

Abstract
Background
Mesenchymal stem cells (MSCs) exhibit immunomodulatory, tissue-protective, and repair-promoting properties in vitro and in animals. Clinical trials in several human conditions support the safety and efficacy of MSC transplantation. Published experience in multiple sclerosis (MS) is modest.
Objective
To assess feasibility, safety, and tolerability, and explore efficacy of autologous MSC transplantation in MS.
Methods
Participants with relapsing remitting (RR) or secondary progressive (SP) MS, Expanded Disability Status Scale score 3.0–6.5, disease activity or progression in the prior two years, and optic nerve involvement were enrolled. Bone-marrow-derived MSCs were culture-expanded, then cryopreserved. After confirming fulfillment of release criteria, 1–2 × 106 MSCs/kg were thawed and administered IV.
Results
Twenty four of 26 screened patients were infused: 16 women and 8 men, 10 RRMS and 14 SPMS, mean age 46.5, mean Expanded Disability Status Scale score 5.2, 25% with gadolinium-enhancing MRI lesions. Mean cell dosage (requiring 1–3 passages) was 1.9 × 106 MSCs/kg (range 1.3–2.0) with post thaw viability uniformly ≥95%. Cell infusion was tolerated well without treatment-related severe or serious adverse events, or evidence of disease activation.
Conclusion
Autologous MSC transplantation in MS appears feasible, safe, and well tolerated. Future trials to assess efficacy more definitively are warranted.
Keywords: multiple sclerosis, clinical trial, mesenchymal stem cells, safety, disability measures, quantitative MRI
Introduction
Multiple sclerosis (MS) pathology is characterized by multifocal central nervous system lesions with perivenular inflammatory cell infiltrates, demyelination, axonal transection, neuronal degeneration, and gliosis. Currently approved therapies are effective in relapsing MS, where they prevent accumulation of focal inflammatory damage, but none has been shown to directly promote repair.
Mesenchymal stem cells (MSCs), also known as multipotent stromal cells, can be isolated from multiple adult tissues including bone marrow, culture-expanded ex vivo, and induced to differentiate into cells of the mesodermal lineage, including osteoblasts, adipocytes, and chondrocytes. Their pleotropic immunomodulatory and repair-promoting effects and ability to traffic to areas of tissue inflammation and damage have led to testing in various inflammatory and tissue injury conditions, and also make them attractive as a potential treatment for MS.1 Pilot studies utilizing cells from several sources and tested in a variety of clinical settings have supported their safety and potential efficacy.1 An uncontrolled phase 1/2 study in secondary progressive (SP) MS suggested tissue repair within the afferent visual system.2 A recent small randomized phase 2 crossover study in relapsing remitting (RR) MS suggested reduced gadolinium-enhancing brain MRI lesions.3 Based on regulatory input on study design, we undertook a phase 1 study of autologous, culture expanded MSC transplantation in MS, focusing on feasibility, safety, and tolerability.
Materials and Methods
Study design
This open-label, phase 1 pre-post comparison study enrolled participants at the Cleveland Clinic Mellen Center for MS Treatment and Research between March 2011 and April 2013. Bone marrow aspiration was performed in the Dahms Clinical Research Unit at University Hospitals Cleveland Medical Center (UHCMC). MSC isolation and culture-expansion were performed in the National Center for Regenerative Medicine Cellular Therapy Laboratory at Case Western Reserve University, as previously described.4 The Cleveland Clinic and UHCMC Institutional Review Boards approved the study. Participants gave written consent before any study-related procedures were performed. The study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice5 and the principles of the Declaration of Helsinki.6 A Steering Committee designed the study and monitored its conduct. A Medical Monitor and independent Data and Safety Monitoring Committee reviewed trial conduct and safety data. Additional administrative details are available in the Supplementary Appendix.
Participants
Eligible participants were 18–55 years of age and had RR or SP MS fulfilling the 2010 McDonald Criteria;7 Expanded Disability Status Scale (EDSS) score 3.0–6.5;8 documented relapse, worsening disability, or MRI lesion activity in the prior two years; evidence of optic nerve involvement clinically, on visual evoked potentials (VEP), or optic coherence tomography (OCT); and brain MRI demonstrating T2-hyperintense lesions fulfilling MS diagnostic criteria.9 Participants could continue interferon-beta or glatiramer acetate, but other disease therapies were discontinued for varying durations prior to screening.
Intervention
After completing required screening, eligible participants underwent posterior iliac crest bone marrow aspiration under local anesthetic to obtain 20–50 ml of marrow. Bilateral aspiration was performed when necessary. MSCs were isolated and culture-expanded using established standardized procedures.4 At each passage after the primary culture, 1 × 106 cells were frozen for laboratory analysis. The cells were propagated in culture for up to eight weeks in low glucose DMEM containing 10% fetal bovine serum (FBS) prescreened for optimal MSC growth and 10 ng/ml human fibroblast growth factor-2 (R&D Systems, Minneapolis, MN). Conditioned culture medium was tested for microbiological contamination at each passage, and the culture terminated if microorganisms were detected. On the day of MSC harvest, the cells were detached from culture flasks using porcine trypsin and washed three times with PlasmaLyte plus 1% human serum albumin. Cell aliquots were evaluated for viability by trypan blue exclusion and cell surface phenotype by cytofluorometry with monoclonal antibodies for CD105, CD73, CD14, and CD45. An aliquot of the final cell product was tested for the presence of mycoplasma, and the conditioned medium was tested for aerobic and anaerobic bacteria, endotoxin, and fungus. MSCs were cryopreserved in a single Cryocyte freezing bag (plus two 1-ml satellite vials for quality assurance/quality control testing if needed), at final concentration of 4 × 106/ml, in PlasmaLyte A containing 10% dimethyl sulfoxide and 5% human serum albumin.
