

Abstract
The APOBEC3 DNA cytosine deaminase family comprises a fundamental arm of the innate immune response and is best known for retrovirus restriction. Several APOBEC3 enzymes restrict HIV-1 and related retroviruses by deaminating viral cDNA cytosines to uracils compromising viral genomes. Human APOBEC3B (A3B) shows strong virus restriction activities in a variety of experimental systems, and is subjected to tight post-translational regulation evidenced by cell-specific HIV-1 restriction activity and active nuclear import. Here we ask whether lysines and/or lysine post-translational modifications are required for these A3B activities. A lysine-free derivative of human A3B was constructed and shown to be indistinguishable from the wild-type enzyme in DNA cytosine deamination, HIV-1 restriction, and nuclear localization activities. However, lysine loss did render the protein resistant to degradation by SIV Vif. Taken together, we conclude that lysine side chains and modifications thereof are unlikely to be central to A3B function or regulation in human cells.
Introduction
Cellular proteins such as APOBECs, TETHERIN, MX2, SERINCs, TRIMs, SAMHD1, and likely others, are called restriction factors because they are able to potently suppress the infectivity of a wide array of pathogenic elements including retroviruses [reviewed by (Harris and Dudley, 2015; Malim and Emerman, 2008; Simon et al., 2015)]. The APOBEC3 family of enzymes constitutes a unique arm of this innate immune defense network because they directly attack single-stranded DNA converting cytosines to uracils, which can result in C-to-T mutations or substrate degradation [reviewed by (Harris and Dudley, 2015; Malim and Emerman, 2008; Simon et al., 2015)]. In the case of retroviruses such as HIV-1, reverse transcription immortalizes cDNA strand C-to-U lesions as genomic strand G-to-A mutations. However, most of the HIV-1 restriction activity of human APOBEC3 enzymes is counteracted by HIV-1 Vif through a poly-ubiquitination and proteasomal degradation mechanism that efficiently eliminates restrictive APOBEC3 enzymes from cells and therefore also from entering nascent viral particles.
In comparison to APOBEC3G (A3G) and other human APOBEC3 enzymes, which have been characterized as robust virus restriction factors, the existing data on APOBEC3B (A3B) are less clear. First, although A3B does not appear to overtly restrict the replication of polyomaviruses and papillomaviruses, the preferred 5′-TC ssDNA target motifs of A3B are depleted from these viral genomes and the predicted 5′-TT product is enriched implying continual selective pressure (Ahasan et al., 2015; Starrett et al., 2016; Starrett et al., 2017; Verhalen et al., 2016; Vieira et al., 2014; Warren et al., 2015). Moreover, both of these small DNA viruses specifically upregulate A3B at the transcriptional level (Mori et al., 2017; Starrett et al., 2017; Verhalen et al., 2016; Vieira et al., 2014). Second, A3B knockdown in liver-derived HepG2 cells results in elevated levels of HBV circular DNA replication intermediates suggesting A3B may have a role in suppressing virus replication (Lucifora et al., 2014), however this result has been debated (Chisari et al., 2014; Ding and Robek, 2014; Meier et al., 2017; Shlomai and Rice, 2014; Xia et al., 2014). Disagreement also exists regarding HBV infection rates in individuals lacking the entire A3B gene due to a common deletion allele [higher: (Prasetyo et al., 2015); unchanged: (Ezzikouri et al., 2013)].
In the case of HIV-1, it has long been hypothesized that, unlike other A3 family members, A3B is not relevant due to the simple fact that it is not a target for proteosomal degradation by the viral protein Vif, indicating that the virus did not evolve a mechanism to escape selective pressures imposed by this enzyme (Bishop et al., 2004; Doehle et al., 2005; Hultquist et al., 2011). However, multiple studies have shown that A3B can potently restrict HIV-1 in 293T cells (Bishop et al., 2004; Doehle et al., 2005; Hultquist et al., 2011), whereas it cannot in various T-cell lines (Hultquist et al., 2011). Further complicating the story, human A3B can be targeted for protesomal degradation by SIV Vif leading to the possibility that A3B may still be relevant in some situations or, at the very least, was relevant in primate ancestors (Land et al., 2015). To better understand how and why A3B is able to restrict HIV-1 in some cell types but not others, and evade some but not all retroviral Vif proteins, we investigated mechanisms responsible for A3B post-translational regulation.
The mechanism of HIV-1 Vif-mediated degradation of APOBEC3 family members occurs through non-specific ubiquitination of lysine residues despite specific engagement of target APOBECs by Vif. Therefore, one hypothesis that could explain why A3B is able to restrict retroviruses in some cell types but not others, is that a lysine modification is part of A3B's post-translational regulatory mechanism, and the modification state depends on the cell type. To determine how lysine residues contribute to the overall regulation of A3B we constructed two lysine-free (K-free) mutant constructs, one fused to eGFP and one untagged, and performed head-to-head functional tests including DNA cytosine deamination activity, localization, and HIV-1 viral restriction assays.
