

Abstract
Pralatrexate is a novel antifolate approved in the USA for the treatment of relapsed or refractory peripheral T‐cell lymphoma. To assess its safety, efficacy, and pharmacokinetics in Japanese patients with this disease, we undertook a phase I/II study. Pralatrexate was given i.v. weekly for 6 weeks of a 7‐week cycle. All patients received concurrent vitamin B12 and folic acid. In phase I, three patients received pralatrexate 30 mg/m2 and none experienced a dose‐limiting toxicity. In phase II, we treated 22 additional patients with that dose. The median number of treatment cycles was 1 (range, 1–9). Nine of 20 evaluable patients (45%) achieved an objective response by central review, including two complete responses. All responses occurred within the first treatment cycle. At the time of data cut‐off, median progression‐free survival was 150 days. Median overall survival was not reached. In the total population, the most commonly reported adverse events included mucositis (88%), thrombocytopenia (68%), liver function test abnormality (64%), anemia (60%), and lymphopenia (56%). Grade 3/4 adverse events included lymphopenia (52%), thrombocytopenia (40%), leukopenia (28%), neutropenia (24%), anemia (20%), and mucositis (20%). The pharmacokinetic profile showed no drug accumulation with repeat dosing. These results indicate that pralatrexate is generally well tolerated and effective in Japanese patients with relapsed or refractory peripheral T‐cell lymphoma. This trial was registered with ClinicalTrials.gov (NCT02013362).
Keywords: Clinical trial, folic acid antagonists, Japanese, peripheral T‐cell lymphoma, pralatrexate
Peripheral T‐cell lymphoma, an aggressive, heterogeneous disease that includes many subtypes of mature T‐ and natural killer‐cell neoplasms, accounts for 5%–10% of non‐Hodgkin lymphomas in North America and Europe.1 The incidence is higher in Asia, where 15%–20% of lymphomas are PTCL or NKTCL.1, 2 Most patients are initially treated with anthracycline‐based therapy, which produces CR rates of 50%–70%, but long‐term survival is poor.1, 3 The disease is characterized by multiple relapses, and primary refractory disease is not uncommon.4 Median survival after first relapse is historically less than 6 months.4 Although there are several guideline‐recommended, single‐agent therapies available for the treatment of relapsed or refractory PTCL in the USA, there are limited options in Europe and Asia.5 In Japan, mogamulizumab, a humanized anti‐CCR4 mAb, is indicated for treatment of relapsed or refractory CCR4‐positive PTCL, and brentuximab vedotin, a CD30‐directed antibody–drug conjugate, is indicated for treatment of relapsed or refractory CD30‐positive ALCL. These are limited in use to patients shown to be CCR4‐positive or CD30‐positive. Recently, forodesine, a purine nucleoside phosphorylase inhibitor, was approved for the treatment of relapsed or refractory PTCL in Japan. The ORR was 22% in patients with relapsed disease, but its effectiveness for refractory disease is unknown.6 Thus, new single‐agent therapies that are effective for both relapsed and refractory PTCL are urgently needed.
Pralatrexate, an antifolate that competitively inhibits dihydrofolate reductase, was the first drug approved in the USA for the treatment of relapsed or refractory PTCL.7, 8 Designed to have high affinity for the reduced folate carrier, a protein that is highly expressed by malignant tissues, and folylpolyglutamate synthase, which converts pralatrexate to its polyglutamated form in the cytosol and increases intracellular retention, pralatrexate has shown superior preclinical activity over other antifolates.9, 10, 11, 12 A single‐arm, open‐label, international phase II study, carried out in North America and Europe (the PROPEL study), reported an ORR of 29%, including 12 CRs (11%), among 109 evaluable patients treated with pralatrexate 30 mg/m2 weekly for 6 weeks of a 7‐week treatment cycle.8 Responses were durable regardless of age, underlying histology, or prior therapy, leading to accelerated approval in 2009 in the USA. Given the need for additional therapies in Japan and the unknown safety and efficacy of pralatrexate in this patient population, the current phase I/II study was undertaken to evaluate the safety, tolerability, efficacy, and pharmacokinetics of the FDA‐approved dosing regimen given with concurrent vitamin B12 and folic acid supplementation to Japanese patients with relapsed or refractory PTCL.
Materials and Methods
Study design and patients
This was an open‐label, non‐randomized, multicenter phase I/II clinical trial. The phase I portion was designed to determine the safety, tolerability, recommended dose, and pharmacokinetics of pralatrexate with concurrent vitamin B12 and folic acid supplementation in Japanese patients with relapsed or refractory PTCL. The phase II portion was designed to evaluate the efficacy, safety, and pharmacokinetics of the recommended dose identified in phase I. The study was carried out in accordance with the Declaration of Helsinki and Ordinance on Good Clinical Practice. The protocol was reviewed and approved by the institutional review board of each participating center, and all patients gave written informed consent.