Release criteria for the post-expansion/pre-cryopreservation MSC product included:
Availability of at least 1 × 106 MSCs/kg participant body weight.
≥90% positivity for CD105/CD73, and <5% positivity for CD14/CD45 surface markers.
Absence of detectable bacteria, fungus, mycoplasma, or endotoxin.
In addition, ≥70% viability by trypan blue exclusion of an aliquot of the infusion product after thaw was required. If a culture was contaminated or provided inadequate cell yield, a second aspirate was requested from the participant.
After completion of the baseline (month 0) visit, participants received a single intravenous (IV) infusion of 1–2 × 106/Kg body weight MSCs without premedication. Vital signs, percutaneous oxygen saturation, electrocardiogram, and adverse events (AEs) were monitored for six hours following infusion.
Procedures
Safety assessments (AE monitoring, concomitant medication review, vital signs, weight, height, percutaneous oxygen saturation by pulse oximetry, general physical examination, blood chemistry, complete blood count, and urinalysis) were performed at screening (median 2.7 months); baseline (month 0); post infusion days 1, 4, 7, 14, 21; and months 1, 2, 3, 6. Clinical immunology studies (thyroid stimulating hormone, anti-thyroglobulin antibodies, anti microsomal antibodies, sedimentation rate, C-reactive protein, anti nuclear antibodies, SSA, SSB, rheumatoid factor, and lymphocyte subsets by cytofluorometry) were performed at screening; baseline; and months 1, 3, and 6. Chest X-ray and electrocardiogram were performed at screening, and months 1 and 6. Participant self reported global well being using a visual analog scale, neurological examination and calculation of EDSS, Multiple Sclerosis Functional Composite (MSFC), high and low-contrast letter acuity using Sloan charts (LCLA), MRI, VEP, and OCT were performed at screening; month-1; baseline; and months 1, 2, 3, and 6. MRI scans were acquired on a 3T Siemens Trio MRI scanner (Erlangen, Germany) according to a standardized protocol (see Supplementary Appendix) and analyzed by the MRI Analysis Center (Biomedical Engineering, Lerner Research Institute, Cleveland Clinic, Cleveland, OH). OCT was performed using a Cirrus spectral domain OCT machine (Carl Zeiss Meditec, Dublin, CA).
Outcomes
The study’s primary objective was to evaluate the feasibility of culturing MSCs, and infusion-related safety and tolerability of autologous MSC transplantation over one month in participants with relapsing forms of MS. Secondary objectives included evaluation of safety and tolerability over six months and effects on disease activity measured by the number of gadolinium-enhancing MRI lesions at one month. Safety measures included AEs, vital signs, oxygen saturation, blood laboratory studies, clinical immunology studies, urinalysis, chest X-ray, and electrocardiogram. AE severity was categorized based on Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.
Pre-specified exploratory efficacy measures included participant self report of global well being using a visual analog scale; MS disease status (relapses, EDSS, MSFC); vision (high-contrast visual acuity, LCLA); brain MRI (gadolinium enhancing lesion number and volume, new/enlarged T2-hyperintense lesion number, T2-hyperintense and T1-hypointense lesion volumes, normalized whole brain volume [brain parenchymal fraction], gray matter fraction, whole brain diffusion tensor imaging [mean diffusivity, fractional anisotropy], whole brain magnetization transfer ratio [MTR]); VEP (P100 latency); and OCT (retinal nerve fiber layer thickness, foveal thickness, macular volume).
MS relapse was defined as new, recurrent, or increased MS symptom(s); developing acutely (evolving over less than 3 months); with onset at least 30 days after the onset of a previous confirmed relapse; lasting at least 24 hours; with new objective neurological findings on the examination; and not associated with infection, fever, increased body temperature for another reason, or metabolic derangement. Confirmed relapses could be treated at the Treating Neurologist’s discretion with IV methylprednisolone 1000 mg/day for 3–5 days without an oral taper.
Statistical methods
Clinically significant treatment-related AEs were expected to be infrequent (<5% of participants), based on accumulating experience with MSC transplantation.10 The trial was conducted under a group sequential monitoring plan, with interim examinations of treatment related AEs after every fourth participant was followed for one month after infusion. Early stopping was to be considered if three participants within the first eight, or four participants overall, experienced treatment related AE CTCAE grade ≥3 or treatment related serious AE at any point during follow-up. This procedure followed an O’Brien-Fleming stopping rule for a nominal α=5% one-sided test of the composite null hypothesis of a 5% or lower risk of such a treatment associated AE against the alternative of a higher risk. This plan gives a ≤3.2% chance of a false finding of harm when true risk is <5%, and 89%, 74%, 50%, and 22% power to detect true risks of 25%, 20%, 15%, and 10%, respectively.
Descriptive methods were chosen appropriate to each variable’s scale (i.e., nominal, ordinal, interval, ratio). Exploratory objectives were to assess the effect of MSC transplantation on the longitudinal course of MS. To this purpose, we fit mixed linear statistical models with random subject effects and, wherever possible, a time series covariance structure, to numerous continuous MS outcome measures, and examined trends using restricted cubic splines and custom tests between specified inter-visit comparisons. Categorical data and nonparametric methods were used for outcomes for which such models were inappropriate. As this was an open label pilot study without a parallel control arm, and not powered to reliably detect temporal trends due to efficacy, p-values were used to describe compatibility of the data with absence of MSC activity, but not for formal hypothesis testing. For most outcomes, the primary exploratory p-value compared slopes or means within three months post-infusion with the 2–3 month pre-infusion period.
