

Abstract
Traumatic brain injury (TBI) triggers endoplasmic reticulum (ER) stress and impairs autophagic clearance of damaged organelles and toxic macromolecules. In this study, we investigated the effects of the post-TBI administration of docosahexaenoic acid (DHA) on improving hippocampal autophagy flux and cognitive functions of rats. TBI was induced by cortical contusion injury in Sprague Dawley rats, which received DHA (16mg/kg in DMSO, interperitonial administration) or vehicle DMSO (1 ml/kg) with an initial dose within 15 minutes after the injury, followed by a daily dose for 3 or 7 days. First, RT-qPCR reveals that TBI induced a significant elevation in expression of autophagy-related genes in the hippocampus, including SQSTM1/p62 (sequestosome 1), Lamp1 and Lamp2 (lysosomal-associated membrane protein 1 and 2), and Ctsd (cathepsin D). Upregulation of the corresponding autophagy-related proteins was detected by immunoblotting and immunostaining. In contrast, the DHA-treated rats did not exhibit the TBI-induced autophagy biogenesis and showed restored CTSD protein expression and activity. T2-weighted images and Diffusion Tensor Imaging (DTI) of ex-vivo brains showed that DHA reduced both grey matter and white matter damage in cortical and hippocampal tissues. DHA-treated animals performed better than the vehicle control group on the Morris water maze test. Taken together, these findings suggest that TBI triggers sustained stimulation of autophagy biogenesis, autophagy flux and lysosomal functions in the hippocampus. Swift post-injury DHA administration restores hippocampal lysosomal biogenesis and function, demonstrating its therapeutic potential.
Keywords: autophagy, cortical contusion injury, docosahexaenoic acid, lysosome, microglial polarization, secondary injury
INTRODUCTION
Traumatic brain injury (TBI) triggers two biochemical repair responses: activation of the ubiquitin-proteasome system and the autophagy pathway. Endoplasmic reticulum (ER) stress and an accumulation of toxic protein derivatives such as unfolded, misfolded, or aggregated proteins stimulate the ubiquitin-proteasome system for degradation of these proteins in neocortical brains after TBI [1–3]. On the other hand, macroautophagy involves the bulk degradation of deviant proteins and organelles through the fusion of double-membrane autophagosomes with lysosomes [4,5]. These pathways are activated after TBI in an attempt to restore conditions to homeostasis. But autophagy flux, a cascade that transports cargo through the autophagy cycle, and lysosome functionality both have been shown to be impaired in neocortical tissues after TBI [2,6]. These studies suggest that increased expression of the autophagy marker protein LC3-II may result from insufficient degradation of autophagosomes but not solely due to an over stimulation of autophagy initiators [6]. The impairment of autophagy flux resulting from lysosomal dysfunction is evidenced by decreased expression and activity of cathepsin D (CTSD) protein in neocortical tissues at 1–24 h after TBI [6], which leads to decreased autophagic clearance of damaged organelles and toxic macromolecules.
Our recent studies have shown that docosahexaenoic acid (DHA) treatment is effective in reducing ER stress and subsequent neocortical brain damage using the controlled cortical impact (CCI) rat model of TBI [1]. DHA treatment diminishes ER stress evidenced by inhibiting the unfolded protein response (UPR) as well as reducing the accumulation of neurodegeneration-associated proteins [1]. Specifically, expression of UPR marker proteins p-elF2α, IRE1α, and ATF4, the proapoptosis protein CHOP, the neurodegenerative amyloid precursor protein (APP) and phosphorylated tau (p-Tau) levels were attenuated in the DHA-treated neocortical brains [1]. DHA has been shown to have neuroprotective effects in ischemic brains via the reduction of proapoptotic proteins and the rapid restoration of ionic homeostasis [7] or by significantly reducing infarct volume and improving neurological score after ischemic stroke [8]. However, no studies have been conducted to investigate the effect of DHA on autophagy and autophagy flux in the hippocampus after TBI. Since the hippocampus is the primary region of the brain responsible for memory and cognitive function [9], this study focuses on the effects of DHA administration on hippocampal function. Therefore, we addressed some of the gaps within the field and explored developing a new treatment for TBI.
In this study, we detected a significant elevation of autophagy-related gene expression in the hippocampus at 3 days after TBI, including SQSTM1/p62, LAMP1, LAMP2 and CTSD. Upregulation of these autophagy-related proteins was confirmed by immunoblotting and immunostaining assays. In contrast, the DHA-treated rats did not exhibit TBI-induced autophagy biogenesis and showed restored hippocampal CTSD protein expression and activity, which was accompanied with improved cognitive function. These findings suggest that TBI stimulates hippocampal autophagy and autophagy flux and post-injury DHA administration accelerates the recovery of the hippocampal lysosomal biogenesis and function.
MATERIALS AND METHODS
Materials
Cis-4, 7, 10, 13, 16, 19-docosahexaenoic acid (DHA) and dimethyl sulfoxide (DMSO) were from SigmaChemicals (St. Louis, MO). Rabbit anti-LC3B polyclonal antibody was from Novus Biologicals (Cat# NB100-2220, Littleton, CO). Rat anti-LAMP1 (Cat#ab25245), rat anti-LAMP2 monoclonal antibody (Cat#ab25339), and Rabbit anti-GFAP monoclonal (Cat#ab33922) antibodies were from Abcam (Cambridge, MA). Rat anti-LAMP1 was from the Developmental Studies Hybridoma Bank deposited by August, J.T. (Cat#DSHB-1D4B, Iowa City, Iowa). Goat anti-cathepsin D (CTSD) polyclonal antibody was from Santa Cruz Biotechnology (Cat#sc-6486, Dallas, Texas). Purified Mouse Anti-p62 Ick ligand (SQSMT1) was from BD Biosciences (Cat#610832, San Jose, CA). Guinea pig anti-p62/SQSTM1 polyclonal antibody was from Progen (Cat#GP62-C, Heidelberg Germany). Donkey anti-goat Alexa Fluor® 488-conjugated IgG, goat anti-rat Alexa Fluor® 488-conjugated IgG, donkey anti-rabbit Alexa Fluor® 546-cojugated IgG, goat anti-rabbit Alexa Fluor® 546-cojugated IgG, and TO-PRO®-3 iodide were from Invitrogen (Carlsbad, CA). Rabbit anti-NeuN monoclonal antibody (Cat#12943) and rabbit anti-β-actin antibody (Cat#4970) were from Cell Signaling Technology (Danvers, MA). Goat anti-rabbit HRP conjugated IgG (H+L) (Cat#172-1019), goat anti-mouse HRP conjugated IgG (H+L) (Cat#170-6516), and rabbit anti-goat IgG (H+L)-HRP conjugated (Cat#172-1034) were from Bio-Rad (Hercules, CA). Goat anti-rat IgG (H+L)-HRP conjugated (Cat#31470) and RIPA buffer (Cat# 89901) were from Thermo Fisher Scientific (Rockford, IL).
Animals
All studies were in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the U.S. Department of Health and Human Services and were approved by the University of Pittsburgh Medical Center Institutional Animal Care and Use Committee.
Male Sprague–Dawley Rats (Envigo, Indianapolis, IN) were group-housed (2 per cage) in standard cages at room temperature (22 ± 2°C) under standard 12-h light/dark cycles (light on at 07:00 AM) with free access to food and water. After injury, rats were housed in separate cages from the uninjured animals under the same conditions as described above. 152 male Sprague–Dawley rats (280–300 g body weight) were used in the study, 39 for immunostaining and MRI, 41 for immunoblotting, 26 for PCR, 26 for CTSD activity analysis and 20 for Morris water maze test).
Surgical procedures and TBI induction
Rats were anesthetized initially with 4% isoflurane with a 2:1 N2O/O2 mixture in a vented anesthesia chamber. Following endotracheal intubation, rats were ventilated mechanically with a 1–1.5% isoflurane mixture. Animals were mounted in a stereotaxic frame on the injury device in a supine position as described before [10] and the core body temperature was monitored continuously by a rectal thermistor probe and maintained at 37 ± 0.5°C with a heating pad. Following a midline incision and dissection of the soft tissues, a 7-mm craniotomy was made between lambda and bregma and centered 5 mm lateral of the central suture. To control for nonspecific effects due to animal handling, anesthesia or surgery procedures, sham control animals underwent identical surgical procedures but did not receive a TBI. To induce TBI, the controlled cortical impact injury (CCI) device, consisting of a small (1.98 cm) bore and an impactor tip (6 mm diameter), was set to produce a tissue deformation of 2.6 mm as described previously (Dixon et al., 1991). The animals received a cortical impact through the craniotomy at a velocity of 4 m/s. Following the surgical procedures, the anesthesia was stopped and the animals were ventilated on 100% oxygen until spontaneous return of respiration. Animal recovery was monitored by recording the times until the return of following reflexes: corneal, pinna, tail pinch, toe pinch, and righting. After this, the rats were monitored until fully recovered from anesthesia and were returned to the housing facility.
DHA administration
TBI or TBI+DHA animals received an initial 1mL/kg i.p. of DMSO or 16mg/mL DHA in DMSO at 10 minutes after the onset of TBI and subsequent daily dose for 3, or 7 days after TBI. Sham control rats received DMSO (1 ml/kg, i.p.) after completion of the surgery procedure and a subsequent daily dose for 3 or 7 days. Naïve control rats did not receive any drugs or surgery procedures. Biochemical assays were performed at 3 or 7 days post-injury.
Quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from brain tissues using TRIzol® reagent (Invitrogen, Carlsbag, CA), according to the manufacturers protocol. For cDNA synthesis, MuLV Reverse Transcriptase (Applied Biosystems, Foster City, CA) was used with 2 μg of total RNA and oligo(dT)18 as a primer. RT-qPCR was carried out using 1:60 dilutions of cDNA, Maxima SYBR Green/ROX qPCR Master Mix (2x) (ThermoFisher, Pittsburgh, PA), and 4 μM primer mix per 10 μL reaction. For gene expression analysis, the following primers were used (IDT, Cralville, IA): Actb, forward 5′-GCTCCGGCATGTGCAAAG-3′ and reverse 5′-CATCACACCCTGGTGCCTA-3′; Ctsd, forward 5′ -TACTCAAGGTATCGCAGGGTG-3′ and reverse 5′-CCAATGAAGACATCGCCCAG-3′; Lamp1, forward 5′-GTAACAACGGAACCTGCCTG-3′ and reverse 5′-TCTGGTCACCGTCTTGTTGT-3′; Lamp2, forward 5′-ATCACTGAGGAGAAGGTGCC-3′ and reverse 5′-TGGCTGTTGTTCAGCCTAAGT-3′; Sqstm1, forward 5′-TGGAGTCGGGAAACTGCTCA-3′ and reverse 5′-CCGGGGATCAGCCTCTGTA-3′. To avoid amplification of genomic DNA and ensure amplification of cDNA, all primers were designed to span exons and negative RT reactions (without reverse transcriptase) were performed as controls. Samples were amplified in triplicate on the 7300 Real Time System (Applied Biosystems, Foster City, CA) using the following program: 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 secs followed by 1 min at 60°C. The 2−δδCT method was used to calculate relative gene expression, where CT corresponds to the cycle at which level of fluorescence crossed the assigned threshold. ΔCT values were calculated as the difference between CT values from the target gene and the housekeeping gene Actb, whereas ΔΔCT values were calculated relative to the contralateral sample within the experiment. Data are represented as fold increase. Error bars represent range of gene expression calculated by subtracting or adding ΔΔCT standard error of the mean (SEM) from the mean δδCT values: 2ˆ(−δδCT−SEM) for low end of the range and 2ˆ(−δδCT+SEM) for high end of the range. p values were calculated with ΔΔCT using standard paired t-test.
Immunofluorescence staining
Animals were deeply anesthetized and transcardially perfused with saline followed by 4% paraformaldehyde (PFA) in 0.1 M PBS (pH 7.4). After perfusion, brains were post fixed in 4% PFA for 12 h, and subsequently cryoprotected with 30% sucrose in 0.1 M PBS for 24–36 h at 4°C as described before (Begum et al., 2014). The brains were frozen in Tissue-Tek O.C.T. compound for 10 min and cut into coronal sections (40 μm thickness) on a freezing microtome (Leica SM 2000R; Leica, Nussloch, Germany). Sections at bregma level −2.64 to −3.84 mm were selected and processed for immunofluorescence staining. Sections were rinsed in 0.1 M Tris-buffered saline (TBS, pH 7.5) for 15 min, and incubated with a blocking solution (0.1% Triton X-100 and 3% goat or donkey serum in 0.1 M TBS) for 60 min at room temperature. Sections were then incubated with LAMP1 (1:100), LAMP2 (1:200) or CTSD (1:50) and rabbit anti-NeuN (1:100) antibody in the blocking solution overnight at 4°C. After rinsing with TBS for 30 min, sections were incubated with the following corresponding secondary antibodies in the blocking solution (1:200) for 1 h at room temperature: goat anti-rat Alexa Fluor 488-conjugated IgG and goat anti-rabbit Alexa Fluor 546-conjugated IgG, or donkey anti-goat Alexa Fluor 488-conjugated IgG and donkey anti-rabbit Alexa Fluor 546-conjugated IgG. After rinsing with TBS for 15 min, sections were incubated with TO-PRO-3 iodide in the blocking solution (1:500) for another 15 min at room temperature. Sections were then mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). For negative controls, brain sections were stained with the secondary antibody only.
Fluorescent images were captured under 63x lens using a Leica DMIRE2 inverted confocal laser scanning microscope (Leica Software, Mannheim, Germany). Samples were excited at 488 nm (argon/krypton), 543 nm, and 633 nm. The emission fluorescence was recorded at 512–548 nm, 585–650 nm, and 650–750 nm, respectively. In a blinded manner, positively stained cells (LAMP1, LAMP2, or CTSD) were counted from CA1, CA2 or CA3 areas in each brain section of the contralateral and ipsilateral hippocampus using ImageJ Software. The total number of cells in the particular area was determined by the TO-PRO-3 staining. Identical digital imaging acquisition parameters were used throughout the study. The number of cells which were positively stained with the anti-LAMP1, the anti-LAMP2 or the anti-CTSD antibody in each area was normalized by the total number of cells in each particular area and data were expressed as an average of the number of positive cells/total cells × 100.
Immunoblotting
Rats were deeply anesthetized with pentobarbital (Nembutal, 100 mg/kg, i.p.) and decapitated. After removal of the brains, the hippocampal tissue was dissected on a chilled ice plate and homogenized in ice-cold sucrose buffer (0.25 M sucrose, 1 mM EDTA, and 10 mM Tris-HCl, pH 7.4), supplemented with protease inhibitor cocktail (1 mg/ml each of aproinin, pepstatin, and leupeptin; 100 mg/ml phenylmethylsulfonyl fluoride and 2 mM sodium orthovanadate). The homogenized samples were centrifuged at 14000 g for 15 min at 4°C. The resulting supernatant was stored at −80°C until use. The protein content was determined by a BCA protein assay kit using a 96-well micro-plate reader (Spectra Max 340; Molecular Devices).
Protein samples (30 μg/lane) and pre-stained molecular weight markers (Bio-Rad) were dissolved in a RIPA buffer diluted sample buffer and separated by SDS PAGE (4–15% gel, Bio-Rad). The resolved proteins were electrophoretically transferred to a PVDF membrane (Millipore). The blots were incubated in 10% nonfat dry milk in tris-buffered saline (TBS) at 4°C. The following primary antibodies and dilutions were used for incubation at 4°C overnight: rabbit anti-LC3B (1:1000), rat anti-LAMP1 (1:1000), rat anti-LAMP2 (1:1000), goat anti-CTSD (1:1000), mouse anti-p62 (1:1000), or rabbit anti-β-actin (1:10000). After rinsing, the blots were incubated with horseradish peroxidase-conjugated secondary IgG for 1 hour at room temperature. Bound antibody was visualized using the enhanced chemiluminescence assay. To assure equal loading of protein content, expression of β-actin was quantified. Relative changes in protein expression were estimated from the mean pixel density of each protein band using ImageJ software. Expression of each interested protein was normalized by β-actin.
T2 and Diffusion Tensor Imaging (DTI) of ex-vivo brains
Three days post-TBI, rats were anesthetized with pentobarbital (100 mg/kg, i.p) transcardially perfused with 4% paraformaldehyde (PFA) and decapitated. For ex-vivo MRI, brains were maintained within the skull to avoid anatomical deformation. After post-fixation in 4% PFA overnight, heads were stored in PBS solution at 4°C. MRI was performed at 500 MHz using a Bruker AV3HD 11.7 T/89 mm vertical bore small animal MRI scanner, equipped with a 30-mm quadrature RF coil and Paravision 6.0.1 software (Bruker Biospin, Billerica, MA). Following positioning and pilot scans, T2-weighted images were acquired using a Rapid Acquisition with Relaxation Enhancement (RARE) sequence, with the following parameters: TE/TR = 20/3500ms, averages = 2, 256 × 256 matrix, 25 slices with a 1 mm slice thickness, a RARE factor = 4, and a field of view (FOV) of 22 × 22 mm. A DTI data set covering the entire brain was collected using a multislice spin echo sequence with 3 A0 and 30 non-collinear diffusion-weighted images with the following parameters: TE/TR = 22/5000ms, 4 averages, matrix size = 192 × 192 reconstructed to 256 × 256, field of view = 22 × 22 mm, 25 axial slices, slice thickness = 1 mm, b-value = 1200 s/mm, and Δ/δ = 10/5 ms.
DTI and T2 datasets were loaded into DSI Studio (http://dsi-studio.labsolver.org/). In a blinded manner, region of interests (ROIs) were drawn segmenting the hippocampus and lesion area in both the ipsilateral and contralateral hemispheres. Hippocampal and lesion volumes, fractional anisotropy (FA) values, radial diffusivity (RD) values, and mean diffusivity (MD) values for the corpus callosum (CC), external capsule (EC), and total white matter (CC+EC) were determined. 3D surface renderings were created using DSI studio.
Morris water maze test (MWM)
Spatial memory acquisition of animals was assessed using the MWM hidden platform at day 14–20 in Sham, TBI vehicle control, and TBI+DHA groups as described previously [10,11]. The MWM consisted of a dark blue plastic pool 180 cm in diameter and 60 cm in depth and the pool was located in a 2.5 × 2.5-meter room with numerous extra-maze visual cues that remained constant throughout the experiment. The pool (a depth of 28 cm) was filled with water and a clear acrylic glass platform (10 cm in diameter and 26 cm high) was positioned 26 cm from the maze wall in the southwest quadrant and held constant for each trial. Testing for spatial learning began on postoperative day 14 and consisted of providing a block of four daily trials (4-min inter-trial interval) for 5 consecutive days (post TBI days 14–18) to locate the platform when it was submerged 2 cm below the water surface (i.e., invisible to the rat). Two additional days (post-TBI days 19–20) were used for animals to locate a visible platform (which was raised 2 cm above the water surface). The visible platform task was used as a control procedure to determine the contributions of non-spatial factors (e.g., sensorimotor performance, motivation, and visual acuity) on MWM performance. For each daily block of trials, the rats were placed in the pool facing the wall at each of the four possible start locations (north, east, south, and west) in a randomized manner. Each trial lasted until the rat climbed onto the platform or until 120 sec had elapsed. Rats that failed to locate the goal within the allotted time were manually guided to the platform. All rats remained on the platform for 30 sec before being placed in a heated incubator between trials. The mean latency scores of the four trials for each rat were used in the statistical analyses. Swim speed and distance was recorded with a video analysis system (Chromatrack; San Diego Instruments, San Diego, CA).
Statistics
Unless otherwise indicated data are expressed as the group means ± standard error (S.E.) of the mean. Statistical significance was determined by paired t-test or ANOVA using the Bonferroni post-hoc test in case of multiple comparisons (SigmaStat, Systat Software, Point Richmond, CA, USA). A significance level of p < 0.05 was used for all tests.