Results
All lysine residues are dispensable for human A3B DNA cytosine deamination activity
Human A3B has 10 lysine residues, with 8 located in the non-catalytic N-terminal domain and 2 in the catalytic C-terminal domain (Fig. 1A). Recent crystal and solution structures show that the 2 lysines in the C-terminal domain are solvent-exposed and located near the catalytic site (Byeon et al., 2016; Shi et al., 2017; Shi et al., 2015) (Fig. 1B). An N-terminal domain structure has yet to be determined. Therefore, Phyre2 was used to develop a structural model of the N-terminal domain, which predicted that 7/8 are located in solvent-exposed regions, including 3 near the pseudo-active site which is likely to regulate enzyme activities including nucleic acid binding.
Fig 1. Lysine residues in human APOBEC3B.
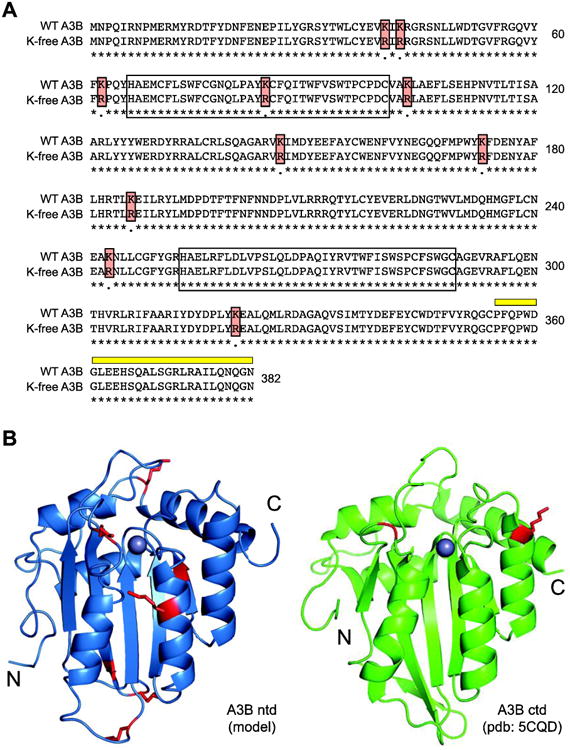
(A) Amino acid alignment of wild-type and K-free A3B. Lysine to arginine substitutions are highlighted with red boxes, N-terminal and C-terminal zinc-coordinating domains by open boxes, and 5210-87-13 mAb epitope by a thick yellow line.
(B) Ribbon structures of A3Bntd (model) and A3Bctd (pdb: 5CQD) with lysines colored red.
To address whether lysine post-translational modification has a role in regulating A3B DNA cytosine deaminase activity, we constructed a non-epitope tagged derivative with all 10 lysines converted to arginines (K-free A3B; Fig. 1). This construct was transfected into 293T cells and expression was assessed by immunoblot with a monoclonal antibody that recognizes a common C-terminal epitope (Leonard et al., 2015) (highlighted in yellow in Fig. 1A). Overall K-free A3B was expressed at levels similar to those of the parental wild-type enzyme, although minor variation was observed between biologically independent experiments (e.g., Fig. 2A versus Fig. 2C). In parallel, whole cell extracts were used in a single-stranded DNA cytosine deaminase activity assay with a 43 deoxy-nucleotide substrate containing a single 5′-TC motif preferred by A3B (Burns et al., 2013; Leonard et al., 2013) (Fig. 2B). Deamination is coupled to uracil excision and abasic site cleavage, which yields a shorter 30 deoxy-nucleotide product. In this assay, extracts expressing wild-type or K-free A3B showed similar activity, which is dependent on the catalytic glutamate of the enzyme (catalytic mutant, CM, in Fig. 2B). As negative controls, 293T extracts expressing a vector control or A3G, which prefers a different dinucleotide motif (5′-CC), showed much lower background levels of DNA deaminase activity.
Fig. 2. Lysines are dispensable for APOBEC3B ssDNA deamination activity.
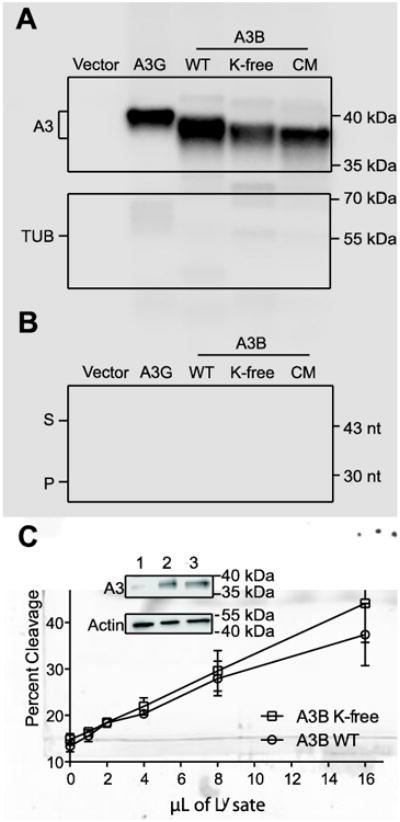
(A) Immunoblot of the indicated proteins expressed in 293T cells with tubulin as a loading control.
(B) Extracts from reactions in panel A tested for deaminase activity using an end-labeled ssDNA substrate (S). Deamination followed by uracil excision and cleavage results in a shorter product (P).
(C) Quantification of ssDNA deaminase activity of increasing amounts of 293T whole cell extracts expressing wild-type or K-free A3B. Each data point is the average +/- SD of 3 independent reactions. Inset immunoblot of lysates expressing vector only (1), wild-type A3B (2), and K-free A3B (3) relative to tubulin as a loading control.