Results
Of 24 participants initially consented, one exhibited gadolinium allergy during the screening MRI and was not enrolled. Another’s culture failed due to slow growth and development of atypical cellular morphology, first noted at the first passage. The participant had no distinguishing past medical history, MS features, or previous or concomitant medications. The participant elected not to undergo a second bone marrow aspiration and discontinued the study. Two replacement participants were consented and enrolled (Figure 1). Demographic, clinical, and imaging characteristics for the 25 enrolled participants are summarized in Table 1. Among the infused participants, yields following the initial Percoll gradient centrifugation, final yields, and kinetics of growth varied considerably, with no apparent relation to participant demographics, MS disease characteristics, or current or prior treatment, and despite adherence to standard protocols for bone marrow harvest and cell culture. Nevertheless, the cell dose was within the target range for all infused participants and for 21 (88%) within 5.5% of 2 × 106/kg (Table 2). All planned visits and assessments were completed except for one blood study for one participant.
Figure 1.
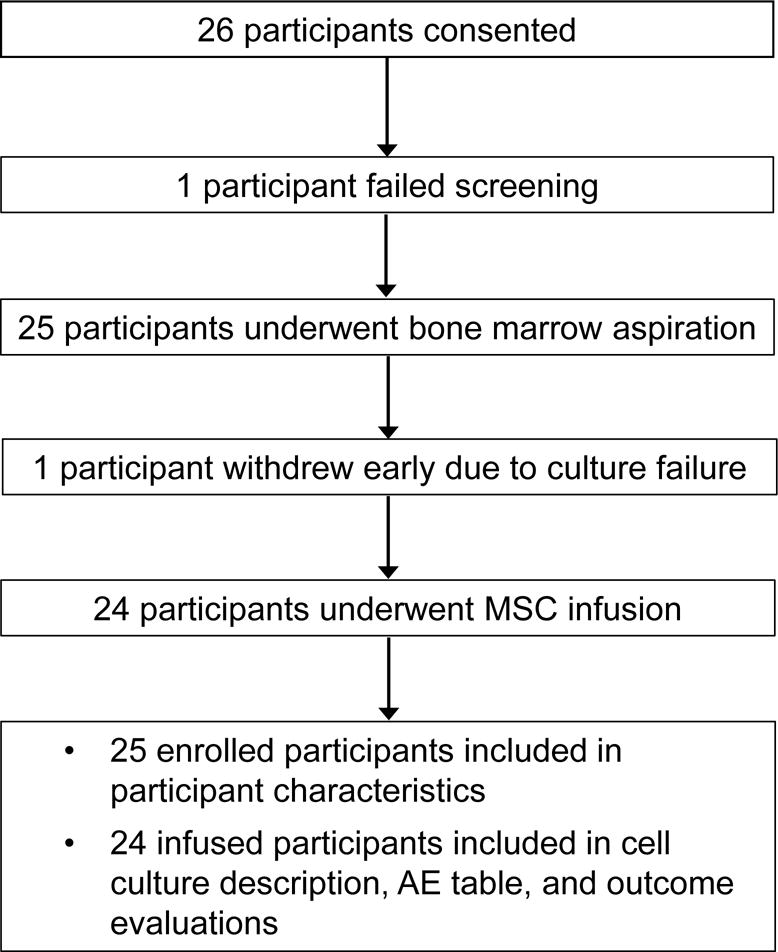
CONSORT diagram.
MSC = mesenchymal stem cell, AE = adverse event
Table 1.
Characteristics of participants who underwent bone marrow aspiration and MSC culture (screening visit)
| Characteristic | Participants (n=2 5) |
|---|---|
| Age, years – mean (SD) | 46.4 (5.2) |
| Sex | 17 female / 8 male |
| Disease course | 11 RR / 14 SP |
| Disease duration, years – mean (SD) | 15.4 (9.0) |
| MS disease activity or worsening in the prior two years – n, (%) a | MS relapse 16 (64.0), EDSS or FSS worsening 11 (44.0), MSFC component worsening 17 (68.0), brain MRI lesion activity 10 (40.0), cervical spine lesion activity 1 (4.0) |
| EDSS – median (range) | 6.0 (3.0–6.5) |
| Current disease treatment – n, (%) | interferon-beta 6 (24.0), glatiramer acetate 7 (28.0), no treatment 12 (48.0) |
| MRI with gadolinium-enhancing lesions – n, (%) | 6 (25.0) |
| T2-hyperintense lesion volume, ml – mean (SD) | 21.5 (12.0) |
| Brain parenchymal fraction – mean (SD) | 0.777 (0.039) |
The total exceeds 100% because some participants qualified based on more than one criterion.
EDSS = Expanded Disability Status Score, FSS = functional system score, MS = multiple sclerosis, RR = relapsing-remitting, SD = standard deviation, SP = secondary progressive
Table 2.
Cell culture kinetics and yield (n=24)
| Parameter | Mean ± SD (range) |
|---|---|
| Initial yield following Percoll centrifugation of bone marrow aspirate, 108 cells | 5.05 ± 2.72 (1.52–11.27) |
| Final yield following cell-expansion, 108 cells | 2.74 ± 1.29 (0.86–5.86) |
| Culture duration, days | 30.0 ± 11.3 (16–62) |
| Percent CD105+/CD73+ | 97.0 ± 2.1 (90.6–99.7) |
| Percent CD45+/CD14+ | 0.36 ± 0.46 (0.00–1.79) |
| Infused dose, 106 cells/kg | 1.9 ± 0.2 (1.3–2.0) |
| Viability: trypan blue exclusion of administered cells | 98.6 ± 0.9 (95.2–99.5) |
There was one culture failure that was terminated after 28 days due to poor growth and atypical cell morphology.
