direct detection of unnatural DNA nucleotides dNaM and d5SICS using the MspA nanopore. PLoS ONE. 2015;10:e0143253. doi: 10.1371/journal.pone.0143253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunniffe SMT, Lomax ME, O’Neill P. An AP site can protect against the mutagenic potential of 8-oxoG when present within a tandem clustered site in E. coli. DNA Repair. 2007;6:1839–1849. doi: 10.1016/j.dnarep.2007.07.003. [DOI] [PubMed] [Google Scholar]
- David SS, Williams SD. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- Delaney S, Jarem DA, Volle CB, Yennie CJ. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radical Res. 2012;46:420–441. doi: 10.3109/10715762.2011.653968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Fleming AM, Burrows CJ. Sequencing the mouse genome for the oxidatively modified base 8-oxo-7,8-dihydroguanine by OG-Seq. J Am Chem Soc. 2017;139 doi: 10.1021/jacs.1026b12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming AM, Burrows CJ. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.1011.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming AM, Ding Y, Burrows CJ. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1619809114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedik CM, Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer-Morrison MK, Kotler JM, Martin BD, Sugden KD. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry. 2003;42:9761–9770. doi: 10.1021/bi034546k. [DOI] [PubMed] [Google Scholar]
- Hosford ME, Muller JG, Burrows CJ. Spermine participates in oxidative damage of guanosine and 8-oxoguanosine leading to deoxyribosylurea formation. J Am Chem Soc. 2004;126:9540–9541. doi: 10.1021/ja047981q. [DOI] [PubMed] [Google Scholar]
- Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Methods. 2015;12:351–356. doi: 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Correa IR, Jr, Romesberg FE. A semi-synthetic organism with an expanded genetic alphabet. Nature. 2014;509:385–388. doi: 10.1038/nature13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshev DA, Dhami K, Quach HT, Lavergne T, Ordoukhanian P, Torkamani A, Romesberg FE. Efficient and sequence-independent replication of DNA containing a third base pair establishes a functional six-letter genetic alphabet. Proc Natl Acad Sci U S A. 2012;109:12005–12010. doi: 10.1073/pnas.1205176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshev DA, Seo YJ, Ordoukhanian P, Romesberg FE. PCR with an expanded genetic alphabet. J Am Chem Soc. 2009;131:14620–14621. doi: 10.1021/ja906186f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SP, Toomire KJ, Strauss PR. DNA modifications repaired by base excision repair are epigenetic. DNA Repair (Amst) 2013;12:1152–1158. doi: 10.1016/j.dnarep.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Ohno M, Miura T, Furuichi M, Tominaga Y, Tsuchimoto D, Sakumi K, Nakabeppu Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006;16:567–575. doi: 10.1101/gr.4769606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK, Hegde ML, Radak Z, Bacsi A, Brasier AR, et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase1-mediated epigenetic regulation of nuclear factor kappaB-driven gene expression. J Biol Chem. 2016;291:25553–25566. doi: 10.1074/jbc.M116.751453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastukh V, Roberts JT, Clark DW, Bardwell GC, Patel M, Al-Mehdi AB, Borchert GM, Gillespie MN. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1367–1375. doi: 10.1152/ajplung.00236.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon O, Sauvaigo S, Gasparutto D, Faure P, Favier A, Cadet J. Effects of 8-oxo-7,8-dihydro-2′-deoxyguanosine on the binding of the transcription factor Sp1 to its cognate target DNA sequence (GC box) Free Radical Res. 1999;31:217–229. doi: 10.1080/10715769900300781. [DOI] [PubMed] [Google Scholar]
- Riedl J, Ding Y, Fleming AM, Burrows CJ. Identification of DNA lesions using a third base pair for amplification and nanopore sequencing. Nat Commun. 2015a;6:8807. doi: 10.1038/ncomms9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Fleming AM, Burrows CJ. Sequencing of DNA lesions facilitated by site-specific excision via base excision repair DNA glycosylases yielding ligatable gaps. J Am Chem Soc. 2015b;138:491–494. doi: 10.1021/jacs.5b11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibel AEP, An N, Jin Q, Fleming AM, Burrows CJ, White HS. Nanopore detection of 8-oxo-7,8-dihydro-2′-deoxyguanosine in immobilized single-stranded DNA via adduct formation to the DNA damage site. J Am Chem Soc. 2010;132:17992–17995. doi: 10.1021/ja109501x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo YJ, Malyshev DA, Lavergne T, Ordoukhanian P, Romesberg FE. Site-specific labeling of DNA and RNA using an efficiently replicated and transcribed class of unnatural base pairs. J Am Chem Soc. 2011;133:19878–19888. doi: 10.1021/ja207907d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- Taghizadeh K, McFaline JL, Pang B, Sullivan M, Dong M, Plummer E, Dedon PC. Quantification of DNA damage products resulting from deamination, oxidation and reaction with products of lipid peroxidation by liquid chromatography isotope dilution tandem mass spectrometry. Nat Protocols. 2008;3:1287–1298. doi: 10.1038/nprot.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 2004;3:483–494. doi: 10.1016/j.dnarep.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Wood ML, Esteve A, Morningstar ML, Kuziemko GM, Essigmann JM. Genetic effects of oxidative DNA damage: comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucleic Acids Res. 1992;20:6023–6032. doi: 10.1093/nar/20.22.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Greenberg MM. Facile quantification of lesions derived from 2′-deoxyguanosine in DNA. J Am Chem Soc. 2007;129:7010–7011. doi: 10.1021/ja072174n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Jiang L, Akatsuka S, Suyama M, Toyokuni S. Genome-wide profiling of 8-oxoguanine reveals its association with spatial positioning in nucleus. DNA Res. 2014;21:603–612. doi: 10.1093/dnares/dsu023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]