Eligible patients were at least 20 years of age and had histologically confirmed PTCL with relapsed or refractory disease after at least one prior antitumor therapy (not including systemic corticosteroid monotherapy). According to the 2008 WHO classification,13 PTCL was defined as: PTCL‐NOS; AITL; ALCL (ALK‐positive or ALK‐negative); extranodal NKTCL, nasal type; enteropathy‐associated TCL; hepatosplenic TCL; subcutaneous panniculitis‐like TCL; or transformed mycosis fungoides. Peripheral T‐cell lymphoma diagnosis for each patient was confirmed by central review. All patients had to have measurable disease >1.5 cm in diameter by CT image according to International Workshop Criteria14 and have an ECOG PS of 0–2. Other eligibility criteria included adequate hematologic, hepatic, and renal function (neutrophil count ≥1000/mm3 without granulocyte colony‐stimulating factor; platelet count ≥100 000/mm3 without blood transfusion; aspartate aminotransferase and alanine aminotransferase <2.5 × upper limit of normal, or <5 × upper limit of normal if hepatic involvement by lymphoma; total bilirubin ≤1.5 mg/dL; and creatinine clearance ≥50 mL/min).
Exclusion criteria included: prior receipt of pralatrexate; receipt of chemotherapy, high‐dose systemic corticosteroid therapy (>10 mg/day prednisolone or equivalent), radiation therapy, phototherapy, or electron beam therapy within the prior 21 days; receipt of antibody therapy or autologous stem cell transplantation within the prior 100 days; history of allogeneic hematopoietic stem cell transplantation; active or history of brain metastasis or central nervous system lesion(s); active concurrent cancers or history of other malignant neoplasm within the last 5 years; severe cardiovascular disease; uncontrolled hypertension or diabetes mellitus despite an adequate therapy; positive CMV or HBV surface antigen test; positive hepatitis C virus or HIV antibody test; positive HBV surface or HBV core antibody test with results above the detection sensitivity of the HBV‐DNA quantitative test; infectious disease currently requiring i.v. antibiotics or antifungal or antiviral treatment; interstitial pneumonia or pulmonary fibrosis; and pregnant or lactating patients or patients of child‐bearing potential who do not intend to use appropriate contraception.
Study design
Pralatrexate (Mundipharma, Tokyo, Japan) was given i.v. over 3–5 min once weekly for 6 weeks, followed by a 1‐week rest period, in repeating 7‐week cycles. Treatment continued until disease progression, initiation of new PTCL therapy or a prohibited concomitant drug or non‐pharmacologic therapy, intolerable AE at the minimum dose (20 mg/m2), drug omissions due to AE(s) for three consecutive visits, pregnancy, withdrawal of consent, or investigator decision. Vitamin supplementation began at least 10 days prior to the first dose of pralatrexate. Vitamin B12 1.0 mg was given i.m. every 8–10 weeks. Folic acid 1.0–1.25 mg was given orally daily and continued for 30 days after the last dose of pralatrexate. Recommended prevention and treatment of mucositis included professional oral care prior to initiation of study treatment, with ongoing consultation during study treatment, self‐care instructions, cryotherapy (oral cooling with ice prior to pralatrexate treatment and for up to 30 min after infusion), and use of oral moisturizing gels. Prophylaxis against Pneumocystis jiroveci pneumonia with sulfamethoxazole–trimethoprim and/or varicella zoster virus infection with an appropriate antiviral drug was permitted at the discretion of the investigator.
Using the 3 + 3 design, during phase I of the study, three patients were initially treated with pralatrexate 30 mg/m2 (Cohort 1) based on the dose approved in the USA.15 If none of the patients experienced a DLT, the trial was to proceed to phase II at this dose level. If one or two patients experienced a DLT, an additional three patients were to be enrolled to Cohort 1. If three or more patients experienced a DLT, Cohort 2 would open at a reduced dose of 20 mg/m2 to evaluate the safety and tolerability, but the trial would not proceed to phase II because the sample size would no longer be adequate. Dose‐limiting toxicities were defined as the following events that were related to pralatrexate during the first treatment cycle: grade 3/4 non‐hematologic toxicity (except nausea, vomiting, and diarrhea); grade 3 nausea, vomiting, or diarrhea persisting for 7 or more days; grade 4 nausea, vomiting, or diarrhea; grade 3/4 febrile neutropenia; grade 4 neutropenia persisting for 7 or more days; grade 4 thrombocytopenia persisting for 7 or more days or thrombocytopenia requiring platelet transfusion; and any AE necessitating omission of more than two doses of pralatrexate. Adverse events were assessed using the NCI's Common Terminology Criteria for Adverse Events, version 3.0.