To more precisely compare the catalytic activity of wild-type and K-free A3B, a dose response experiment was performed with increasing amounts of whole cell lysates expressing similar levels of either wild-type or K-free A3B. PAGE was used to analyze three independent experiments, as described above, and Image J was used to quantify the percentage of DNA deamination (substrate accumulation). Both enzymes showed similar activities at all concentrations (Fig. 2C; p=0.536 by Welch two-tailed t-test), and had similar levels of protein expression (immunoblot in graph inset). These data indicate that all 10 lysines are dispensable for the steady state single-stranded DNA cytosine deaminase activity of A3B.
Lysine residues are dispensable for A3B-catalyzed restriction and hypermutation of Vif-deficient HIV-1
The overall ability of an APOBEC enzyme to restrict Vif-deficient HIV-1 is due to multiple intrinsic properties including packaging into the core of nascent viral particles and binding and deaminating single-stranded DNA. Virus infectivity therefore provides a sensitive biological measure of overall APOBEC functionality. To test if any of these properties may be affected by lysine modification, the restriction activity of wild-type and K-free A3B was assessed in dose response experiments against Vif-deficient HIV-1 IIIB in comparison to A3G as a positive control (Fig. 3A). As expected from prior studies, A3G restricts Vif-deficient HIV-1 in a strong, dose-responsive manner. Interestingly, both wild-type and K-free A3B restricted Vif-deficient HIV-1 to similar levels, and as strong as those due to A3G restriction. The majority of the anti-viral activity of wild-type A3B requires an intact catalytic glutamate, as evidenced by higher viral infectivity in the presence of the E255A catalytic mutant (CM), particularly at lower protein concentrations. Some deaminase-independent effects were observed at higher protein concentrations, reminiscent of prior work for A3G (Browne et al., 2009; Newman et al., 2005; Schumacher et al., 2008). In addition, despite higher levels of A3B relative to A3G in cell lysates, less wild-type or K-free A3B protein was packaged into viral particles suggesting the antiviral effects of this enzyme may be greater than those of A3G (immunoblots in Fig. 3A). To exclude possible strain-specific effects, an analogous experiment was done with HIV-1 LAI and similar results were obtained for both wild-type and K-free A3B (Fig. 3B). Overall, the results of these virus infectivity experiments indicate that neither lysines nor lysine modifications are essential for A3B-mediated restriction of Vif-deficient HIV-1 in 293T cells.
Fig. 3. Lysine Free APOBEC3B shows wild-type levels of HIV-1 restriction.
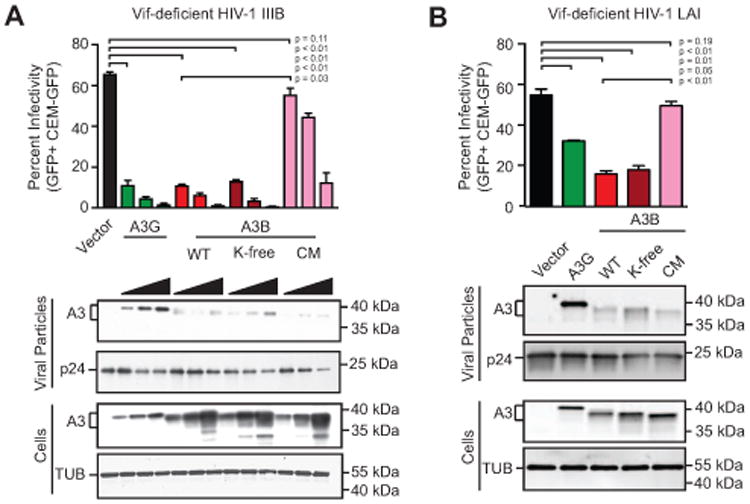
(A) Infectivity of Vif-deficient HIV-1IIIB produced in 293T cells expressing increasing amounts of the indicated expression constructs (0 ng, 25 ng, 50 ng, or 100 ng of A3-coding plasmid balanced with empty vector up to 100 ng to ensure equal DNA amounts). Each histogram bar is the average +/- SD of technical triplicate infections (p-values obtained by Welch two-tailed t-test). Immunoblots of the indicated cellular and viral proteins in whole cell extracts and virus-like particles with tubulin and p24 as loading controls, respectively.
(B) Infectivity of Vif-deficient HIV-1LAI produced in 293T cells expressing a single amount of the indicated expression constructs (25 ng). Each histogram bar is the average +/- SD of technical triplicate infections (p-values obtained by Welch two-tailed t-test). Immunoblots of the indicated cellular and viral proteins in whole cell extracts and virus-like particles with tubulin and p24 as loading controls, respectively.
