

Abstract
More than 100 different mutations in the RPE65 gene are associated with inherited retinal degeneration. Although some missense mutations have been shown to abolish isomerase activity of RPE65, the molecular mechanisms leading to loss of function and retinal degeneration remain incompletely understood. Here we show that several missense mutations resulted in significant decrease in expression level of RPE65 in human retinal pigment epithelium (RPE) cells. The 26S proteasome non-ATPase regulatory subunit 13 (PSMD13), a newly identified negative regulator of RPE65, mediated degradation of mutant RPE65s, which were misfolded and formed aggregates in the cells. Many mutations, including L22P, T101I, and L408P, were mapped on non-active sites. Enzyme activities of these mutant RPE65s were significantly rescued at low temperature, whereas mutant RPE65s with a distinct active site mutation could not be rescued under the same conditions. 4-phenylbutyrate (PBA) and glycerol displayed a significant synergistic effect on the low temperature-mediated rescue of the mutant RPE65s. Our results suggest that a low temperature eye mask and PBA, a FDA-approved oral medicine, may provide a promising “protein repair therapy” that can enhance the efficacy of gene therapy.
1. Introduction
RPE65 is a key retinoid isomerase (Jin et al., 2005) in the visual cycle necessary for regenerating 11-cis retinal (11cRAL) chromophore, which functions as a molecular switch for activating opsins in response to light stimulation. The significance of RPE65 in retinal health is reflected by the effect of its mutations, over 100 of which are associated with retinal degenerative diseases (Human Gene Mutation Database). Among these mutations, more than 70 are missense mutations. Although most of these mutations have not been studied for their pathogenicity, some mutations have been shown to severely eliminate isomerase activity of RPE65 (Jin et al., 2005; Redmond et al., 2005; Takahashi et al., 2006; Philp et al., 2009). The activities of mutant RPE65s measured in the laboratory were related to whether or not they were disease-causing in the patients (Redmond et al., 2005; Philp et al., 2009). Several missense mutations resulted in rapid degradation of RPE65 in HEK cell lines (Chen et al., 2006; Takahashi et al., 2006; Philp et al., 2009). The molecular basis for the rapid degradation is unknown.
Recent gene therapy trials showed improvement in vision in some patients with RPE65 mutations (Cideciyan et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008; Jacobson et al., 2012). However, a subsequent study showed that gene therapy could not stop the progressive retinal degeneration (Cideciyan et al., 2013). In general, gene therapy can confer isomerase activity to RPE of patients by expressing wild-type RPE65, but it cannot stop the degenerative component of the disease process. Recently, a dominant mutation in the RPE65 gene has been found in patients with retinitis pigmentosa (Bowne et al., 2011), suggesting that this mutated allele has a dominant pathogenic effect. Misfolding, mislocalization, and aggregation of mutant RPE65 (Chen et al., 2006; Takahashi et al., 2006; Li et al., 2014) may cause cytotoxic effects. To enhance the gene therapy effect, it is important to develop a strategy that can rescue the enzyme activity but also reduce cytotoxic effects of mutant RPE65s. In this study, we investigated the common properties of several disease-causing RPE65s with regard to their pathogenic mechanism and rescue of their function.
2. Materials and Methods
2.1 Immunohistochemistry and Immunoblot analysis
All animal experiments were approved by the Institutional Animal Care and Use Committee for the Louisiana State University Health Sciences Center and performed according to guidelines established by the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Retinal and cellular immunostaining as well as immunoblot analysis were performed as described previously (Sato et al., 2013; Li et al., 2014).
2.2 Cell Culture, Transfection and Knockdown of PSMD13
Primary human RPE (Hu and Bok, 2001), ARPE-19 (Dunn et al., 1996), and 293T-LC (Jin et al., 2005) cells were maintained as described previously. PolyJet (SignaGen) or Lipofectamine 2000 (Invitrogen) was used for transfection. To reduce the expression level of endogenous RPE65 in the human RPE cells, transfected RPE was maintained in plastic culture plates instead of Millicell-HA chambers (Hu and Bok, 2001). Knockdown of the 26S proteasome non-ATPase regulatory subunit 13 (PSMD13) in ARPE-19 cells was performed by transfecting PSMD13 siRNA (OriGene Technologies, Inc.).
2.3 Retinoid Isomerase Assay
The 293T-LC cells transfected with wild-type (WT) or mutant RPE65 constructs (Philp et al., 2009; Li et al., 2014) were incubated with 5 μM all-trans-retinol (atROL) for 16 h at 30 or 37°C. Retinoids extracted from the cells were saponified (Jin et al., 2005) and analyzed by HPLC (Jin et al., 2009).