RESULTS
Effects of DHA on TBI-mediated changes of hippocampal biogenesis of the autophagy-related genes and proteins
Our RT-qPCR data in Figure 1 (a) show that TBI selectively stimulated biogenesis of autophagy-related genes in the injured ipsilateral (IL) hippocampi of the TBI+Vehicle rats at 3 days post-TBI. Compared to sham-treated IL, the IL hippocampi of TBI+Vehicle treated rats show ~5-fold increase in Ctsd mRNA levels (4.72 ± 0.88 fold increase; average ΔΔCt=−2.27±0.30, p < 0.05). There are ~3-fold upregulation in p62/SQSTM1 mRNA (3.33 ±1.10 fold increase; average ΔΔCt = −1.74 ± 0.58, p < 0.05). Lamp1 and Lamp2 mRNA demonstrated ~ 3-fold increase as well (3.27 ± 0.78 and 3.31±1.46 fold increase respectively; average ΔΔCt = −1.71 ± 0.40 and −1.73 ± 0.84 respectively, p < 0.05 for Lamp1; Lamp2 is not significant). In contrast, the p62, Lamp1, Lamp2 or Ctsd mRNA levels in the IL hippocampus of TBI+DHA rats were similar with Sham, or decreased. There was no statistically significant increase in these RNA in the non-lesion contralateral (CL) hippocampi (data not shown). These data suggest that the vehicle-treated and DHA-treated rats exhibit different biogenesis patterns of the autophagy-related genes in hippocampus in response to TBI.
Figure 1. Changes of autophagy-related gene transcription and protein expression in hippocampus 3 days after TBI.
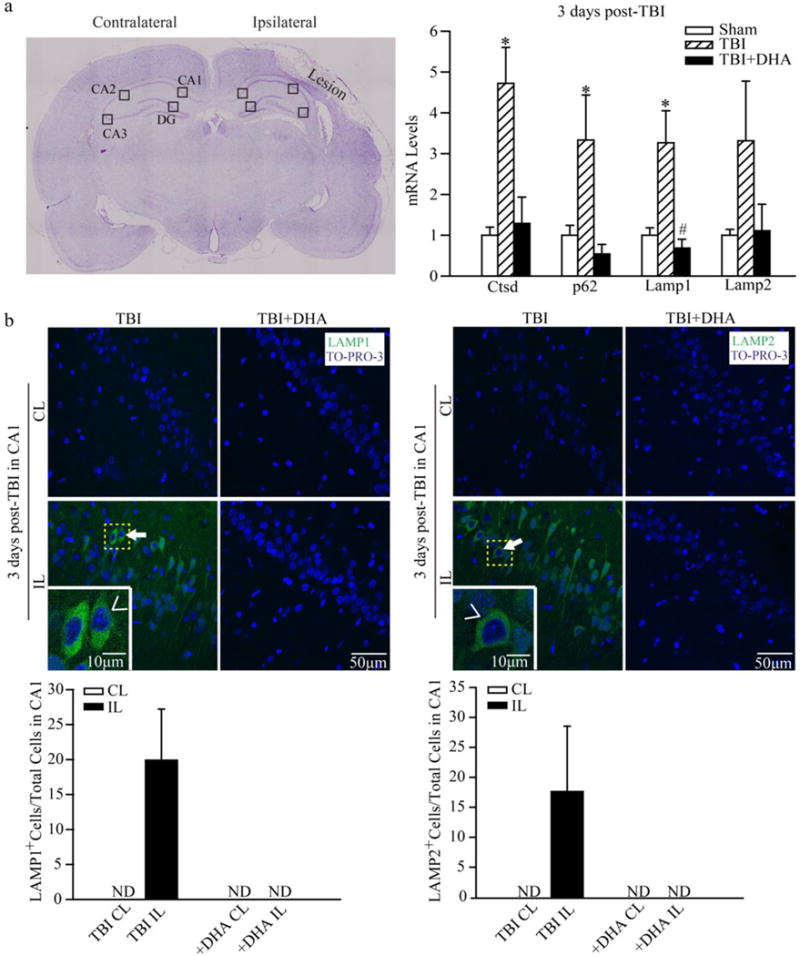
(a) TBI-mediated brain damage was shown in the representative cresyl violet stained coronal section. Region of interests in CA1, CA2, CA3, and DG were illustrated. For RT-qPCR study, the ipsilateral (IL) and controlateral (CL) hippocampi from Sham, TBI + Vehicle, or TBI + DHA-treated rats were harvested at 3 days post-TBI. Ctsd, SQSTM1/p62, Lamp1, and Lamp2 mRNA were determined by RT-qPCR. The relative changes in the IL hippocampal tissues were normalized by Sham hippocampal controls. Data are the mean ± 2ˆ(ΔΔCT range) as discussed in the Methods section (n = 5). * p < 0.05 vs. Sham; # p< 0.05 vs. TBI.
(b) Representative confocal immnunofluorescence images show selective elevation of LAMP1 and LAMP2 protein expression in CA1 in the TBI + Vehicle rats at 3 days post-TBI. No such a change was detected in the TBI + DHA-treated rats. Arrow: Increased expression of Lamp1 or Lamp2 in the IL hippocampal tissues. Arrowhead: enlarged images in inset. Values are mean ± S.E (n = 3). ND = not detected.
We subsequently evaluated changes of the autophagy-related protein expression in the hippocampus of TBI+Vehicle and TBI+DHA rats. Figure 1(b) shows low LAMP1 protein was expression in the CL hippocampus at 3 days post-TBI. However, there is a profound increase in LAMP1 expression in the ipsilateral CA1 region at 3 days post-TBI (Figure 1b, arrow). LAMP1 expression appears to be in neurons, and is not located in the GFAP-positive (GFAP+) reactive astrocytes (data not shown). No LAMP1 immunoreactive signals were detected in the IL of CA2, CA3, or dentate gyrus regions in the TBI+Vehicle rats (data not shown). LAMP2 expression is also significantly upregulated in the IL hippocampus in the CA1 region at 3 days post-TBI (Figure 1b, arrow). In contrast, in the TBI+DHA rats, no elevation of LAMP1 expression was detected in the IL CA1, CA2, and CA3 regions. DHA treatment also prevented the upregulation of LAMP2 protein expression in CA1 region.
Immunoblotting quantification of denatured LAMP1 and LAMP2 proteins in hippocampal homogenates in post-TBI brains
A 69 kDa and a 100 kDa LAMP1 protein bands were detected by immunoblotting (Figure 2a). Sham rats exhibit low level expression of LAMP1 protein in both IL and CL hippocampus. TBI triggered a significant increase in LAMP1 protein expression (100 kDa) in the IL hippocampus (47.6 ± 21.0 fold, p < 0.05) at 3 days after TBI, compared with the CL hippocampus (3.6 ± 0.9 fold). The 100 kDa Lamp1 protein was lower in the IL hippocampus of the TBI+DHA rats (28.1 ± 4.1 fold, p < 0.05). The LAMP1 core protein expression (69 kDa) was also higher in the IL than in the CL hippocampus of the TBI + vehicle rats (37.9 ± 11.6 fold vs. 3.1 ± 0.8 fold, p < 0.05). The TBI+DHA treated rats exhibited higher elevated levels of LAMP1 core protein in the IL hippocampus, compared to the CL hippocampus (73.7 ± 29.7 vs. 2.0 ± 0.4 fold, p < 0.05). LAMP2 protein expression shows similar trends in the TBI+vehicle and TBI+DHA groups (Figure 2b). These data suggest that TBI triggered a significant elevation of LAMP1 and LAMP2 protein expression. DHA treatment did not significantly reduce hippocampal LAMP1 and LAMP2 protein expression in the immunoblotting assay.
Figure 2. Denatured LAMP1 and LAMP2 protein expression in hippocampus of 3 day post-TBI brains.
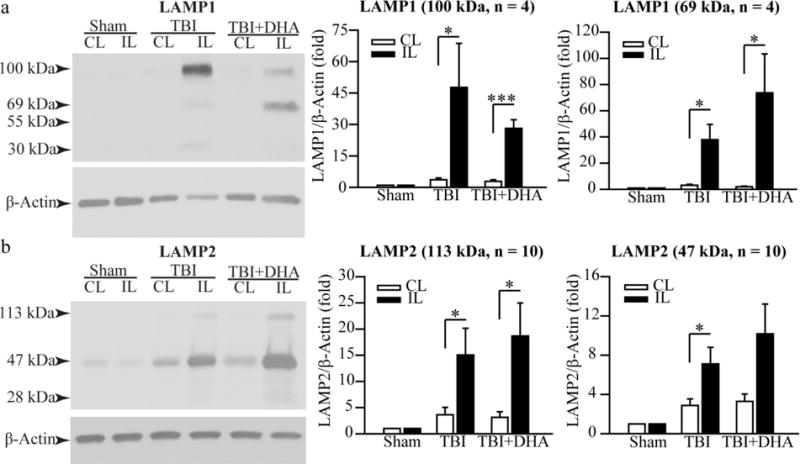
(a) Representative Western blots of LAMP1 expression in homogenates isolated from the CL and IL hippocampi at 3 days post-TBI. The 100 kDa and 69 kDa Lamp1 were detected. The same blot was probed with the antibody against β-actin as a loading control. Summary data expressed as a ratio of LAMP1 and β-actin band intensity. Data are the mean ± S.E. (n = 4). *p < 0.05, ***p < 0.001. TBI triggers significant increases in both mature LAMP1 and its lower molecular weight precursor in the IL hippocampi. DHA treatment shows the similar results.
(b) Representative Western blots of LAMP2 expression in hippocampal homogenates. The 113 kDa and 45 kDa Lamp2 band were detected. The same blot was probed with the antibody against β-actin as a loading control. Summary data expressed as the ratio of LAMP2 and β-actin band intensity. The data were the mean ± SE (n = 10). *p < 0.05
DHA treatment prevents TBI-induced CTSD protein downregulation in hippocampus and improves its activity
We further investigated lysosomal function by probing changes in CTSD protein expression and activity. The Western blot in Figure 3a displays CTSD precursor protein (43 kDa) and mature form of CTSD (30 kDa). There are no significant differences in hippocampal 43 kDa CTSD protein expression between Sham, TBI+vehicle, or TBI+DHA rats. However, the 30 kDa mature CTSD expression was significantly lower in the IL than the CL hippocampus (0.5 ± 0.08 vs. 0.9 ± 0.07 fold, p < 0.01) in the TBI + vehicle rats at 3 days post-TBI. In contrast, the IL and the CL hippocampus of TBI+DHA rats did not show significant differences in the mature CTSD (30 kDa) expression (0.6 ± 0.1 fold vs. 0.8 ± 0.09 fold, p > 0.05). These findings indicate that TBI causes a decrease in CTSD protein expression in hippocampus and DHA treatment prevented such a change.