SIV Vif-mediated degradation of human A3B is only partly affected by lysine loss
Despite the inability of HIV-1 Vif to degrade human A3B, the Vif protein of multiple SIV strains is active for human A3B degradation (Land et al., 2015). Vif-mediated degradation of A3 enzymes occurs mainly by poly-ubiquitination of lysine residues and subsequent processing by the 26S proteasome [reviewed by (Harris and Dudley, 2015; Malim and Bieniasz, 2012; Simon et al., 2015)]. Therefore, we sought to determine whether K-free A3B would become resistant to SIV Vif-mediated degradation. This experiment was done by co-transfecting 293T cells with an HA-tagged SIVmac239-Vif construct (abbreviated SIV Vif hereafter) and either untagged wild-type or K-free A3B followed by measurement of steady state A3B levels via immunoblotting (Fig. 4A). As a positive control, SIV Vif caused the degradation of human A3G, and this effect could be partly reversed by MG132 which blocks proteosomal processing. SIV Vif also mediated the degradation of human A3B, as reported (Land et al., 2015), and this effect could also be suppressed by MG132. In contrast, K-free A3B was unaffected by SIV Vif expression or MG132 treatment. These data indicate first that wild-type A3B levels in the cell are not likely to be regulated by polyubiquitination and proteomsomal degradation (little difference in band intensities with/without MG132), and second they confirm that SIV Vif-mediated degradation of wild-type A3B most likely occurs by the canonical lysine polyubiquitination and proteasomal degradation mechanism.
Fig. 4. SIV-Vif mediated degradation occurs through lysines in human A3B.
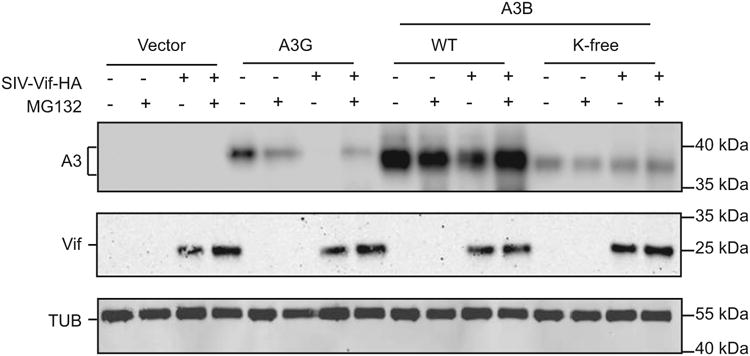
Immunoblot of the indicated APOBEC proteins in the presence or absence of SIV-Vif and with or without 10 μM MG132 (18 hrs). The corresponding blots below show Vif (anti-HA) and tubulin.
The hallmark nuclear localization activity of A3B does not require lysine residues or lysine post-translational modifications
The most obvious activity that sets A3B apart from related human APOBEC family members is constitutive nuclear localization (Bogerd et al., 2006; Burns et al., 2013; Lackey et al., 2012; Lackey et al., 2013; Stenglein and Harris, 2006). To determine whether lysine residues or lysine post-translational modifications contribute to the nuclear localization activity of A3B, immunofluorescent microscopy was used to assess the subcellular localization of wild-type and K-free A3B was assessed in HeLa and 293T cells (HeLa data in Fig. 5A and 293T data not shown). As expected, wild-type A3B was predominantly nuclear and, as a contrasting control, A3G was largely cytoplasmic. K-free A3B also localized to the nuclear compartment, indicating that lysine amino acid side chains and/or modifications are not likely to be part of the A3B nuclear localization mechanism.
Fig. 5. Nuclear localization of APOBEC3B does not require lysines.
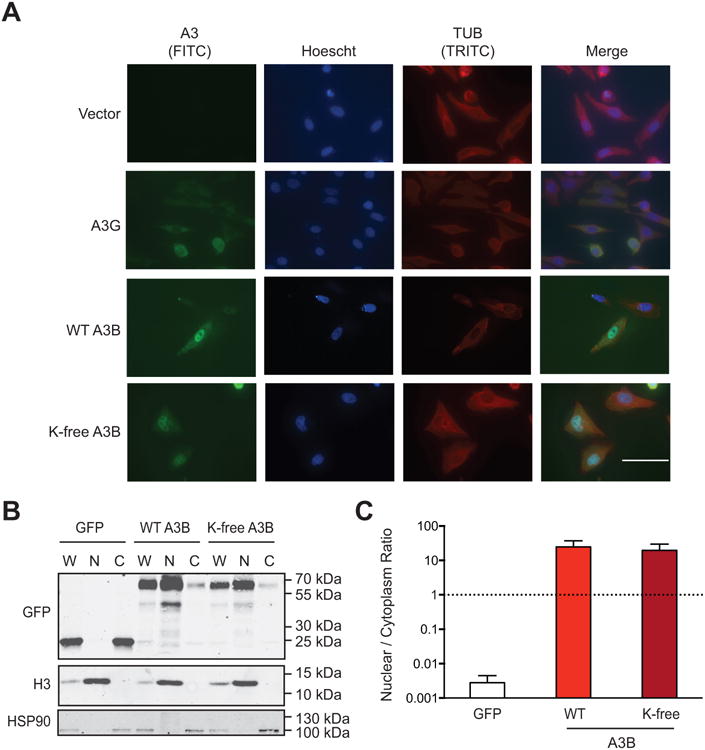
(A) Representative images of HeLa cells expressing an empty vector, A3G (untagged), wild-type A3B (untagged), or K-free A3B (untagged). The rabbit mAb 5210-87-13 was used to detect each A3 protein through a nearly identical C-terminal epitope (anti-rabbit IgG-FITC as secondary). 100 μM scale applies to all images.