There were no severe (CTCAE grade ≥3) or serious AEs related to study treatment. Table 3 lists AEs experienced by ≥2 participants from infusion through follow-up. One participant with SP MS died eight months after completing the study. After study follow-up, this patient had progressive neurologic disability, with further worsening after intrathecal baclofen pump placement requiring two revisions, and died in hospice with severe spastic quadriplegia but no unusual infections or other medical condition. A second participant with SP MS died 40 months after completing the study after choking on food. We are unaware of other participants with unexpected AEs after study completion.
Table 3.
Adverse events during or after autologous MSC infusion among participants who underwent infusion (n=24)
| Events | Number (%) of participants |
|---|---|
| Any event | 22 |
| Any severe eventa | 3 (12.5)c |
| Any event leading to discontinuation of trial participation | 0 |
| Any serious event | 4 (16.7)c |
| Deaths | 0d |
| Most frequently reported events b | |
| Muscle spasticity | 11 (45.8) |
| Urinary tract infection | 10 (41.7) |
| Multiple sclerosis relapse | 7 (29.2) |
| Fall | 7 (29.2) |
| Multiple sclerosis | 5 (20.8) |
| Vision blurred | 5 (20.8) |
| Upper respiratory tract infection | 5 (20.8) |
| Balance disorder | 4 (16.7) |
| Nasopharyngitis | 3 (12.5) |
| Depression | 3 (12.5) |
| Diarrhoea | 3 (12.5) |
| Blood thyroid stimulating hormone decreased | 2 (8.3) |
| Back pain | 2 (8.3) |
| Fatigue | 2 (8.3) |
| Paraesthesia | 2 (8.3) |
| Anxiety | 2 (8.3) |
| Migraine | 2 (8.3) |
| Headache | 2 (8.3) |
CTCAE grade ≥3
reported by ≥2 participants
Events classified as both severe and serious included: hospitalizations of two participants for treatment of a urinary tract infection with associated temporary worsening of neurologic symptoms, two hospitalizations of one participant for depression, hospitalization of one participant for management of hyperglycemia following IV methylprednisolone treatment of an MS relapse. None were considered related to mesenchymal stem cell transplantation.
Two deaths occurred, 6 months and 40 months after completing the study, not considered related to mesenchymal stem cell transplantation.
No participant developed clinical manifestations or serologic studies indicating autoimmune phenomena. There was no clinical or radiographic evidence of paradoxical MS disease activation (Table 4). Although an active MRI was not required at enrollment, 24–50% of MRIs at the three pre infusion time points demonstrated gadolinium enhancing-lesions. Post-infusion, there was no evidence of activation or inhibition of gadolinium enhancement or new/enlarged T2 hyperintense MRI lesion accumulation. Among 24 infused participants, 18 (75%) were free of relapse, three (12.5%) experienced a single relapse during post-infusion follow-up, and three (12.5%) experienced two relapses (two pre-infusion, four post infusion). All relapses but one occurred in participants with RR MS. The single relapse in a participant with SP MS (but with a gadolinium-enhancing lesion at screening) was documented at the final visit. Among participants with RR MS, annualized relapse rates were 0.81 (95% CI 0.10–2.93) pre-infusion, 1.85 (95% CI 0.50–4.73) in months 1–3 post infusion, and 0.90 (95% CI 0.11–3.24) in months 4–6 post-infusion (p=0.58).
Table 4.
Exploratory efficacy results (n=24 for all values unless otherwise specifieda)
| Screening | Month-1 | Baseline | Month 1 | Month 2 | Month 3 | Month 6 | |
|---|---|---|---|---|---|---|---|
| Participant self-reported outcome | |||||||
| Global well-being by visual analog scale, mm from the left | |||||||
| Mean ± SD | 73.2 ± 17.2 | NA | 66.6 ± 15.9 | 67.9 ± 15.0 | 67.8 ± 16.0 | 58.6 ± 21.8 | 68.2 ± 17.0 |
| Clinician-assessed outcomes | |||||||
| Expanded Disability Status Scale | |||||||
| Mean ± SD | 5.2 ± 1.4 | 5.3 ± 1.4 | 5.3 ± 1.3 | 5.1 ± 1.5 | 4.9 ± 1.8 | 5.1 ± 1.7 | 5.0 ± 1.7 |
| Median (range) | 6.0 (3.0–6.5) | 6.0 (3.0–6.5) | 6.0 (3.0–6.5) | 6.0 (2.0–6.5) | 6.0 (1.5–6.5) | 6.0 (2.0–6.5) | 6.0 (2.0–6.5) |
| Multiple Sclerosis Functional Composite-4b | |||||||
| Mean ± SD | −0.17 ± 0.60 | −0.13 ± 0.62 | −0.14 ± 0.61 | −0.09 ± 0.57 | −0.07 ± 0.57 | −0.12 ± 0.56 | −0.13 ± 0.58 |
| Timed 25-foot walk, sec | |||||||
| Mean ± SD | 9.8 ± 4.9 | 10.2 ± 5.5 | 10.1 ± 5.0 | 9.8 ± 4.9 | 10.2 ± 5.0 | 11.6 ± 6.8 | 11.4 ± 6.9 |