Figure 3. TBI-mediated changes of hippocampal cathepsin D protein expression and activity at 3 days post-TBI.
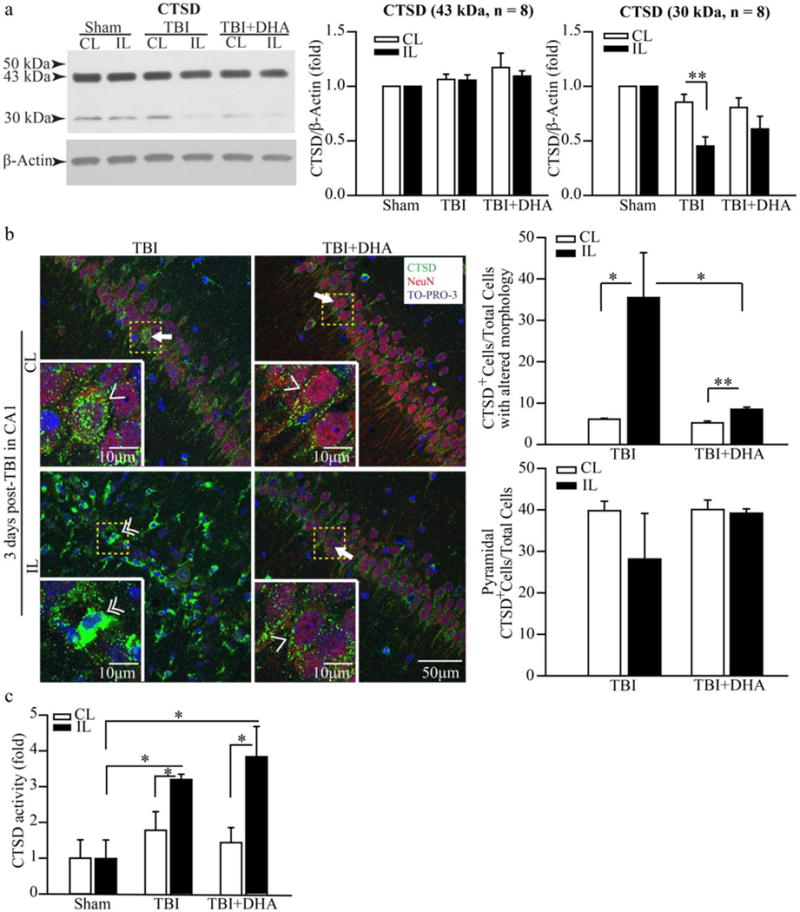
(a) Representative Western blots of cathepsin D (CTSD) expression in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA-treated rats at 3 days post-TBI were shown. Both 43 kDa proprotein and cleaved 30 kDa CTSD core proteins were detected. The same blot was probed with the antibody against β-actin as a loading control. The data were normalized with β-actin. Values are mean ± S.E (n = 8). *p < 0.05, **p < 0.01. There are no significant changes in 43 kDa CTSD proprotein expression but 30 kDa CTSD was significantly decreased in the IL hippocampi of TBI group, but not in the TBI+DHA group.
(b) Representative confocal immnunofluorescence images show selective elevation of CTSD protein expression in CA1 region in the TBI + Vehicle rats at 3 days post-TBI. No such a change was observed in the TBI + DHA-treated rats. Summary data expressed as percentage of CTSD-positive (CTSD+) cells with normal pyramidal morphology or with the altered morphology. Values are mean ± S.E. (n = 4). *p < 0.05, **p < 0.01. Arrow & Single arrowhead: pyramidal neurons with normal morphology and punctate CTSD expression pattern. Double arrowhead: cells with atypical pyramidal morphology, absence of NeuN and increased CTSD expression in a clustered and aggregated format.
(c) CTSD enzymatic activity in hippocampal tissues was determined in Sham, TBI + Vehicle and TBI + DHA-treated rat at 3 days post-TBI. The values are the mean ± S.E. (n = 3–5). *p < 0.05.
We further examined changes of hippocampal CTSD protein expression in the TBI + vehicle and TBI+DHA rats. Figure 3 (b) shows that pyramidal neurons in CA1 regions expressed abundant CTSD protein (punctate signals, arrow & single arrowhead) in the CL hippocampi of the TBI + vehicle and the TBI+DHA treated rats. CTSD protein was localized in neurons, which were localized in the same cells stained for the neuronal marker protein NeuN (arrow). In contrast, in the IL hippocampus of TBI rats at 3 days after TBI, pyramidal neurons in CA 1 regions lost its classical punctate CTSD immunoreactive staining pattern. Instead, the CTSD+ cells exhibited irregular shape and often formed clusters, and were not stained for NeuN (double arrowhead, Figure 3 b). We speculate that these CTSD immunoreactive signals represent microglia-mediated phagocytosis. On the other hand, the IL hippocampus of the TBI+DHA rat brains shows normal CTSD expression pattern (punctate, arrow) in CA1 regions. No changes in CTSD expression pattern were detected in CA2 or CA3 of hippocampus in the TBI + Vehicle or TBI+DHA rat brains (Supplemental Figure 1). These findings are consistent with our Western blotting data and further support that DHA treatment preserves lysosomal function.
To test this speculation, CTSD activity was measured in the Sham, the TBI + Vehicle and the TBI+DHA rat hippocampal tissues. Figure 3 (c) shows that the CL hippocampi from the Sham, the TBI + Vehicle and the TBI+DHA rats had similar levels of CTSD activity. In contrast, the IL hippocampus of the TBI + Vehicle rat exhibited ~ 3-fold increase in the CTSD activity and similar changes were detected in the IL hippocampus of the DHA-treated brains.
The surgical procedures may have impact on autophagy and lysosomal functions. We compared key autophagy protein expression in hippocampus tissues from naïve control rats (without any surgical procedures) and Sham-operated control rats. Supplemental Figure 2c illustrates that no differences of LAMP1 expression was detected between the naïve and Sham-operated hippocampal control tissues. In addition, immunostaining data revealed that the hippocampal CA1 neurons exhibited similar expression pattern of CTSD in the naïve control and Sham-operated control brains (Supplemental Figure 2b). Elevation of LAMP1 protein expression was only detected in the hippocampal CA1 neurons of TBI brains but not in the naïve control and Sham-operated control (Supplemental Figure 2a). Taken together, using immunoblotting and immunostaining methods, we did not detect significant effects of the anesthesia and surgical procedures on autophagy-related protein expression.
TBI-induced changes of hippocampal SQSTM1/p62 protein expression
We also investigated the protein expression of sequestosome-1 or ubiquitin-binding protein p62. In Figure 4a of 3 days post-TBI hippocampus data, a basal p62 protein (30 kDa) and a higher molecular weight p62 band (53 kDa) were detected. The 53 kDa band was significantly increased in the IL hippocampus than in the CL hippocampus (16.5 ± 4.8 fold vs. 4.2 ± 1.0 fold, p < 0.01) in the TBI + Vehicle group. In the TBI + DHA rats, 53 kDa band remains higher in the IL hippocampus than in the CL hippocampus (23.5 ± 7.9 fold vs. 4.9 ± 1.5, p < 0.05). TBI also triggers an increase in the 30 kDa band in the IL hippocampus, compared to the CL hippocampus (17.8 ± 6.4 fold vs. 4.5 ± 1.2 fold, p < 0.05, Figure 4a). The TBI+DHA treatment group shows a slightly higher 30 kDa expression in the IL hippocampus (27.4 ± 10.3 fold, Figure 4b). Interestingly, in the 7 day post-TBI hippocampus, no 53 kDa p62 band was detected in the hippocampal tissues of either Sham, the TBI + Vehicle or the TBI+DHA rats (p <0.05). In addition, the 30 kDa p62 band was reduced in the IL hippocampus of both the TBI + Vehicle and the TBI+DHA rats (Figure 4b).
Figure 4. Changes of hippocampal p62 protein expression after TBI.
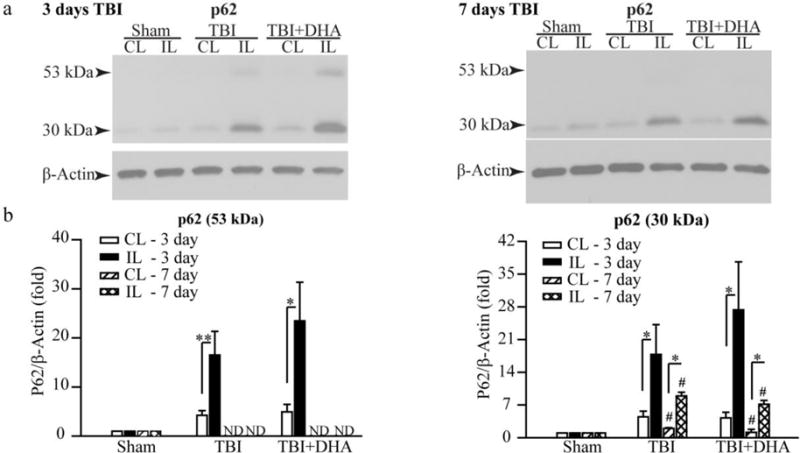
(a) Representative Western blots of p62 expression in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA rats at 3 or 7 days post-TBI. Both 53 kDa precursor and 30 kDa mature p62 proteins were detected in the 3 day post-TBI samples. Only a 30 kDa mature p62 protein was detected in the 7 day post-TBI samples. The same blot was probed with the antibody against β-actin as a loading control.
(b) The data were normalized with β-actin. Values are mean ± S.E (n = 6–10) *p < 0.05, **p < 0.01. # p <0.05 vs 3 days post-TBI.
Supplemental Figure 3 shows that TBI did not increase LC3-II expression but reduced the amount of LC3-I in the hippocampus. Expression of ubiquinated protein in hippocampus was significantly elevated at 3 days post-TBI.