(B) Immunoblot of 293T whole cell (W), nuclear (N), and cytoplasmic (C) extracts expressing eGFP, wild-type A3B-eGFP, or K-free A3B-eGFP. Fractionation controls are anti-Histone H3 for nuclear components, and anti-HSP90 for cytoplasmic components.
(C) Quantification of the fractionation data from panel B and 2 biologically independent experiments (not shown). Each histogram bar reports the mean +/- SEM nuclear to cytoplasmic ratio for the indicated constructs.
A subcellular fractionation procedure was used to confirm the K-free A3B nuclear localization result (Fig. 5B-C). 293T cells were transfected with relevant A3 constructs, processed into whole cell, cytoplasmic, and nuclear extracts, and subjected to analysis by immunoblotting with HSP90 and Histone H3 as cytoplasmic and nuclear controls, respectively. Representative immunoblots revealed no difference between the nuclear localization of wild-type and K-free A3B (Fig. 5B). Immunoblot signals were quantified from 3 biologically independent experiments and, again, the nuclear localization activities of wild-type and K-free A3B were indistinguishable (Fig. 5C). Independent experiments with HeLa cells yielded similar data (not shown). Overall, these microscopy and subcellular fractionation results combined to show that lysine side chains and modifications are unlikely to be an essential part of the nuclear localization mechanism of A3B.
Discussion
Our results indicate that neither lysine residues nor lysine post-translational modifications are required for several activities of A3B. Single-stranded DNA cytosine deamination is the hallmark activity of the APOBEC family, and titration experiments showed no difference in catalytic activity between wild-type and K-free A3B. Restriction of Vif-deficient HIV-1 is to-date the most sensitive bioassay for APOBEC activity, as it provides a quantitative read-out of several enzyme activities including encapsidation (likely RNA binding activity) and viral cDNA deamination. A3B catalytic activity is largely required for restriction of HIV-1 produced using 293 cells (regardless of Vif), as shown by several groups (Bogerd et al., 2006; Doehle et al., 2005; Lackey et al., 2012; Ooms et al., 2012). This result parallels prior reports for A3G in which the bulk of HIV-1 restriction activity is dependent upon catalytic activity and some deaminase-independent effects are observed at higher expression levels (Browne et al., 2009; Miyagi et al., 2007; Newman et al., 2005; Schumacher et al., 2008). However, despite observing clear deamination-dependent effects, no measurable differences were seen between the Vif-deficient HIV-1 restriction activities of wild-type A3B and K-free A3B. In addition, even steady-state nuclear localization, which is the only APOBEC activity unique to A3B, was unaffected by changing all 10 lysines into arginines. We conclude that the DNA cytosine deamination, HIV-1 restriction, and nuclear localization activities of human A3B do not require lysines or lysine post-translational modifications. However, we note that lysine post-translational modification could still play an important role in A3B regulation in other cell types, or in normal or cancerous cells in vivo.
The lysine residues of human A3B are required for SIV Vif-mediated degradation evidenced by MG132 blocking degradation of wild-type A3B and the 10 lysine to arginine substitutions rendering the protein resistant to degradation. These results indicate that, despite the cross-species comparison, SIV Vif is still using the canonical poly-ubiquitination and proteasome degradation pathway to purge cells of human A3B. These results mirror prior studies on human A3G and A3F, where 20 and 19 lysines were simultaneously converted to arginine, and the K-less variants resisted Vif-mediated degradation (Albin et al., 2013; Dang et al., 2008; Shao et al., 2010). Since A3B may very well restrict the replication of other viruses, including HBV, HPV, and HTLV (Ahasan et al., 2015; Lucifora et al., 2014; Ooms et al., 2012; Starrett et al., 2016; Starrett et al., 2017; Verhalen et al., 2016; Vieira et al., 2014; Warren et al., 2015), it is likely that additional A3B counteraction strategies exist and that the functional K-free construct described here may be useful as a mechanistic probe. The K-free enzyme may also be useful for future studies investigating A3B cellular interactions.
Materials and Methods
Structures and modeling
The A3B C-terminal domain x-ray structure depicted in Fig. 1 is derived from pdb 5CQD (Shi et al., 2015). Phyre2 was used to draw upon existing pdb information to develop a composite structural model of the N-terminal domain (Kelley et al., 2015).
Plasmids
The untagged wild-type and eGFP tagged APOBEC3B (NM_004900.4) constructs were made by changing threonine 146 to lysine using a previously described untagged APOBEC3B construct [UniProtKB/Swiss-Prot: Q9UH17.1; described in (Hultquist et al., 2011)] as a template for site-directed mutagenesis. The K-free A3B construct was ordered as a G-Block from IDT where all 10 lysine residues were mutated to arginines and subsequently cloned into an untagged expression vector pcDNA 3.1 (+) or into pEGFP-N3 (Clontech). The untagged A3B catalytic mutant (CM) construct encoding two glutamate to alanine amino acid substitutions (E68A/E255A) was PCR amplified from existing pEGFP-N3 (Clontech) plasmids described previously (Burns et al., 2013; Hultquist et al., 2011; Lackey et al., 2013; Land et al., 2015) using primers NNNAAGCTTACCGCCATGAATCCACAGATC and NNNNGCGGCCGCTCAGTTTCCCTGATTCTGGAGAATGG. PCR products were then digested with restriction enzymes HindIII-HF and NotI-HF in CutSmart Buffer (NEB #R3104S and #R3189S respectively). After digestion, the backbone and insert fragments were gel extracted using the Epoch GenCatch Gel Extraction Kit protocol (Epoch #2160250). The backbone and insert fragments were ligated with T4 DNA ligase (NEB #M0202S). Plasmid DNA was isolated utilizing the Thermo Fisher Scientific GeneJet Plasmid Miniprep Kit protocol (Thermo #K0503). The DNA sequences of all plasmid constructs were confirmed by Sanger sequencing using sequencing primers CGTGTACGGTGGGAGGTCTA, GCCCGCGTGACGATCATGGACTATGA, and GGAAAGCAAAATCTCAGGCTTTGAGG and alignment with Sequencher. Larger plasmid preparations utilized the Thermo Fisher Scientific PureLink HiPure Plasmid Maxiprep Kit (Thermo #K210007).