| 9-hole peg test, sec | |||||||
| Right, mean ± SD | 28.4 ± 11.6 | 28.5 ± 12.0 | 30.3 ± 21.1 | 29.7 ± 15.9 | 28.8 ± 17.4 | 28.1 ± 14.7 | 27.8 ± 14.1 |
| Left, mean ± SD | 27.8 ± 9.7 | 28.8 ± 11.7 | 28.3 ± 9.6 | 27.9 ± 9.7 | 27.6 ± 8.8 | 26.9 ± 8.7 | 27.9 ± 9.7 |
| Paced auditory serial addition test, n correct | |||||||
| Mean ± SD | 45.3 ± 11.4 | 49.0 ± 10.0 | 47.9 ± 10.2 | 49.5 ± 10.3 | 49.8 ± 9.2 | 51.1 ± 8.6 | 50.8 ± 8.8 |
| Sloan low contrast letter acuity, binocular, n correct | |||||||
| 100%, mean ± SD | 56.6 ± 4.6 | 56.3 ± 4.1 | 56.4 ± 5.2 | 56.7 ± 4.6 | 55.9 ± 5.5 | 55.7 ± 5.7 | 56.3 ± 5.2 |
| 2.5%, mean ± SD | 32.5 ± 9.6 | 31.3 ± 8.3 | 31.3 ± 9.7 | 31.1 ± 9.2 | 29.0 ± 9.2 | 30.7 ± 8.7 | 30.8 ± 9.4 |
| 1.25%, mean ± SD | 17.0 ± 12.5 | 15.5 ± 11.6 | 16.5 ± 12.1 | 16.6 ± 11.4 | 16.5 ± 9.8 | 14.0 ± 9.0 | 13.9 ± 9.4 |
| MRI outcomes | |||||||
| Gadolinium enhancing lesions, n | |||||||
| Mean ± SD | 0.58 ± 1.53 | 0.63 ± 1.20 | 0.75 ± 0.90 | 0.96 ± 1.57 | 0.92 ± 1.89 | 0.67 ± 1.37 | 0.79 ± 1.41 |
| Median (range) | 0 (0–7) | 0 (0–5) | 0.5 (0–3) | 0 (0–6) | 0 (0–6) | 0 (0–5) | 0 (0–5) |
| Proportion of scans with gadolinium-enhancing lesions | |||||||
| n (%) | 6 (25) | 8 (33) | 12 (50) | 9 (38) | 8 (33) | 7 (29) | 8 (33) |
| Number of new or enlarged T2-hyperintense lesions (compared to the Screening MRI), n | |||||||
| Mean ± SD | NA | 0.63 ± 0.92 | 0.92 ± 1.25 | 1.29 ± 1.78 | 1.38 ± 1.81 | 1.54 ± 2.23 | 1.79 ± 2.48 |
| Median (range) | NA | 0 (0–3) | 0.5 (0–3) | 0 (0–6) | 0 (0–6) | 0 (0–8) | 0 (1–10) |
| T2-hyperintense lesion volume, ml | |||||||
| Mean ± SD | 21.6 ± 12.2 | 21.8 ± 12.5 | 22.2 ± 13.0 | 22.5 ± 13.0 | 22.1 ± 13.0 | 22.2 ± 13.0 | 22.0 ± 12.8 |
| T1-hypointense lesion volume, ml | |||||||
| Mean ± SD | 3.44 ± 3.20 | 3.36 ± 3.11 | 3.41 ± 3.13 | 3.36 ± 3.10 | 3.32 ± 3.00 | 3.32 ± 3.02 | 3.31 ± 2.98 |
| Normalized brain volume (brain parenchymal fraction) | |||||||
| Mean ± SD | 0.776 ± 0.039 | 0.775 ± 0.040 | 0.775 ± 0.039 | 0.774 ± 0.039 | 0.773 ± 0.040 | 0.774 ± 0.040 | 0.772 ± 0.041 |
| Gray matter fraction | |||||||
| Mean ± SD | 0.460 ± 0.020 | 0.460 ± 0.020 | 0.460 ± 0.020 | 0.459 ± 0.020 | 0.459 ± 0.021 | 0.457 ± 0.021 | 0.458 ± 0.021 |
| Whole brain mean diffusivity, ×10−6 mm2/S | |||||||
| Mean ± SD | 836.7 ± 33.8 | 841.1 ± 33.0a | 840.9 ± 29.7 | 845.1 ± 32.6a | 844.3 ± 33.3 | 843.9 ± 35.3a | 845.4 ± 33.4 |
| Whole brain fractional anisotropy | |||||||
| Mean ± SD | 282.8 ± 11.3 | 282.5 ± 10.4a | 281.5 ± 10.4 | 281.8 ± 10.6a | 279.9 ± 11.7 | 282.2 ± 11.9a | 282.0 ± 10.5 |
| Whole brain magnetization transfer ratio | |||||||
| Mean ± SD | 37.51 ± 2.56 | 37.49 ± 2.60 | 37.44 ± 2.66 | 37.50 ± 2.61 | 37.52 ± 2.62 | 37.62 ± 2.58 | 37.64 ± 2.59 |
| Visual evoked potentials | |||||||
| P100 latency, average of both eyes, msec | |||||||
| Mean ± SD | 133.3 ± 17.3 | 135.5 ± 19.1 | 133.1 ± 19.8 | 135.0 ± 19.2 | 135.3 ± 18.3 | 135.9 ± 18.5 | 137.1 ± 18.8 |
| Optical coherence tomography | |||||||
| Retinal nerve fiber layer thickness, average of both eyes, microns | |||||||
| Mean ± SD | 72.7 ± 7.6 | 72.8 ± 7.6 | 72.9 ± 8.2 | 72.6 ± 7.8 | 72.0 ± 7.7 | 72.3 ± 7.8 | 72.2 ± 7.7 |
| Macular volume, average of both eyes, micron3 | |||||||
| Mean ± SD | 9.38 ± 0.46 | 9.36 ± 0.47 | 9.34 ± 0.50 | 9.34 ± 0.47 | 9.35 ± 0.44 | 9.35 ± 0.45 | 9.31 ± 0.51 |
| Foveal thickness, average of both eyes, microns | |||||||
| Mean ± SD | 261.78 ± 12.86 | 261.20 ± 12.58 | 260.68 ± 13.95 | 260.50 ± 12.96 | 260.89 ± 12.26 | 261.01 ± 12.50 | 259.72 ± 14.14 |
SD = standard deviation, NA = not available, because not measured at that visit
n=23
Timed 25-foot walk, 9-hole peg test, paced auditory serial addition test, low-contrast letter acuity
Most other exploratory efficacy outcomes were generally stable over the course of the study (Table 4). Descriptive p values fell below the conventional 5% for three outcomes, compatible with two expected by chance alone based on number of outcomes studied. These outcomes were whole brain MTR (p=0.03) and MTR peak height (p=0.008), both of which trended upwards in post infusion months 1–3 relative to pre-infusion, and EDSS, for which three post-infusion measurements worsened while 17 were improved from baseline (signed rank p=0.001), with others stable. Improvement in the EDSS reflected improvements in ambulation, and cerebral and sensory subsystems.