3. Results
3.1 PSMD13 Promoted Degradation of Disease-Associated RPE65 Proteins
To analyze the impact of disease-causing mutations on expression of RPE65 in RPE, we transfected WT and mutant RPE65 constructs into primary human RPE cells. As shown in Fig. 1a, expression levels of all tested mutant RPE65s were significantly lower than that of WT RPE65. Coexpression of PSMD13 exacerbated the decrease in expression levels of mutant RPE65s with an L22P, T101I, or L408P mutation, whereas knockdown of PSMD13 increased expression levels of these mutant RPE65s (Fig. 1b). Immunohistochemistry revealed that PSMD13 expresses in mouse RPE (Fig. 1c).
Fig. 1.
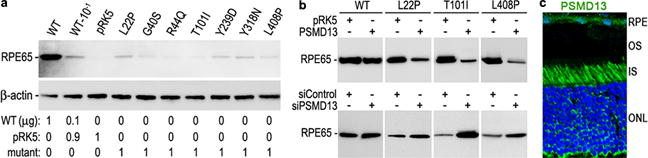
PSMD13 mediates degradation of mutant RPE65s. a. Immunoblot analysis of human RPE cells transfected with the indicated amount of pRK5 mock vector and constructs for WT or disease-causing RPE65. Beta actin was used as a loading control. b. Immunoblot analysis of WT and mutant RPE65s in ARPE-19 cells co-transfected with the indicated vector or siRNA. c. Mouse retinal immunohistochemistry for PSMD13. Nuclei were stained with DAPI.
3.2 Rescue of Enzyme Activity of Disease-causing RPE65s with Nonactive Site Mutations
By mapping disease-causing mutation sites onto the crystal structure of the bovine RPE65 (Kiser et al., 2009), we found that many mutations are non-active site mutations (Fig. 2). This observation prompted us to test whether low temperature can rescue enzyme activity of mutant RPE65s. As shown in Table 1, isomerase activities of three mutant RPE65s with non-active site mutations (L22P, T101I, and L408P) are significantly increased at 30°C, whereas mutant RPE65s with active site (H180R, H313R) or near active site (Y239D, C330Y, E417Q) mutations could not be rescued under the same conditions.
Fig. 2.
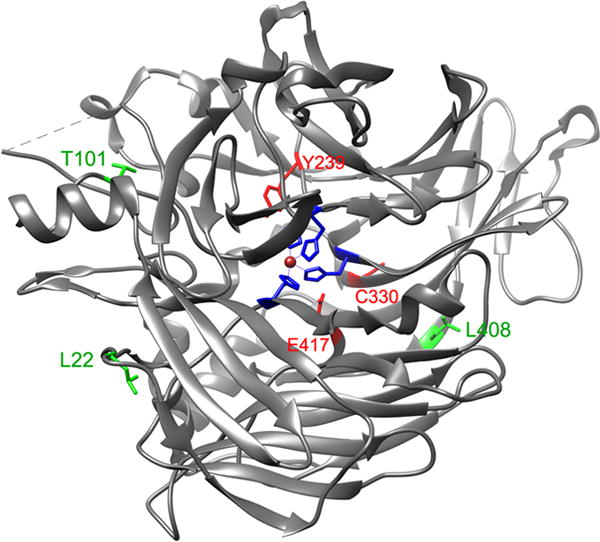
Mapping of disease-causing mutation sites on the crystal structure of bovine RPE65. The catalytic site containing Fe2+ (brown sphere) is in the center of RPE65. The three mutation sites (L22, T101, & L408) shown in green are mapped in the non-active sites, whereas the other three mutation sites (Y239, C330, & E417) shown in red are close to the active site cavity. The four iron-binding histidines (H180, H241, H313, & H527) are shown in blue.
Table 1. Synthesis of 11cROL (pmol/mg protein).