Cell culture
HeLa and 293T cells were grown in HyClone RPMI 1640 media supplemented with 10% heat inactivated fetal bovine serum, penicillin and streptomycin (100U) on adhesion cell culture plates and incubated at 37°C (5% CO2). Semi-confluent cells were transfected using the Mirus TransIT- LT1 transfection reagent protocol (Mirus #MIR 3205). Cells were harvested 48 hrs after transfection by disrupting adhesion with 0.05% Trypsin-EDTA (Gibco, #25300-054) diluted 1:10 in PBS-EDTA.
Gel electrophoresis and immunoblotting
Protein samples were boiled in 6× Laemmli sample buffer and incubated at 98°C for 10 min. Proteins were separated by SDS-PAGE (4% stacking, 12.5% resolving) in SDS-PAGE running buffer at 90V for 20 min and 150V for 90 min. The proteins were transferred to a Licor PVDF membrane at 90 V for 2 hrs. Membranes were blocked with 5% powdered milk in tween 20-PBS (PBST) followed by incubation for 16-24 hrs at 4°C with primary antibodies: mouse α TUBULIN (Sigma-Aldrich B512); rabbit α HA (Cell Signaling, #3724); mouse α HSP90 (Thermo Fisher Scientific, PA3-012); rabbit α H3F3A (Thermo Fisher Scientific, E.960.2); rabbit α APOBEC3B (RSH #10.87.13 (Leonard et al., 2015)). The membranes were washed in PBST for 10-30 min and incubated with a secondary antibody: TUBULIN - 800 α mouse IgG (Licor 926-32350); HSP90 - 680 α Mouse IgG (Licor 926-68020); HA and HISTONE3 - 800 α Rabbit IgG (Licor 926-32211); APOBEC3B - HRP α Rabbit IgG (Jackson Immunodiagnostics, # 111035144) for 45 min at room temperature. The membranes were washed with PBST for 30 min and antibody detection was performed using the Licor Fc for the fluorescent antibodies and the Denville HyGLO Chemiluminescent HRP reagent.
DNA cytosine deaminase oligo cleavage assay
5.0×105 293T cells were transiently transfected with 500 ng of DNA in a 6-well plate and harvested 48 hrs later. Pellets from transfected cells were resuspended in 500uL HED buffer (25 mM HEPES, 5mM, 10% glycerol, 1mM DTT and 1 EDTA-free Proteasome inhibitor tablet) and placed on ice. The samples were sonicated at the lowest setting with the Misonix Sonicator system for three 5-second pulses and kept on ice between pulses. They were centrifuged at 13,000 rpm for 20 min at 4°C and the supernatant was transferred to cold fresh tubes. Deamination reactions were prepared with 25 μl RNase A, 2 μl 10× UDG buffer, .25 μl UDG, 1 μl 4uM ATTATTATTATTCTAATGGATTTATTTATTTATTTATTTATTT-fluorescein, and 16.5uL cleared lysate. Reactions were incubated at 37°C for 1 hour and subsequently treated with 2uL 1M NaOH and incubated at 98°C for 10 min. 22 μl 2× formamide buffer was added to each reaction and they were heated at 98°C for 5 min. 10 μl of each sample was loaded onto a 15% Urea-TBE gel in 1× TBE buffer and electrophoresed at 150V for 60 min. Oligos were imaged using fluorescence detection via a Typhoon FLA 7000 fluorescent imager (GE Healthcare Life Sciences) laser scanner. To quantify the percent of substrate cleavage, ImageJ was used to perform densitometry from 3 independent deamination reactions and plotted versus an increasing titration of lysate (Burns et al., 2013; Carpenter et al., 2012; Leonard et al., 2013; Shi et al., 2015; Vieira et al., 2014).
Subcellular localization
5.0×105 HeLa cells were transiently transfected with 500 ng of plasmid. Protein was harvested at 48 hrs as described in “REAP: A two-minute cell fractionation method” (Suzuki et al., 2010). After harvest cells were washed with 1 mL of phosphate buffered saline (PBS), 300 μl of the whole cell lysate was placed in a cold 1.5 mL microfuge tube and kept on ice. The remaining 600 μl was pulse spun to maximum speed of 14,800 rpm for 10 seconds. 300 μl of the supernatant was placed in a cold 1.5 mL microfuge tube as the cytosolic fraction and kept on ice; the remaining supernatant was discarded. The nuclear fraction was resuspended in 180 μl 1× Laemmli sample buffer. 100 μl of 5× Laemmli sample buffer was added to the whole cell and cytosolic fractions. All fractions were sonicated twice for 5 seconds at the lowest setting of the Misonix Sonicator system and kept on ice in between sonications. All samples were incubated at 98°C for 10 min and centrifuged at high speed for 10 seconds after cooling. The fractions were analyzed by gel electrophoresis and immunoblotting.