Hippocampal autophagy flux recovery at 7 days post-TBI
Interestingly, at 7 days post-TBI, the TBI + Vehicle treated rats showed distinctly different profiles of hippocampal LAMP1, LAMP2, (Figure 5), or CTSD expression (Figure 6). First, no mature Lamp1 (100 kDa) or Lamp2 (113 kDa) expression was detected, which is in contrast to the 3 day post-TBI data. Instead, smaller molecular weight Lamp1 proteins (55 kDa and 30 kDa) and Lamp2 (47 kDa and 28 kDa) were elevated. The TBI+DHA-treated rats exhibited similar changes, except showing significantly lower levels of 47 kDa Lamp2 proteins (Figure 5b). Additionally, only ~11% of cells in CA1 region shows upregulation of CTSD expression in a clustered pattern at day 7 post-TBI (Figure 6), which is significantly lower than the ~35% at 3 days post-TBI (p < 0.05). On the other hand, the IL hippocampus of the DHA+TBI-treated rats exhibits punctate CTSD expression in the NeuN+ pyramidal neurons, which is similar to the CL normal pyramidal neurons. These findings suggest that DHA treatment accelerated hippocampal autophagy function recovery at day 7 post-TBI. This speculation is supported by the findings that elevation of the CTSD activity was only detected in the TBI + Vehicle treated hippocampus but not in the TBI+DHA-treated hippocampus (Figure 6b). In addition, no significant increases in the autophagy-related genes were detected in the IL hippocampi of either the TBI + Vehicle or the TBI+DHA-treated rats in our RT-qPCR assays (data not shown).
Figure 5. Changes of autophagy-related protein expression in hippocampus 7 days after TBI.
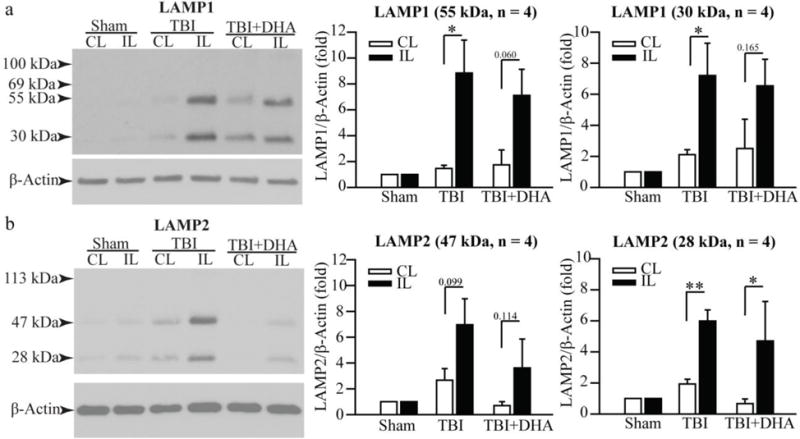
(a) Representative Western blots of LAMP1 expression in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA rats at 7 days post-TBI. Only a 55 kDa and 30 kDa Lamp1 were detected. The same blot was probed with the antibody against β-actin as a loading control. Summary data expressed as the ratio of LAMP1 and β-actin band intensity. Values are mean ± S.E. (n = 4). *p < 0.05.
(b) Representative Western blots of LAMP2 expression in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI+DHA rats at 7 days post-TBI. A 47 kDa and 28 kDa Lamp2 were detected. The same blot was probed with the antibody against β-actin as a loading control. Summary data expressed as the ratio of LAMP2 and β-actin band intensity. Values are mean ± S.E. (n = 4). *p < 0.05, **p < 0.001.
Figure 6. TBI-mediated changes of hippocampal cathepsin D protein expression and activity at 7 days post-TBI.
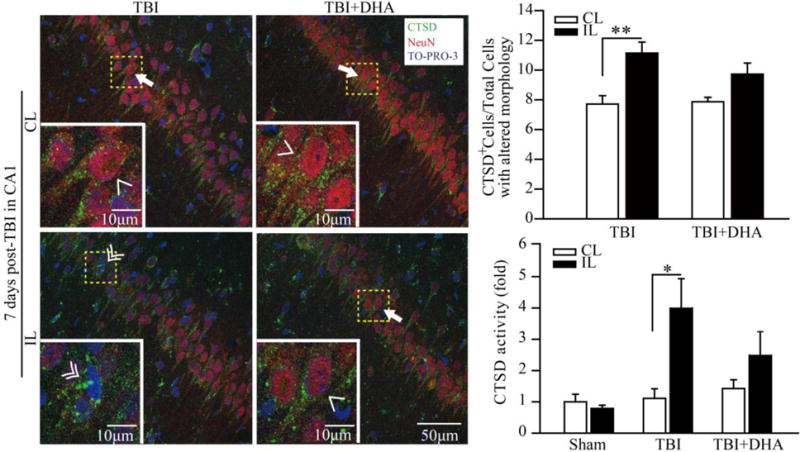
(a) Representative confocal immnunofluorescence images show selective elevation of CTSD protein expression in CA1 region in the TBI + Vehicle and TBI+DHA rats at 3 days post-TBI. Arrow & Single arrowhead: pyramidal neurons with normal morphology and punctate CTSD expression pattern. Double arrowhead: cells with atypical pyramidal morphology and increased in CTSD expression in a clustered and aggregated fashion.
(b) Summary data expressed as percentage of CTSD-positive (CTSD+) cells with the altered morphology and absence of NeuN staining. Values are mean ± S.E. (n = 4). *p < 0.05, **p < 0.01.
(c) CTSD enzymatic activity was determined by fluorometric assay in Sham, TBI+Vehicle and TBI+DHA-treated rat hippocampal tissues at 7 days post-TBI. The values are the mean ± S.E. (n = 3–5). *p < 0.05, **p < 0.01.
DHA treatment reduces TBI-induced hippocampal damage and white matter injury
We also conducted ex-vivo MRI studies of brains from Sham, TBI + Vehicle-treated and TBI + DHA-treated rats at 3 days post-TBI. Representative 3D surface renderings in Figure 7 (b and c) show that DHA treatment reduced the overall lesion volume by ~ 40% (p < 0.05). Representative T2-weighted images and Directionally Encoded Color (DEC) maps shown in Figure 7(a and d) further illustrates the reduced TBI-induced damage in the TBI + DHA-treated hippocampus when compared to the TBI + Vehicle-treated brains. Surface renderings of the hippocampal volume are shown in Figure 7(e), while Figure 7(f), shows that DHA-treated animals had preserved hippocampal volumes, comparable to shams, but the TBI + Vehicle-treated animals had decreased ipsilateral hippocampal volumes, albeit not significant. A significant decrease in mean RD, AD and MD values (~18%) was detected in the IL hippocampus of the TBI+DHA-treated brains (p < 0.05), while FA was not different between IL and CL hippocampi. Similar decreases in diffusivity (9~13%) were observed in the TBI + Vehicle-treated rats (p > 0.05, data not shown). To further confirm that the DHA treatment reduces white matter damage, we examined DTI data from the corpus callosum (CC) and external capsule (EC) axonal tracts. Figure 8 (a–d) illustrated that the TBI + Vehicle-treated rats exhibited significant reductions in the mean FA values in the EC tract and a concurrent increase in the mean RD values, both represent a loss of white matter integrity. In contrast, the TBI+DHA-treated brains showed better preserved white matter microstructures of the EC, which was reflected by a significantly less FA reduction and lower MD values. Taken together, these findings imply that the post-TBI administration of DHA reduces white matter injury in TBI brains.
Figure 7. DHA treatment reduces TBI-mediated cortical and hippocampal damage.
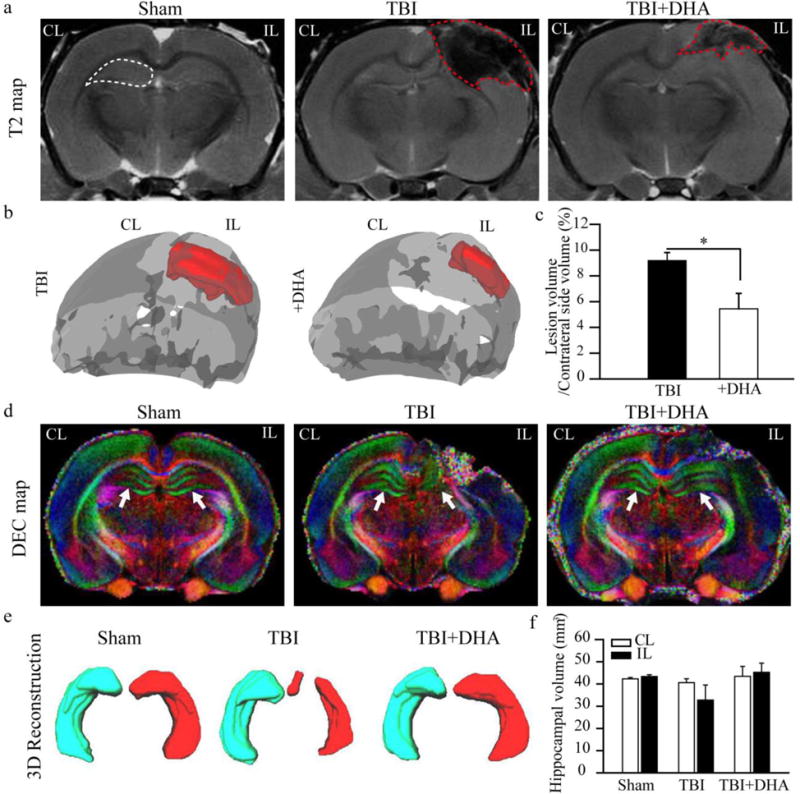
(a) Representative T2-weighted images maps of ex vivo brains. The ROIs show the hippocampus (white dash line) and lesioned areas (red dash line).
(b) 3D surface renderings of the lesioned areas (red) of TBI + Vehicle and TBI+DHA brains.
(c) Lesion volume. Values are mean ± S.E. (n=4). *p < 0.05.
(d) Representative DEC maps of ex vivo brains. The color of the DEC maps represents the diffusion direction of the white matter fibers in which red is left-right, green is dorsalventral, and blue is head-foot. Arrow: hippocampus
(e) 3D reconstructions of the CL and IL hippocampi of Sham, TBI + Vehicle-treated and TBI + DHA-treated rats.
(f) Hippocampal volume. Values are mean ± S.E. (n=4).
Figure 8. DHA-treated rats show less white matter lesion and less demyelination after TBI.