Discussion
Previous experience in other disorders and limited published experience in MS suggested MSC transplantation would be safe and well tolerated. However, unanticipated autoimmune AEs have occurred with other MS therapies, e.g. alemtuzumab,11 altered peptide ligands,12 and tumor necrosis factor blockade.13 Therefore, based on regulatory input on study design, we conducted a pilot study focusing on feasibility, safety, and tolerability. During our study’s start-up preparations, the MSCT Study Group recommended a phase 2 design,14 stimulating the multinational Mesenchymal Stem Cells for Multiple Sclerosis (MESEMS) trial from which preliminary results recently were reported by a participating center.3
Our phase I study results support the feasibility, safety, and tolerability of IV administration of autologous, culture-expanded, bone marrow derived MSCs in MS. No treatment-related severe or serious AEs occurred during the follow-up period. The deaths of two participants after completing the trial did not appear to be related to MSC transplantation. Since the study was relatively small with only six-month follow up, we cannot rule out the possibility of rare or late-appearing AEs.
Exploratory efficacy measures demonstrated no substantial evidence of inhibition of disease activity, tissue repair, or recovery of function comparing 2–3 months pre infusion to six months post-infusion. Given the lack of comparison group, small sample size, and relatively short follow-up, demonstration of benefit was not anticipated. There was a suggestion of possible benefit on EDSS. This result must be interpreted most cautiously, particularly without evidence of benefit on other disability measures. In the 4.0–5.5 range, where improvement was seen, the EDSS score is determined primarily by distance a participant walks. A participant’s motivation and degree of examiner encouragement can be affected by expectation effects, as can the cerebral and sensory system scores that also improved. The benefit on two MTR measures could be random false positives among the dozens of outcome variables examined.
Lack of inhibition of gadolinium enhancing lesions was surprising given the pleotropic anti-inflammatory actions of MSCs. One possibility is MSCs lack MS-relevant anti inflammatory activity in vivo despite suggestive data from in vitro and animal studies. Or, possibly, the cell dose was insufficient. There were few data to guide dose selection when this study was designed. The target dose, 2 × 106 MSCs/kg body weight, was based on a dose effective in treating graft-versus-host disease following allogeneic hematopoietic stem cell transplantation,15 and was anticipated to be safe and reliably achievable based on our experience with MSC transplantation for several conditions.16 Also, multiple administrations might be more effective.17
Third, there is no current consensus concerning optimal delivery route. We elected to administer cells IV because of ease of administration and data supporting ability of MSCs to traffic from blood to injured or inflamed central nervous system in animal models of stroke, brain tumor, traumatic brain injury, spinal cord demyelination, and MS.1 This route has been employed in most studies of MSC transplantation in MS.1 Although the lungs trap many cells following IV injection, numerous cells reach systemic circulation and become widely distributed in experimental models and human studies.18 Intra-carotid delivery of MSCs, which has been explored in multiple system atrophy19 but not in MS to date, raises microembolization concerns. Some studies have utilized an intrathecal, or combined IV and intrathecal routes.20,22 Finally, we utilized cryopreserved cells thawed just before infusion. Although viability of the infused cell product exceeded 95% in all cases, recent studies suggest that, immediately after thawing, cryopreserved MSCs have cytoskeletal changes,23 impaired immunomodulatory function,24,25 altered in vivo trafficking,26 and increased T-cell mediated apoptosis.23 Thus, harvesting cells directly from culture or allowing recovery time after thawing appears prudent.
Another cell production issue in designing the planned phase 2 study is use of FBS in our culture protocol. Although we received regulatory approval based on our previous experience, validation of a xenon-free culture protocol for future studies is underway. Preliminarily, MSCs from five normal donors have been culture-expanded in parallel in low glucose DMEM containing 10% FBS or 5% human platelet lysate (PLUS supplement). Mean yields after three passages were more than tripled and culture times nearly halved with platelet lysate, with equivalent purity.27
We utilized bone marrow-derived MSCs based on our previous experience, but important differences might exist between MSCs from different tissue sources, e.g. adipose tissue and bone marrow, a point not yet systematically investigated. A related issue is whether MSCs should be autologous or can/should be isolated from donors without MS. We used autologous cells to avoid potential concerns with transmitting infection or cancer from the donor. Also, although MSCs are relatively non-immunogenic permitting allogeneic administration,15,28 some studies indicate repeated administration stimulates rejection29 – an issue if multi-dosing is planned. Conversely, “universal donor” MSCs have the advantage of an “off the shelf” reagent with consistent properties, and would not be subject to putative deficient immunomodulatory or repair-promoting properties associated with MS. While some studies indicate that MSCs isolated from MS subjects and non-MS controls exhibit similar growth in culture, differentiation potential, surface antigen expression, and immunomodulatory properties,30–32 others report notable functional differences,33–35 suggesting administration of MSCs from a donor without MS might be preferable. This key issue is unresolved and is the subject of ongoing ancillary studies.