Immunofluorescent microscopy experiments
2.5×104 HeLa cells were plated into 6-well plates containing glass coverslips, transfected with 500 ng of plasmid DNA at 80% confluency, and incubated at 37°C for 48 hrs. Cells were then washed 3 times with PBS and fixed with 4% paraformaldehyde in PBS. Cells were washed again in PBS and subsequently incubated with primary antibodies rabbit α A3B [Harris lab 5210-87-13 (Leonard et al., 2015)] (1:50) and mouse α TUBULIN (Sigma-Aldrich, #B512) (1:500) rocking at 4°C overnight. The cells were washed 5 times with PBS and incubated with secondary antibodies rabbit αFITC (Abcam, #ab6717) (1:500) and mouse αTRITC (Abcam, #ab6786) (1:500) for 1 hour at room temperature and washed in PBS. The glass coverslips were extracted and excess liquid was removed. 15 μl of 50% glycerol with .01% DAPI was placed on the center of a glass microscope slide and the cell covered surface of the coverslip was placed on the glycerol and sealed with clear nail polish. Imaging was performed using a Nikon Inverted TiE Deconvolution Microscope System [based on protocols described in (Starrett et al., 2016)].
SIV Vif-mediated degradation of A3B
5.0×105 293T cells were co-transfected with pcDNA3.1-derived constructs encoding wild-type A3B, K-free A3B, A3G, or empty vector and SIV Vif or additional empty vector. After 48 hrs incubation, cells were treated with 10 μM MG132 (Stem Cell Technologies), or mock-treated as a negative control, for an additional 18 hrs, prior to harvesting for immunoblot experiments [based on protocol described in (Land et al., 2015)].
HIV-1 single cycle experiments
Single cycle experiments were performed as described (Hultquist et al., 2011). Infectious particles were produced by cotransfecting 5.0×105 293T cells with an HIV-1IIIB A200C molecular clone (pIIIB) and an expression construct for A3G, A3B, K-free A3B, or vector control. Supernatants containing viruses were harvested after 48 hrs, filtered (0.45 μm), and used to infect CEM-GFP reporter cells. After an additional 48 hrs incubation, infectivity was quantified by flow cytometry and reported as the percentage of GFP-positive cells. 1 mL of the virus-containing supernatants was concentrated by centrifugation (16,000g, 2 hrs, RT) and used for immunoblotting.
Acknowledgments
We thank Nadine Shaban for modeling A3Bntd, Matthew Jarvis for assistance with statistics, and Jiayi Wang for helpful comments on the manuscript. This work was supported by NIAID R37 AI064046 and NCI R21 CA206309. A.M.M. and C.M.R. received salary support from NIAID T32-AI83196, G.J.S. from a NSF Graduate Research Fellowship 00039202, and B.D.A. from NIAID F31 AI116305. R.S.H. is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished McKnight University Professor, and an Investigator of the Howard Hughes Medical Institute. R.S.H. is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc.
Footnotes
The other authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahasan MM, Wakae K, Wang Z, Kitamura K, Liu G, Koura M, Imayasu M, Sakamoto N, Hanaoka K, Nakamura M, Kyo S, Kondo S, Fujiwara H, Yoshizaki T, Mori S, Kukimoto I, Muramatsu M. APOBEC3A and 3C decrease human papillomavirus 16 pseudovirion infectivity. Biochem Biophys Res Commun. 2015;457:295–299. doi: 10.1016/j.bbrc.2014.12.103. [DOI] [PubMed] [Google Scholar]
- Albin JS, Anderson JS, Johnson JR, Harjes E, Matsuo H, Krogan NJ, Harris RS. Dispersed sites of HIV Vif-dependent polyubiquitination in the DNA deaminase APOBEC3F. J Mol Biol. 2013;425:1172–1182. doi: 10.1016/j.jmb.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binka M, Ooms M, Steward M, Simon V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J Virol. 2012;86:49–59. doi: 10.1128/JVI.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne EP, Allers C, Landau NR. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology. 2009;387:313–321. doi: 10.1016/j.virol.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon IJ, Byeon CH, Wu T, Mitra M, Singer D, Levin JG, Gronenborn AM. Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry. 2016;55:2944–2959. doi: 10.1021/acs.biochem.6b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SM, Bohn MF, Schiffer CA, Brown WL, Harris RS. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J Biol Chem. 2012;287:34801–34808. doi: 10.1074/jbc.M112.385161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari FV, Mason WS, Seeger C. Virology. Comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA”. Science. 2014;344:1237. doi: 10.1126/science.1254082. [DOI] [PubMed] [Google Scholar]
- Dang Y, Siew LM, Zheng YH. APOBEC3G is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J Biol Chem. 2008;283:13124–13131. doi: 10.1074/jbc.M708728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Robek MD. Cytidine deamination and cccDNA degradation: A new approach for curing HBV? Hepatology. 2014;60:2118–2121. doi: 10.1002/hep.27386. [DOI] [PubMed] [Google Scholar]
- Doehle BP, Schäfer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Ezzikouri S, Kitab B, Rebbani K, Marchio A, Wain-Hobson S, Dejean A, Vartanian JP, Pineau P, Benjelloun S. Polymorphic APOBEC3 modulates chronic hepatitis B in Moroccan population. J Viral Hepat. 2013;20:678–686. doi: 10.1111/jvh.12042. [DOI] [PubMed] [Google Scholar]
- Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479-480:131–145. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS. APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol. 2012;419:301–314. doi: 10.1016/j.jmb.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle. 2013;12:762–772. doi: 10.4161/cc.23713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AM, Wang J, Law EK, Aberle R, Kirmaier A, Krupp A, Johnson WE, Harris RS. Degradation of the cancer genomic DNA deaminase APOBEC3B by SIV Vif. Oncotarget. 2015;6:39969–39979. doi: 10.18632/oncotarget.5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, Li Y, Zhang Y, Maurer MJ, Oberg AL, Cunningham JM, Shridhar V, Bell DA, April C, Bentley D, Bibikova M, Cheetham RK, Fan JB, Grocock R, Humphray S, Kingsbury Z, Peden J, Chien J, Swisher EM, Hartmann LC, Kalli KR, Goode EL, Sicotte H, Kaufmann SH, Harris RS. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73:7222–7231. doi: 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, Burns MB, McDougle RM, Parker PJ, Brown WL, Harris RS. The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015;75:4538–4547. doi: 10.1158/0008-5472.CAN-15-2171-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Hüser N, Durantel D, Liang TJ, Münk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Bieniasz PD. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb Perspect Med. 2012;2:a006940. doi: 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Emerman M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Meier MA, Suslov A, Ketterer S, Heim MH, Wieland SF. Hepatitis B virus covalently closed circular DNA homeostasis is independent of the lymphotoxin pathway during chronic HBV infection. J Viral Hepat. 2017 doi: 10.1111/jvh.12689. [DOI] [PubMed] [Google Scholar]
- Miyagi E, Opi S, Takeuchi H, Khan M, Goila-Gaur R, Kao S, Strebel K. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J Virol. 2007;81:13346–13353. doi: 10.1128/JVI.01361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Takeuchi T, Ishii Y, Yugawa T, Kiyono T, Nishina H, Kukimoto I. Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J Virol. 2017;91 doi: 10.1128/JVI.02413-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- Ooms M, Krikoni A, Kress AK, Simon V, Münk C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J Virol. 2012;86:6097–6108. doi: 10.1128/JVI.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasetyo AA, Sariyatun R, Reviono, Sari Y, Hudiyono, Haryati S, Adnan ZA, Hartono, Kageyama S. The APOBEC3B deletion polymorphism is associated with prevalence of hepatitis B virus, hepatitis C virus, Torque Teno virus, and Toxoplasma gondii co-infection among HIV-infected individuals. J Clin Virol. 2015;70:67–71. doi: 10.1016/j.jcv.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Schumacher AJ, Haché G, Macduff DA, Brown WL, Harris RS. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J Virol. 2008;82:2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q, Wang Y, Hildreth JE, Liu B. Polyubiquitination of APOBEC3G is essential for its degradation by HIV-1 Vif. J Virol. 2010;84:4840–4844. doi: 10.1128/JVI.01911-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K, Carpenter MA, Banerjee S, Shaban NM, Kurahashi K, Salamango DJ, McCann JL, Starrett GJ, Duffy JV, Demir Ö, Amaro RE, Harki DA, Harris RS, Aihara H. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat Struct Mol Biol. 2017;24:131–139. doi: 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K, Carpenter MA, Kurahashi K, Harris RS, Aihara H. Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J Biol Chem. 2015;290:28120–28130. doi: 10.1074/jbc.M115.679951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomai A, Rice CM. Virology. Getting rid of a persistent troublemaker to cure hepatitis. Science. 2014;343:1212–1213. doi: 10.1126/science.1252186. [DOI] [PubMed] [Google Scholar]
- Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol. 2015;16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, Carpenter MA, Harris RS. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun. 2016;7:12918. doi: 10.1038/ncomms12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starrett GJ, Marcelus C, Cantalupo PG, Katz JP, Cheng J, Akagi K, Thakuria M, Rabinowits G, Wang LC, Symer DE, Pipas JM, Harris RS, DeCaprio JA. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. MBio. 2017;8 doi: 10.1128/mBio.02079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006;281:16837–16841. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K. REAP: A two minute cell fractionation method. BMC Res Notes. 2010;3:294. doi: 10.1186/1756-0500-3-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhalen B, Starrett GJ, Harris RS, Jiang M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J Virol. 2016;90:6379–6386. doi: 10.1128/JVI.00771-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, Lee D, Soares MA, Lambert PF, Howley PM, Harris RS. 3Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio. 2014;5 doi: 10.1128/mBio.02234-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, Lambert PF, Santiago ML, Pyeon D. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol. 2015;89:688–702. doi: 10.1128/JVI.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Lucifora J, Reisinger F, Heikenwalder M, Protzer U. Virology. Response to Comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA”. Science. 2014;344:1237. doi: 10.1126/science.1254083. [DOI] [PubMed] [Google Scholar]