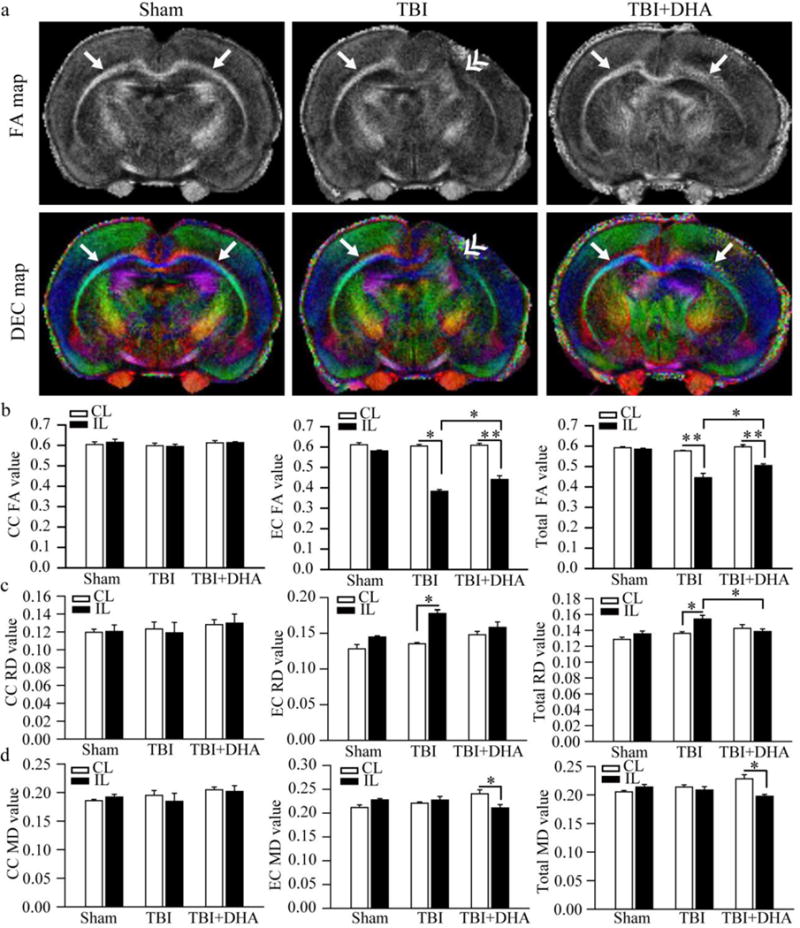
(a) Representative FA and DEC maps of ex vivo brains of Sham, TBI + Vehicle-treated and TBI+DHA-treated rats. Arrow: intact external capsule (EC). Double arrowhead: damaged EC.
(b) FA values of white matter tissues [corpus callosum (CC), EC or total white matter]. Values are mean ± S.E. (n=4). *p < 0.05, **p < 0.01
(c) RD values of white matter tissues. Values are mean ± S.E. (n=4). *p < 0.05.
(d) MD values of white matter tissues. Values are mean ± S.E. (n=4). *p < 0.05.
DHA treatment improves long-term neurological function after TBI
At last, we also examined whether DHA improves long-term cognitive deficits at day 14–20 after TBI. MWM test was used to assess the spatial learning and memory deficits. As shown in Figure 9, the TBI+DHA rats exhibited improved memory and spatial learning (with a hidden platform) at day 14–18 after TBI, and also with the cued MWM test at day 20 after TBI (* p < 0.05 vs. TBI + vehicle control). These findings clearly illustrate that post-TBI administration of DHA can improve the cognitive neurological function in the chronic recovery period.
Figure 9. DHA improves neurological function after TBI.
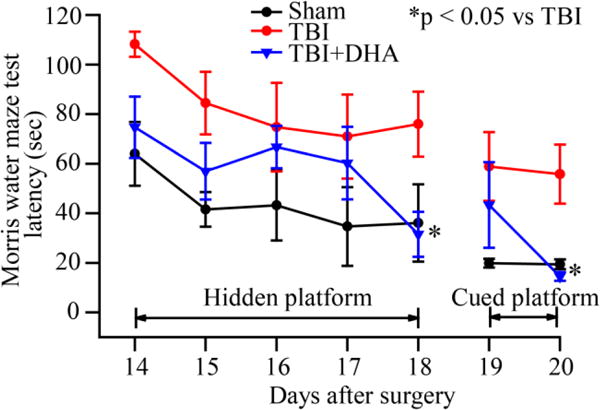
Sham, TBI + vehicle control, and TBI + DHA-treated animals were used to conduct Morris water maze (MWM) test at day 14–20 after surgery. Data are mean ± SE (n=6–7). *p < 0.05 vs. TBI + vehicle control. TBI + DHA-treated animals performed better than untreated TBI vehicle control animals.
DISCUSSION
TBI impairs autophagy through disruption of autophagy flux
It has been known for many years that autophagy is activated after TBI. In 2007, Bingren Hu and colleagues demonstrated that autophagy was induced as soon as 4 hours after TBI through the observation of increased autophagy-related ATG12-ATG5 complex and LC3-II in the cortex [5]. The ATG12-ATG5 complex is a protein conjugate responsible for the formation of double membraned autophagosomes that transport cellular detritus to lysosomes for degradation and recycling. LC3-II is also a marker of autophagy and has become a hallmark for measuring autophagy because it is present only within autophagosomes. The authors of this initial study proposed that autophagy served as a protective mechanism to help cells recover after TBI. A recent study by Sarkar et al more adequately addressed this issue [6]. They demonstrated that increased expression of autophagy markers such as LC3-II alone is not sufficient to conclude activation of autophagy. Instead, the authors proposed that the autophagosomal formation, maturation and degradation cycle, known as autophagy flux, may be impaired to create the accumulation of autophagosomes and their marker proteins. Since SQSTM1/p62 is concentrated within autophagosomes and is normally degraded after lysosome fusion, their accumulation showed disruption of autophagy flux. Although these two studies were conducted years apart, there are still key similarities between them. First, accumulation of LC3-II peaks at around 3 days post TBI before decreasing. Secondly, these effects are most prominently found within the neocortex. While Sarkar et al provided evidence that the hippocampus shares many of the similar results, the hippocampus does have slight differences such as a non-statistically significant elevation of autophagy initiator protein ULK1 after TBI. Therefore, in our study, we sought to examine effects of TBI on the hippocampal autophagy flux disruption at the peak time, day 3–7 post-TBI.
TBI-mediated changes in hippocampal autophagy and autophagy flux
Sarkar et al observed that autophagy flux impairment peaks at 3 days post-TBI within the cortex, after which LC3-II levels begin to decrease as CTSD levels begin to increase [6]. However, it is not well studied whether lysosomal functions are altered in hippocampal tissues after TBI. We report here that TBI alters autophagy and autophagy flux in hippocampus with the following evidence. First, TBI did not increase LC3-II expression but reduced the amount of LC3-I in the hippocampus at 3 days post-TBI. LC3-I is a precursor to LC3-II, and thus low LC3-I levels would indicate normal autophagic behavior [12]. However, the absence of LC3-II expression implies that autophagy induction may be increased after TBI, but the amount of LC3 II is low and cannot be detected or it is effectively cleared by autophagy flux. When examining SQSTM1/p62 mRNA levels and protein expression, we contend that autophagy is stimulated in hippocampus after TBI. Under normal conditions, ubiquinated proteins are transported to the autophagosome through p62, and both the ubiquinated protein and p62 are degraded after the autophagosome fuses with the lysosome [13]. In our study, expression of ubiquinated protein and p62 in hippocampus was significantly elevated at 3 days post-TBI. Additionally, mRNA analysis shows p62 mRNA was increased by ~ 3 fold at 3 days post-TBI, which may be the cell’s response to increase autophagy transport proteins for autophagic degradation (Figure 10). These findings suggest that autophagy induction is stimulated by TBI, leading to increased expression of all autophagy related genes and proteins. However, p62 elevation may also result from impaired autophagy flux. The lack of LC3-II buildup suggests that TBI did not cause complete autophagy flux disruption in the hippocampus at day 3 post-TBI.
Figure 10. Schematic illustration of ER stress and autophagy flux after TBI.
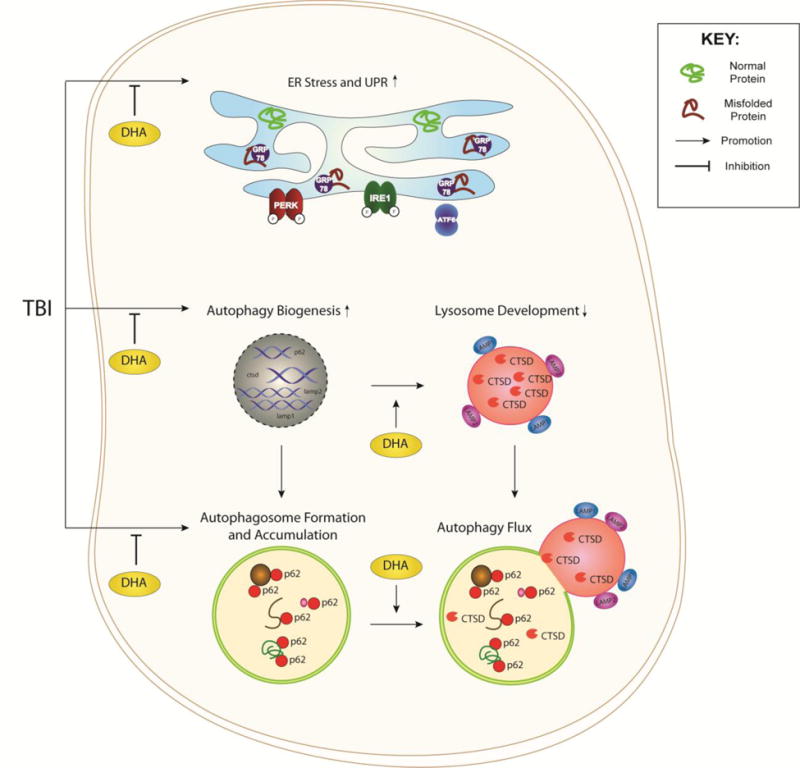
TBI induces ER stress through a surge of damaged and misfolded proteins within the ER. The UPR is triggered when the ER chaperone protein GRP78/BIP is dissociated from the ER-transmembrane sensors (PERK, IRE1, and ATF6) and bind to the misfolded proteins. TBI also stimulates autophagy biogenesis through the transcription of the related p62, ctsd, lamp1, and lamp2 genes and formation and accumulation of autophagosomes. UPR and autophagy stimulation actions are interconnected by the additional autophagic demand to recycle damaged protein and organelle detritus or when the efficiency of autophagy flux is compromised. In TBI brains, lysosomal development and function in neurons is diminished by a reduced mature CTSD expression or activity despite of its increased transcription. Autophagy flux is thus disrupted by the inefficiency in clearing cargo, leading to the observed accumulation of p62. DHA reduces ER stress, promotes CTSD efficiency and development of lysosomes and autophagy flux.