In summary, this phase 1 trial supports the feasibility, safety, and tolerability of autologous MSC transplantation in MS. Future trials to assess efficacy more definitively are warranted.
Supplementary Material
Acknowledgments
We thank Cynthia Mackey, RN, BSN, MSN; Vinette Zinkand, RN; Robert Fox, RN; the staff of the Dahms Clinical Research Unit, Seidman Cancer Center; the Advisory Committee (Supplementary Appendix); and the Data Safety Monitoring Committee (Supplementary Appendix) for their assistance with the study. Preliminary results of this study were presented at the 2014 Joint ACTRIMS-ECTRIMS Meeting in Boston, MA USA September 10–13, 2014.
Funding
This trial was funded by Department of Defense Grant W81XWH 10-1-0270 and National Institutes of Health Grant RO1 NS074787, National Multiple Sclerosis Society Pilot Grant PP-1752, and Cleveland Clinic RPC Grant 2012–2017. The Dahms Clinical Research Unit is funded by the Clinical and Translational Science Collaborative of Cleveland UL1000439 from the National Center for Advancing Translational Sciences component of the National Institutes of Health and NIH Roadmap for Medical Research. Manuscript contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Trial registration: clinicaltrials.gov identifier: NCT00813969
Author contributions
Study conception or design: JA Cohen, PB Imrey, SM Planchon, E Fisher, A Bar-Or, J Reese Koc, SL Gerson, HM Lazarus. Study supervision or coordination: all authors. Analysis or interpretation of data: all authors. Statistical analysis: PB Imrey, M Karafa, S Morrison. Drafting and revising the manuscript for content: all authors.
Declaration of Conflicting Interests
JA Cohen reports consulting fees from Adamas, Merck, Mallinkrodt, Novartis, and Receptos and compensation as Co-Editor of Multiple Sclerosis Journal – Experimental, Translational and Clinical. RA Bermel reports consulting fees from Biogen, Genentech, Genzyme, and Novartis. E Fisher is an employee of Biogen. RJ Fox reports consulting fees from Actelion, Biogen, Genentech, Mallinkrodt, MedDay, Novartis, and Teva and research funding from Novartis. A Bar-OR reports consulting fees from Biogen Merck, Novartis, Receptos, Roche, Sanofi-Genzyme, and Teva. PB Imrey, SM Planchon, SL Sharp, T Skaramagas, P Jogodnik, M Karafa, S Morrison, J Reese Koc, SL Gerson, and HM Lazarus have nothing to disclose.
Contributor Information
Jeffrey A Cohen, Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Peter B Imrey, Department of Quantitative Health Sciences, Lerner Research Institute, and Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Sarah M Planchon, Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Robert A Bermel, Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Elizabeth Fisher, Department of Biomedical Engineering, Lerner Research Institute, Cleveland Clinic, Cleveland, OH USA.
Robert J Fox, Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Amit Bar-Or, Montreal Neurological Institute, McGill University, Montreal, Canada.
Susan L Sharp, Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Thomai T Skaramagas, Mellen Center, Neurological Institute, Cleveland Clinic, Cleveland, OH USA.
Patricia Jagodnik, Department of Biomedical Engineering, Lerner Research Institute, Cleveland Clinic, Cleveland, OH USA.
Matt Karafa, Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH USA.
Shannon Morrison, Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic, Cleveland, OH USA.
Jane Reese Koc, Case Comprehensive Cancer Center and National Center for Regenerative Medicine, Case Western Reserve University and Seidman Cancer Center, University Hospitals Cleveland Medical Center, Cleveland, OH USA.
Stanton L Gerson, Case Comprehensive Cancer Center and National Center for Regenerative Medicine, Case Western Reserve University and Seidman Cancer Center, University Hospitals Cleveland Medical Center, Cleveland, OH USA.
Hillard M Lazarus, Case Comprehensive Cancer Center and National Center for Regenerative Medicine, Case Western Reserve University and Seidman Cancer Center, University Hospitals Cleveland Medical Center, Cleveland, OH USA.