Retinoid isomerase activities of WT and the indicated mutant RPE65s were determined by measuring synthesis of 11-cis retinol (11cROL) at 30°C or 37°C. Numbers indicate 11cROL content (pmol ± SD, n = 3) in 1 mg of cellular protein (middle columns) or ratio of the isomerase activities at 30°C to those at 37°C (right column). NA, no activity.
| Mutation | Activity at 30°C | Activity at 37°C | Ration 30/37°C |
|---|---|---|---|
| L22P | 22 ± 3 | 4 ± 1 | 5.5 |
| T101I | 12 ± 2 | 2 ± 1 | 6.0 |
| L408P | 26 ± 3 | 5 ± 1 | 5.2 |
| H180R | NA | NA | – |
| H313R | NA | NA | – |
| Y239D | 1.3 ± 0.3 | 1.3 ± 0.5 | 1.0 |
| C330Y | 1.8 ± 0.3 | 1.6 ± 0.3 | 1.1 |
| E417Q | 1.2 ± 0.4 | 1.1 ± 0.4 | 1.1 |
| WT | 124 ± 10 | 138 ± 10 | 0.9 |
3.3 Low Temperature Inhibited Aggregate Formation of Mutant RPE65s
Based on the above results, we hypothesized that misfolding is the main molecular basis for loss of enzymatic function of nonactive site mutant RPE65s. To test this possibility, we performed immunocytochemistry. As shown in Fig. 3, the non-active site mutant RPE65s formed numerous aggregates in ARPE-19 cells incubated at 37 °C. These aggregates were significantly reduced in ARPE-19 cells grown at 30°C (Fig. 3).
Fig. 3.
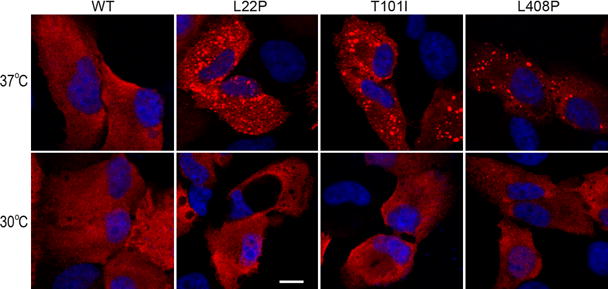
Low temperature reduced aggregation of mutant RPE65s. ARPE-19 cells expressing WT or the indicated mutant RPE65 were incubated at 37°C or 30°C, stained with RPE65 antibody, and observed using a confocal microscope. Scale bar denotes 10 μm.
3.4 PBA Enhanced Low Temperature Rescue of the Nonactive Site Mutant RPE65s
4-phenylbutyrate (PBA) has been shown to help proper folding of other mutant proteins (Bonapace et al., 2004; Li et al., 2013a). We therefore tested whether PBA and low-temperature display synergistic effects on rescue of mutant RPE65s. As shown in Fig. 4, activity of L22P RPE65 was increased approximately 10-fold at 30°C in the presence of PBA compared to its activity at 37°C.
Fig. 4.
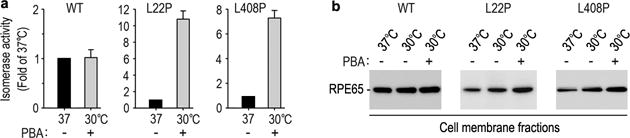
PBA enhanced low temperature-mediated rescue of mutant RPE65s. Relative retinoid isomerase activities of the indicated mutant RPE65s at 30°C in the presence of PBA are shown as fold of their activities at 37°C. Error bars show SD (n = 3).
4. Discussion
The role of PSMD13 in vision and retinal health remains poorly understood. In our previous study, we identified PSMD13 as a negative regulator of RPE65 and found that PSMD13 inhibits RPE65 function (Li et al., 2013b). This might be due to the slight promotion of RPE65 degradation by PSMD13 co-expression (Fig. 1b). Its abundant expression in RPE (Fig. 1c) suggests that PSMD13 could regulate RPE65 function by regulating degradation of RPE65. Importantly, PSMD13 strongly promoted degradation of disease-causing RPE65s. Knockdown of PSMD13 significantly increased expression levels of mutant RPE65s (Fig. 1b), indicating that PSMD13 mediates degradation of mutant RPE65s in the proteasome. The results also suggest that PSMD13 may play a critical role in regulation of pathogenicity of mutant RPE65s.
Low temperature has been shown to restore function to mutated proteins and reduce cellular damage by promoting proper folding of many mutated proteins (Denning et al., 1992; Li et al., 2013a). In this study, we observed that low temperature reduced aggregation of mutant RPE65s in the cells (Fig. 3). Moreover, it significantly rescued isomerase activity of disease-causing RPE65s with different mutations in the nonactive sites (Fig. 2 and Table 1). Under the same experimental conditions, RPE65s with mutations in the active or near the active sites could not be rescued (Fig. 2 and Table 1). Although the biochemical attributes of amino acid residue mutated are important in determining the enzyme activity of a mutant RPE65 (Nikolaeva et al., 2010), our results also suggest that the relative spatial distance between a mutation site and the catalytic site is a critical factor in determining whether the mutant RPE65 can be rescued. Importantly, many disease-causing missense mutations are non-active site mutations. Further studies are needed to test whether these mutations can also be rescued at low temperature.