At present, the TFEB/TFE3 family of transcription factors are the best candidates for the lysosomal biogenesis. First, all four genes upregulated in our studies belong to the family of genes regulated by the TFEB/TFE3 transcription factors (the lysosomal biogenesis network) [14]. Second, TFEB has been shown to be activated by oxidative stress or by the factors associated with oxidative stress in the recent studies by Peña and Kiselyov (2015) and Zhang et al (2016) [15–16]. Oxidative stress is involved in TBI pathology and additional study is required to investigate changes of TFEB expression and regulation after TBI.
Lysosomal function in neurons and microglia after TBI
Sarkar et al proposed that autophagy flux stoppage may be caused by damage to lysosomes, which may prevent successful fusion of lysosomes and autophagosomes [6]. CTSD is a lysosomal enzyme that can be used as a marker to gauge the state of lysosomes and has a two-step production of the mature, active isoform of ~30 kDa from a ~53 kDa precursor into a ~47 kDa intermediate, which is matured within the lysosome into the 30 kDa active form [17]. Our Western blot analysis data show a significant decrease in 30 kDa CTSD in the IL hippocampus at 3 days post-TBI, compared to sham and contralateral hippocampus. Additionally, our data show no significant changes in the amount of intermediate ~43 kDa CTSD. Thus, the reduced lysosomal function in the hippocampus after TBI may lead to stimulation of autophagy biogenesis. Moreover, we detected an elevation of CTSD protein expression in NeuN negative cells of CA1 region, which may present upregulation of CTSD in the activated microglia. This may contribute to a significant increase in CTSD activity in the IL hippocampi. Collectively, these data suggest that TBI stimulates lysosome activity in the hippocampus.
Maturation of the lysosomal proteases is a complex process involving numerous steps of proteolysis executed by the lysosomal proteases [18, 19]. As an example, Colletti et al (2012) reported that the lysosomal dysfunction in the mucolipidosis type IV model results in a leakage of under-processed pro-cathepsin b isoform into the cytoplasm, makes pro-cathepsin b isoform appeared to accumulate in the cells [20]. Therefore, TBI-induced lysosomal dysfunction could contribute to cathepsin misprocessing and accumulation of the isoforms in damaged hippocampal tissues. Moreover, nitric oxide released from microglia/macrophages is involved in stimulating autolysosome formation and increased phagocytic activity [21]. We speculate that increasing CTSD expression and activity in microglia/macrophages may compensate for the reduced CTSD function in neurons. This is consistent with our data on elevation of CTSD protein expression in non-pyramidal cells, likely microglia/macrophages, in the CA1 regions at day 3 post-TBI.
A significant elevation in Lamp1 and Lamp2 mRNA levels was detected in the IL hippocampus at 3 days post TBI. Western blot analysis also shows a significant increase in LAMP1 and LAMP2 protein expression in the IL hippocampus at 3 days post-TBI. This expression can be visualized most clearly in the CA1 regions of the brain with the immunostaining assay. As elucidated by Carlsson et al, mature LAMP1 and LAMP2 are glycosylated with a size of around ~120 kDa but are formed from their respective precursors, which each has a polypeptide core of around ~40 kDa [22]. Moreover, the glycoprotein intermediates may have radically different molecular weights based on differing amounts of polylactosaminoglycan in cell types or conditions. In our study, the lower molecular weight LAMP1 is the precursor while the higher molecular weight of 100 kDa or 113 kDa is the mature form of Lamp1 or Lamp2. At day 3 post-TBI, there was an increase in both mature and precursor LAMP1 and LAMP2 expression, which may intend to increase the number and activity of lysosomes, especially under a condition that lysosomal efficacy is weakened from reduced mature CTSD expression. This induction of LAMP1 and LAMP2, an essential component of the lysosomal double membrane, may facilitate additional lysosomal formation or increase the size of lysosomes.
DHA treatment improves lysosomal function recovery after TBI
There are few studies on the direct effects of DHA on autophagy after TBI but several studies focused on its effects on autophagy in non-brain cell types. Within the colon of mice, DHA has been found to enhance autophagy through inhibition of the mTOR pathway [23]. Moreover, DHA induces p62 expression that is not derived from inhibition of autophagy flux in both human colon cancer cells and human retinal epithelia cells [24, 25]. We report here that post-TBI DHA administration stimulates autophagy as well as improvement of lysosomal functions. Our data follow the trend that DHA induces increased p62 expression, even though it was not statistically significant compared to the TBI + Vehicle brains. Moreover, DHA treatment improved CTSD protein expression, and restored CTSD mRNA expression and activity in hippocampus after TBI. Immunostaining revealed restored punctate lysosomal CTSD expression pattern in the CA1 hippocampal regions. These findings suggest that DHA treatment facilitates improvement of lysosomal function after TBI.
We have shown that post-TBI DHA treatment reduces ER stress as well as decreases neuroinflammation through less activation of proinflammatory microglia and macrophages [1, 26]. Furthermore, DHA treatment protects neural cells against oxidative stress [27]. Studies have shown that autophagy can help protect the cell from ER stress and oxidative stress [28]. Thus, DHA may improve autophagy by reducing these various stressors.
Lastly, we did not detect Lamp1 and Lamp2 mRNA elevation or increased LAMP1 and LAMP2 protein expression by immunostaining assay in the TBI+DHA hippocampus. In contrast, our Western blotting data revealed a slight increase in LAMP1 and LAMP2 protein expression after DHA treatment at 3 days post-TBI. At 7 days post-TBI, no 100 kDa Lamp1 or 113 kDa Lamp2 proteins were detected in hippocampus. The cause for this discrepancy is unclear and the data may reflect the localization and/or structural changes of LAMP1 and LAMP2 proteins. The LAMP family of transmembrane proteins have been found to localize not only in lysosomes, but also the plasma membrane, and furthermore, their cellular surface expression is common in inflammatory cells [29]. Lacking Lamp1 and Lamp2 protein expression in the DHA-treated hippocampus in the immunostaining study may reflect a decrease in the plasma membrane expression of these proteins. We speculate that DHA enhances lysosomal function recovery to the point that cellular overstimulation of autophagic pathways are no longer necessary.
DHA treatment is able to reduce hippocampal injury and improves cognitive function after TBI
Our findings suggest that hippocampus autophagy and lysosome disruption peak at 3 days post-TBI and are consistent with the same conclusion reached by Sarkar et al [6]. Additionally, we show that DHA administration not only accelerated autophagy function recovery in the hippocampus, but also lessened hippocampal damage after TBI which was reflected by the reduction of hippocampal tissue loss. Better preservation of white matter microstructures was also detected in the EC of the DHA-treated brains. As evidenced in our Morrison water maze test, DHA-treated rats performed better than non-treated rats even 20 days post-TBI (Figure 9). We hypothesize that the 3–7 days after TBI are most critical in determining the extent of the recovery and the natural clearance of cellular detritus. Our observation on the DHA-mediated protection of white matter microstructures and improvement of cognitive function of TBI rats is consistent with a recent report by Schober et al on the effects of dietary DHA in the immature rat after traumatic brain injury [30].
Conclusion
The effect of TBI of autophagy and lysosome function is time dependent and peaks at 3 days post-TBI. Post-TBI administration of DHA in rats is shown to mitigate these effects and reduces hippocampal damage and improves their cognitive function recovery. In this study, we detected a significant elevation of autophagy-related genes in the rat hippocampus at 3 days after TBI, including SQSTM1/p62, Lamp1, Lamp2 and Ctsd. Upregulation of these autophagy-related proteins was confirmed by immunoblotting and immunostaining assays. In contrast, the DHA-treated rats did not exhibit the TBI-induced autophagy biogenesis and showed restored hippocampal CTSD protein expression and activity, which was accompanied with the improved cognitive function. Our findings suggest that TBI stimulates hippocampal autophagy and autophagy flux and post-injury DHA administration restores the hippocampal lysosomal biogenesis and function. Therefore, stimulating autophagy flux after TBI plays an important role in reducing brain tissue damage and promoting cognitive function recovery.
Supplementary Material
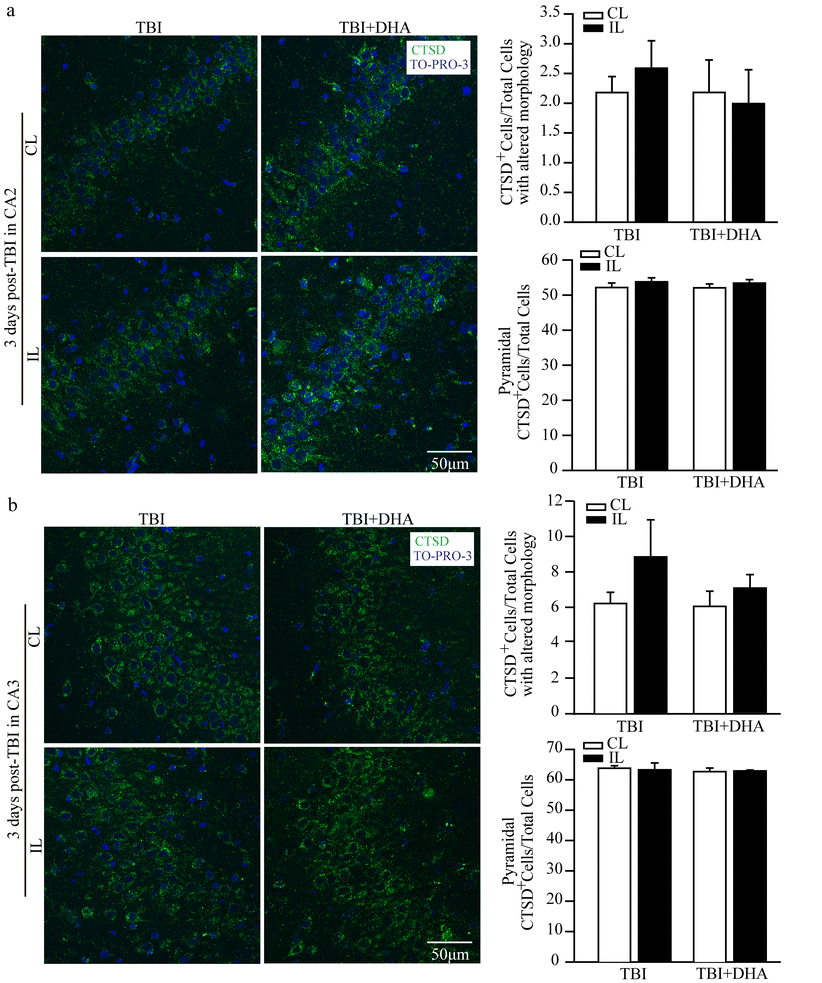
Supplemental Figure 1. No changes of CTSD protein expression in CA2 or CA3 regions after TBI
Representative confocal immnunofluorescence images show no changes in CTSD protein expression in CA2 or CA3 regions in the TBI + Vehicle and TBI + DHA rats at 3 days post-TBI.