References
- 1.Cohen JA. Mesenchymal stem cell transplantation in multiple sclerosis. J Neurol Sci. 2013;333:43–49. doi: 10.1016/j.jns.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connick P, Kolappan M, Crawley C, et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open label phase 2a proof-of-concept study. Lancet Neurology. 2012;11:150–156. doi: 10.1016/S1474-4422(11)70305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Llufriu S, Sepulveda M, Blanco Y, et al. Randomized placebo-controlled phase II trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE. 2014;9:e113936. doi: 10.1371/journal.pone.0113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazarus HM, Haynesworth SE, Gerson SL, et al. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. 1995;16:557–564. [PubMed] [Google Scholar]
- 5.ICH harmonised tripartite guideline-guideline for good clinical practice: E6(R1) Geneva: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; Jun 10, 1996. (Accessed 28 September 2014, at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf.) [Google Scholar]
- 6.World Medical Association Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. doi: 10.1191/0969733002ne486xx. Available at: http://www.wma.net/en/30publications/10policies/b3/index.html. Accessed 22 Aug 2016. [DOI] [PubMed]
- 7.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald Criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 9.Swanton JK, Rovira A, Tintore M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: a multicentre retrospective study. Lancet Neurol. 2007;6:677–686. doi: 10.1016/S1474-4422(07)70176-X. [DOI] [PubMed] [Google Scholar]
- 10.Lalu MM, McIntyre L, Pugliese C, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS ONE. 2012;7:e47559. doi: 10.1371/journal.pone.0047559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coles AJ, Wing M, Smith S, et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet. 1995;354:1691–1695. doi: 10.1016/S0140-6736(99)02429-0. [DOI] [PubMed] [Google Scholar]
- 12.Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 13.The Lenercept Multiple Sclerosis Study Group, The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS. Results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53:457–465. [PubMed] [Google Scholar]
- 14.Freedman MS, Bar-Or A, Atkins HL, et al. The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: consensus report of the International MSCT Study Group. Mult Scler. 2010;16:503–510. doi: 10.1177/1352458509359727. [DOI] [PubMed] [Google Scholar]
- 15.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft versus host disease: a phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 16.Koc ON, Day J, Nieder M, et al. Mesenchymal stem cells Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS IH) Bone Marrow Transplant. 2002;30:215–222. doi: 10.1038/sj.bmt.1703650. [DOI] [PubMed] [Google Scholar]
- 17.Harris VK, Yan QJ, Vyshkina T, et al. Clinical and pathological effects of intrathecal injection of mesenchymal stem cell derived neural progenitors in an experimental model of multiple sclerosis. J Neurol Sci. 2012;313:167–177. doi: 10.1016/j.jns.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 18.Cogle CR, Yachnis AT, Laywell ED, et al. Bone marrow transdifferentiation in brain after transplantation: a retrospective study. Lancet. 2004;363:1432–1437. doi: 10.1016/S0140-6736(04)16102-3. [DOI] [PubMed] [Google Scholar]
- 19.Lee PH, Lee JE, Kim HS, et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol. 2012;72:32–40. doi: 10.1002/ana.23612. [DOI] [PubMed] [Google Scholar]
- 20.Karussis D, Karageorgiou C, Vaknin Dembinsky A, et al. Safety and immunologic effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:1187–1194. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moyeddin Bonab M, Yazdanbakhsh S, Loft J, et al. Does mesenchymal stem cell therapy help multiple sclerosis patients? Iran J Immunol. 2007;4:50–57. [PubMed] [Google Scholar]
- 22.Yamout B, Hourani R, Salti H, et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: A pilot study. J Neuroimmunol. 2010;227:185–189. doi: 10.1016/j.jneuroim.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Chinnadurai R, Copland IB, Garcia MA, et al. Cryopreserved mesenchymal stromal cells are susceptible to T-cell mediated apoptosis which is partly rescued by IFNgamma licensing. Stem Cells. 2016;34:2429–2442. doi: 10.1002/stem.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francois M, Copland IB, Yuan S, et al. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat shock response and impaired interferon gamma licensing. Cytotherapy. 2012;14:147–152. doi: 10.3109/14653249.2011.623691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moll G, Alm JJ, Davies LC, et al. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells. 2014;32:2430–2442. doi: 10.1002/stem.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chinnadurai R, Garcia MA, Sakurai Y, et al. Actin cytoskeletal disruption following cryopreservation alters the biodistribution of human mesenchymal stromal cells in vivo. Stem Cell Reports. 2014;3:60–72. doi: 10.1016/j.stemcr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reese JS, Hooper BM, Lazarus HM, et al. Identification of an alternative to fetal bovine serum for the culture expansion of human mesenchymal stem cells for use in clinical studies (P901) Mult Scler J. 2014;20(Suppl 1):461. [Google Scholar]
- 28.Rasmusson I, Uhlin M, Le Blanc K, et al. Mesenchymal stem cells fail to trigger effector functions of cytotoxic T lymphocytes. J Leukoc Biol. 2007;82:887–893. doi: 10.1189/jlb.0307140. [DOI] [PubMed] [Google Scholar]
- 29.Eliopoulos N, Stagg J, Lejeune L, et al. Allogeneic marrow stromal cells are immune rejected by MHC class I-and class II-mismatched recipient mice. Blood. 2005;106:4057–4065. doi: 10.1182/blood-2005-03-1004. [DOI] [PubMed] [Google Scholar]
- 30.Papadaki HA, Tsagournisakis M, Mastorodemos V, et al. Normal bone marrow hematopoietic stem cell reserves and normal stromal cell function support the use of autologous stem cell transplantation in patients with multiple sclerosis. Bone Marrow Transplant. 2005;36:1053–1063. doi: 10.1038/sj.bmt.1705179. [DOI] [PubMed] [Google Scholar]
- 31.Mazzanti B, Aldinucci A, Biagioli T, et al. Differences in mesenchymal stem cell cytokine profiles between MS patients and healthy donors: Implications for assessment of disease activity and treatment. J Neuroimmunol. 2008;199:142–150. doi: 10.1016/j.jneuroim.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Mallam E, Kemp K, Wilkins A, et al. Characterization of in vitro expanded bone marrow-derived mesenchymal stem cells from patients with multiple sclerosis. Mult Scler. 2010;16:909–918. doi: 10.1177/1352458510371959. [DOI] [PubMed] [Google Scholar]
- 33.de Oliveira GLV, de Lima KWA, Colombini AM, et al. Bone marrow mesenchymal stromal cells isolated from multiple sclerosis patients have distinct gene expression profile and decreased suppressive function compared with healthy counterparts. Cell Transplant. 2015;24:151–165. doi: 10.3727/096368913X675142. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar P, Redondo J, Kemp K, et al. Reduced expression of mitochondrial fumarate hydratase contributes to impaired MSC mediated neuroprotection in multiple sclerosis (139) Mult Scler J. 2016;22(S3):38–39. doi: 10.1177/13524585211060686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redondo J, Sarkar P, Kemp K, et al. Alterations in the secretome of MSCs isolated from patients with MS are in keeping with their reduced neuroprotective potential under conditins of oxidative stress (P669) Mult Scler J. 2016;22(S3):321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.