In both phases, treatment omissions and dose reductions were mandated by protocol for the development of grade ≥2 oral mucositis, grade ≥3 non‐hematologic toxicity (other than oral mucositis), platelet count <50 000/mm3, and neutrophil count <1000/mm3 (Table S1).
Study assessment
In phase I, DLT assessment and the recommended dose were confirmed by the Efficacy and Safety Evaluation Committee. In phase II, the primary end‐point was ORR based on CT image evaluation by central review. Secondary end‐points included ORR based on CT image evaluation by the investigator, ORR based on FDG‐PET/CT review by central review and by investigator, OS, PFS, time to response, and duration of response. An exploratory analysis included ORR within clinically relevant subgroups, including age, sex, ECOG PS, histology, stage, number of prior therapies, response to and time from most recent therapy, and at‐baseline LDH level. Response was assessed by CT and FDG‐PET/CT at week 7 of odd‐numbered cycles according to International Workshop Criteria and Revised Response Criteria for Malignant Lymphoma, respectively.14, 16 Time‐to‐event analyses were carried out using the Kaplan–Meier method.
Statistical considerations
Based on an ORR of 29% (95% CI, 21%–39%) reported in the previous international phase II study (PROPEL), the expected ORR in phase II of this trial was set at 30%. The sample size was estimated as 18 patients to provide 80% statistical power to detect that an observed ORR was above an alternative threshold of 10%, with a one‐sided alpha error of 0.1 by means of binomial testing. The target enrollment was set at 20 patients to ensure at least 18 evaluable patients for the efficacy analysis. The protocol specified that the data cut‐off for an efficacy and safety analysis would occur when all patients in phase II of the study had completed three treatment cycles. Efficacy was evaluated in the full analysis set consisting of patients who received at least one dose of pralatrexate, had a post baseline efficacy assessment and met major eligibility criteria, including PTCL confirmed by central review.
Pharmacokinetic analysis
Plasma and urine samples were collected from the first six patients at the following time points: plasma samples at before, immediately after, and 0.5, 1, 3, 5, 8, 12, 24, 48, and 72 h after each pralatrexate administration on visit 1 and visit 6 during cycle 1; urine samples at before, 0–24, 24–48, and 48–72 h after pralatrexate administration on cycle 1, visit 1. Plasma and urine pralatrexate concentrations were measured using the liquid chromatography–tandem mass spectrometry method. Because pralatrexate is a 1:1 racemic mixture of stereoisomers at the C10 position, the concentrations of both pralatrexate‐10a (the S‐stereoisomer) and pralatrexate‐10b (the R‐stereoisomer) were included. Pharmacokinetic parameters were estimated using non‐compartmental methods with Phoenix WinNonlin 6.4 (Certara, Tokyo, Japan).
Results
Patients
A total of 25 patients (Cohort 1: phase I, n = 3; phase II, n = 22) were enrolled to the study from March 2014 to September 2015 and received at least one dose of pralatrexate. None of the phase I patients discontinued treatment during the DLT evaluation period, and all three consented to continue receiving the study drug after the DLT evaluation period. All 25 patients were included in safety analyses. Two patients in phase II were excluded from efficacy analyses due to lack of centrally confirmed PTCL. None of the phase I patients and four of the phase II patients were still receiving treatment as of the data cut‐off date of December 28, 2015.
Table 1 shows the baseline characteristics of the enrolled patients. All three patients in phase I were male and ranged in age from 54 to 64 years. They were heavily pretreated (median of 6 prior systemic therapies; range, 5–8), and only one had responded to the most recent prior therapy. Across the total population, most patients were male (68%). The median age was 71 years (range, 42–83 years), and most patients were elderly (age ≥65 years, 72%). The most common histologic diagnosis was PTCL‐NOS (48%) followed by AITL (36%) and ALCL, ALK‐negative (8%). The median number of prior therapies (not including systemic corticosteroid monotherapy) was 3 (range, 1–8). All patients had received prior chemotherapy and two patients in phase II had undergone prior autologous stem cell transplantation. Almost half of patients did not have evidence of response to the most recent therapy (56%), and were enrolled to the study within 3 months after the end of the prior therapy (52%). All patients had a good performance status (ECOG PS 0–1).
Table 1.