Supplemental Figure 2. No differences of LAMP1 and CTSD protein expression in naive and SHAM hippocampus
Representative confocal immnunofluorescence images show changes in LAMP1 (a) or CTSD (b) protein expression in CA1 regions in the naïve, SHAM, or TBI + Vehicle rats at 3 days post-TBI. Representative Western blots of LAMP1 (c) expression in the CL and IL hippocampi of naïve, Sham, or TBI + Vehicle-treated rats at 3 days after TBI. The same blot was probed with the antibody against β-actin as a loading control.
Supplemental Figure 3. Changes of hippocampal protein ubiquitination and microtubule-associated light chain 3 protein after TBI
(a) Representative Western blots of LC3 expression in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA-treated rats at 3 days after TBI. The same blot was probed with the antibody against β-actin as a loading control. Summary data expressed as the band intensity ratio between LC3I and β-actin. The values are the mean ± SE (n = 6). * p < 0.05. No significant changes in LC3 were observed in the TBI+ Vehicle and TBI+DHA rats.
(b) Representative Western blots of protein ubiquitination in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA-treated rats at 3 days TBI. The intensity of proteins in a range of molecular weights from 250 kDa to 50 kDa was calculated. The same blot was probed with the antibody against β-actin as a loading control. Summary data were normalized with β-actin. The values are the mean ± SE (n = 8). **p < 0.01 vs Sham, ***p < 0.001 vs Sham. No significant changes in protein ubiquitination were observed in the TBI+ Vehicle and TBI+DHA rats.
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported in part by the National Institutes of Health [R01NS089051] (DS) and the United States Department of Veterans Affairs VA [RR&D#B6761R] (CED). YY was supported by the Chinese Dalian Municipal Bureau Study Abroad Research Award.
Footnotes
AUTHOR CONTRIBUTION
Q.H.Y. and C.E.D conducted CCI. Y.Y and I.Y.A performed immunostaining and quantified immunofluorescent images. Y. Y. performed CTSD activity assays. E.L. conducted immunoblotting assays. L.A.A and K.K. performed and quantified RT-PCR assays. T.K.H and L.M.F conducted MRI experiment. Y.Y. and D.S. designed the experiments. Y.Y. G.S., and D.S. completed the manuscript writing.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No conflicts of interest are declared by the authors.
References
- 1.Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci. 2014;34:3743–3755. doi: 10.1523/JNEUROSCI.2872-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park Y, Liu C, Luo T, Dietrich WD, Bramlett H, Hu B. Chaperone-mediated autophagy after traumatic brain injury. J Neurotrauma. 2015;32:1449–1457. doi: 10.1089/neu.2014.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao X, Liu J, Lee E, Ling G, McCabe J. Cullin 5 gene expression in the rat cerebral cortex and hippocampus following traumatic brain injury (TBI) Neurosci Lett. 2006;409:65–69. doi: 10.1016/j.neulet.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Lipinski MM, Wu J, Faden AI, Sarkar C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid Redox Signal. 2015;23:565–577. doi: 10.1089/ars.2015.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. J Cerb Blood Flow Metab. 2008;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10:2208–2222. doi: 10.4161/15548627.2014.981787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayurasakorn K, Williams JJ, Ten VS, Deckelbaum RJ. Docosahexaenoic acid: brain accretion and roles in neuroprotection after brain hypoxia and ischemia. Curr Opin Clin Nutr Metab Care. 2011;14:158–167. doi: 10.1097/MCO.0b013e328342cba5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belayev L, Khoutorova L, Atkins KD, Bazan NG. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient focal cerebral ischemia. Stroke. 2009;40:3121–3126. doi: 10.1161/STROKEAHA.109.555979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariza M, Serra-Grabulosa JM, Junque C, Ramirez B, Mataro M, Poca A, Bargallo N, Sahuquillo J. Hippocampal head atrophy after traumatic brain injury. Neuropsychologia. 2006;44:1956–1961. doi: 10.1016/j.neuropsychologia.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 11.Dixon CE, Kraus MF, Kline AE, Ma X, Yan HQ, Griffith RG, et al. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restor Neurol Neurosci. 1999;14:285–94. [PubMed] [Google Scholar]
- 12.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–18. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62SQSTM1 binds directly to Atg8LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 14.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 15.Peña KA, Kiselyov K. Transition metals activate TFEB in overpexpressing cells. Biochem J. 2015;470(1):65–76. doi: 10.1042/BJ20140645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, Delling M, Marugan J, Ferrer M, Xu H. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gieselmann V, Pohlmann R, Hasilik A, Von Figura K. Biosynthesis and transport of cathepsin D in cultured human fibroblasts. J Cell Biol. 1983;97:1–5. doi: 10.1083/jcb.97.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudenko G, Bonten E, d’Azzo A, Hol WG. Three-dimensional structure of the human ‘protective protein’: structure of the precursor form suggests a complex activation mechanism. Structure. 1995;3(11):1249–1259. doi: 10.1016/s0969-2126(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 19.Rosenfeld MG, Kreibich G, Popov D, Kato K, Sabatini DD. Biosynthesis of lysosomal hydrolases: their synthesis in bound polysomes and the role of co-and post-translational processing in determining their subcellular distribution. J Cell Biol. 1982;93(1):135–143. doi: 10.1083/jcb.93.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colletti GA, Miedel MT, Quinn J, Andharia N, Weisz OA, Kiselyov K. Loss of Lysosomal Ion Channel Transient Receptor Potential Channel Mucolipin-1 (TRPML1) Leads to Cathepsin B-dependent Apoptosis. J Biol Chem. 2012;287(11):8082–8091. doi: 10.1074/jbc.M111.285536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakanishi H, Zhang J, Koike M, Nishioku T, Okamoto Y, Kominami E, von Figura K, Peters C, Yamamoto K, Saftig P, Uchiyama Y. Involvement of nitric oxide released from microglia-macrophages in pathological changes of cathepsin D-deficient mice. J Neurosci. 2001;21:7526–7533. doi: 10.1523/JNEUROSCI.21-19-07526.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlsson SR, Roth J, Piller F, Fukuda M. Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp- 1 and h-lamp-2. J Biol Chem. 1988;263:18911–18919. [PubMed] [Google Scholar]
- 23.Zhao J, et al. Docosahexaenoic acid attenuated experimental chronic colitis in interleukin 10-deficient mice by enhancing autophagy through inhibition of the mtor pathway. JPEN J Parenter Enteral Nutr. 2015;20:1–6. doi: 10.1177/0148607115609308. [DOI] [PubMed] [Google Scholar]
- 24.Pettersen K, Monsen VT, Hakvåg Pettersen CH, et al. DHA-induced stress response in human colon cancer cells – Focus on oxidative stress and autophagy. Free Radic Biol Med. 2016;90:158–172. doi: 10.1016/j.freeradbiomed.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 25.Johansson I, Monsen VT, Pettersen K, et al. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy. 2015;11:1636–1651. doi: 10.1080/15548627.2015.1061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harvey LD, Yin Y, Attarwala IY, et al. Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN Neuro. 2015;7:1–15. doi: 10.1177/1759091415618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calandria JM, Asatryan A, Balaszczuk V, et al. NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neural cell survival. Cell Death Diff. 2015;22:1363–1377. doi: 10.1038/cdd.2014.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogata M, Hino S, Saito A, et al. Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 30.Schober ME, Requena DF, Abdullah OM, Casper TC, Beachy J, Malleske D, Pauly JR. Dietary docosahexaenoic acid improves cognitive function, tissue sparing, and magnetic resonance imaging indices of edema and white matter injury in the immature rat after traumatic brain injury. J Neurotraum. 2016;33:390–402. doi: 10.1089/neu.2015.3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. No changes of CTSD protein expression in CA2 or CA3 regions after TBI
Representative confocal immnunofluorescence images show no changes in CTSD protein expression in CA2 or CA3 regions in the TBI + Vehicle and TBI + DHA rats at 3 days post-TBI.
Supplemental Figure 2. No differences of LAMP1 and CTSD protein expression in naive and SHAM hippocampus
Representative confocal immnunofluorescence images show changes in LAMP1 (a) or CTSD (b) protein expression in CA1 regions in the naïve, SHAM, or TBI + Vehicle rats at 3 days post-TBI. Representative Western blots of LAMP1 (c) expression in the CL and IL hippocampi of naïve, Sham, or TBI + Vehicle-treated rats at 3 days after TBI. The same blot was probed with the antibody against β-actin as a loading control.
Supplemental Figure 3. Changes of hippocampal protein ubiquitination and microtubule-associated light chain 3 protein after TBI
(a) Representative Western blots of LC3 expression in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA-treated rats at 3 days after TBI. The same blot was probed with the antibody against β-actin as a loading control. Summary data expressed as the band intensity ratio between LC3I and β-actin. The values are the mean ± SE (n = 6). * p < 0.05. No significant changes in LC3 were observed in the TBI+ Vehicle and TBI+DHA rats.
(b) Representative Western blots of protein ubiquitination in the CL and IL hippocampi of Sham, TBI + Vehicle, and TBI + DHA-treated rats at 3 days TBI. The intensity of proteins in a range of molecular weights from 250 kDa to 50 kDa was calculated. The same blot was probed with the antibody against β-actin as a loading control. Summary data were normalized with β-actin. The values are the mean ± SE (n = 8). **p < 0.01 vs Sham, ***p < 0.001 vs Sham. No significant changes in protein ubiquitination were observed in the TBI+ Vehicle and TBI+DHA rats.