PBA, a FDA-approved safe oral medication, has also been shown to reverse cellular mislocalization and rescue function of many mutated proteins (Rubenstein and Zeitlin, 1998; Bonapace et al., 2004; Li et al., 2013a). We observed that PBA and low temperature exhibited a significant synergistic effect on rescue of the non-active site mutant RPE65s (Fig. 4). Since low temperature can inhibit aggregate formation (Fig. 3), our results suggest that low temperature and PBA not only can restore enzymatic function to nonactive mutant RPE65s but also can reduce the cytotoxic effect of misfolded RPE65s. Continuing retinal degeneration in patients who received gene therapy (Cideciyan et al., 2013) suggests that a combinatorial therapy is needed to improve vision and to prevent or delay retinal degeneration. A low temperature eye mask and PBA functioning as a “protein repair therapy” may be a promising option for combinatorial therapy.
Acknowledgments
This work was supported by NIH grants (EY021208 to M. J., EY017280 to S. G. J., and GM103340 to LSU Neuroscience COBRE facility), a Macula Vision Research Foundation grant (to D. B. and to S.G.J.), and a Research to Prevent Blindness grant (to LSU Department of Ophthalmology).
References
- Bonapace G, Waheed A, Shah GN, et al. Chemical chaperones protect from effects of apoptosis-inducing mutation in carbonic anhydrase IV identified in retinitis pigmentosa 17. Proc Natl Acad Sci USA. 2004;101:12300–12305. doi: 10.1073/pnas.0404764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19:1074–1081. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Moiseyev G, Takahashi Y, et al. Impacts of two point mutations of RPE65 from Leber’s congenital amaurosis on the stability, subcellular localization and isomerohydrolase activity of RPE65. FEBS Lett. 2006;580:4200–4204. doi: 10.1016/j.febslet.2006.06.078. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA. 2013;110:E517–525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, et al. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Bok D. A cell culture medium that supports the differentiation of human retinal pigment epithelium into functionally polarized monolayers. Mol Vis. 2001;7:14–19. [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Moghrabi WN, et al. Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell. 2005;122:449–459. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Nusinowitz S, et al. The role of interphotoreceptor retinoid-binding protein on the translocation of visual retinoids and function of cone photoreceptors. J Neurosci. 2009;29:1486–1495. doi: 10.1523/JNEUROSCI.3882-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc Natl Acad Sci USA. 2009;106:17325–17330. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yang Z, Hu J, et al. Secretory defect and cytotoxicity: the potential disease mechanisms for the retinitis pigmentosa (RP)-associated interphotoreceptor retinoid-binding protein (IRBP) J Biol Chem. 2013a;288:11395–11406. doi: 10.1074/jbc.M112.418251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Lee J, Zhou Y, et al. Fatty acid transport protein 4 (FATP4) prevents light-induced degeneration of cone and rod photoreceptors by inhibiting RPE65 isomerase. J Neurosci. 2013b;33:3178–3189. doi: 10.1523/JNEUROSCI.2428-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Izumi T, Hu J, et al. Rescue of Enzymatic Function for disease-associated RPE65 proteins containing various missense mutations in non-active sites. J Biol Chem. 2014;289:18943–18956. doi: 10.1074/jbc.M114.552117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaeva O, Takahashi Y, Moiseyev G, et al. Negative charge of the glutamic acid 417 residue is crucial for isomerohydrolase activity of RPE65. Biochem Biophys Res Commun. 2010;391:1757–1761. doi: 10.1016/j.bbrc.2009.12.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp AR, Jin M, Li S, et al. Predicting the pathogenicity of RPE65 mutations. Hum Mutat. 2009;30:1183–1188. doi: 10.1002/humu.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, Poliakov E, Yu S, et al. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci USA. 2005;102:13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Use of protein repair therapy in the treatment of cystic fibrosis. Curr Opin Pediatr. 1998;10:250–255. doi: 10.1097/00008480-199806000-00005. [DOI] [PubMed] [Google Scholar]
- Sato K, Li S, Gordon WC, et al. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J Neurosci. 2013;33:17458–17468. doi: 10.1523/JNEUROSCI.1380-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Chen Y, Moiseyev G, et al. Two point mutations of RPE65 from patients with retinal dystrophies decrease the stability of RPE65 protein and abolish its isomerohydrolase activity. J Biol Chem. 2006;281:21820–21826. doi: 10.1074/jbc.M603725200. [DOI] [PubMed] [Google Scholar]