Baseline characteristics of Japanese patients with relapsed/refractory peripheral T‐cell lymphoma enrolled in a phase I/II study of pralatrexate
| Characteristic | Phase I (n = 3) | Phase II (n = 22) | Total (N = 25) |
|---|---|---|---|
| Age, years | |||
| Median (range) | 56 (54–64) | 71.5 (42–83) | 71 (42–83) |
| <65, n (%) | 3 (100) | 4 (18) | 7 (28) |
| ≥65, n (%) | 0 (0) | 18 (82) | 18 (72) |
| Sex, n (%) | |||
| Male | 3 (100) | 14 (64) | 17 (68) |
| Female | 0 (0) | 8 (36) | 8 (32) |
| ECOG PS, n (%) | |||
| 0 | 1 (33) | 11 (50) | 12 (48) |
| 1 | 2 (67) | 11 (50) | 13 (52) |
| 2 | 0 (0) | 0 (0) | 0 (0) |
| Histology by central review, n (%) | |||
| PTCL‐NOS | 2 (67) | 10 (45) | 12 (48) |
| AITL | 0 (0) | 9 (41) | 9 (36) |
| ALCL, ALK‐negative | 1 (33) | 1 (5) | 2 (8) |
| Others (not PTCL)† | 0 (0) | 2 (9) | 2 (8) |
| Ann Arbor Staging at baseline, n (%) | |||
| I | 0 (0) | 2 (9) | 2 (8) |
| II | 0 (0) | 0 (0) | 0 (0) |
| III | 2 (67) | 10 (45) | 12 (48) |
| IV | 1 (33) | 10 (45) | 11 (44) |
| Prior systemic therapies‡ | |||
| Median (range) | 6 (5–8) | 2 (1–8) | 3 (1–8) |
| 1, n (%) | 0 (0) | 4 (18) | 4 (16) |
| 2, n (%) | 0 (0) | 8 (36) | 8 (32) |
| 3, n (%) | 0 (0) | 2 (9) | 2 (8) |
| 4 or more, n (%) | 3 (100) | 8 (36) | 11 (44) |
| Response to most recent therapy, n (%) | |||
| CR | 1 (33) | 7 (32) | 8 (32) |
| PR | 0 (0) | 3 (14) | 3 (12) |
| SD | 1 (33) | 5 (23) | 6 (24) |
| PD | 0 (0) | 2 (9) | 2 (8) |
| UE/NE | 1 (33) | 5 (23) | 6 (24) |
| Time from most recent therapy, n (%) | |||
| <3 months | 2 (67) | 11 (50) | 13 (52) |
| ≥3 months | 1 (33) | 11 (50) | 12 (48) |
| LDH level at baseline, n (%) | |||
| ≤ULN | 2 (67) | 11 (50) | 13 (52) |
| >ULN | 1 (33) | 11 (50) | 12 (48) |
†Including one patient with classical Hodgkin lymphoma and one patient with malignant melanoma. ‡Not including systemic corticosteroid monotherapy. AITL, angioimmunoblastic T‐cell lymphoma; ALCL, anaplastic large‐cell lymphoma; ALK, anaplastic lymphoma kinase; CR, complete response; LDH, lactate dehydrogenase; NOS, not otherwise specified; PD, progressive disease; PR, partial response; PS, performance status; SD, stable disease; UE/NE, unestimable/not evaluable; ULN, upper limit of normal.
Tolerability and safety
No DLTs were observed among three patients in Cohort 1 of phase I, and the recommended dose was determined to be 30 mg/m2.
In phase I, the median number of cycles was 8 (range, 6–11). Two patients withdrew from the study for confirmation of progressive disease, and one withdrew due to development of AE in cycle 11. In phase II, the median number of cycles was 1 (range, 1–9). Eighteen of 22 patients withdrew from the study, most commonly for progressive disease (n = 10) or AEs (n = 5). One patient each withdrew for decision to institute new treatment for PTCL to maintain response, necessity to initiate a prohibited concomitant drug, and patient request. Table 2 further summarizes the extent of exposure to pralatrexate.
Table 2.
Exposure of Japanese patients with relapsed/refractory peripheral T‐cell lymphoma to pralatrexate in phase I/II study of pralatrexate
| Exposure | Phase I (n = 3) | Phase II (n = 22) | Total (N = 25) |
|---|---|---|---|
| Number of cycles | |||
| Median (range) | 8 (6–11) | 1 (1–9) | 2 (1–11) |
| Cumulative cycles, n (%) | |||
| 1 | 3 (100) | 22 (100) | 25 (100) |
| 2 | 3 (100) | 10 (45) | 13 (52) |
| 3 | 3 (100) | 8 (36) | 11 (44) |
| 4 | 3 (100) | 6 (27) | 9 (36) |
| 5 | 3 (100) | 3 (14) | 6 (24) |
| 6 | 3 (100) | 3 (14) | 6 (24) |
| 7 | 2 (67) | 3 (14) | 5 (20) |
| Number of doses | |||
| Median (range) | 34 (30–49) | 4 (1–48) | 7 (1–49) |
| Treatment duration, days | |||
| Median (range) | 347 (282–547) | 49 (12–445) | 81 (12–547) |
| Discontinued due to AE n (%) | 1 (33) | 5 (23) | 6 (24) |
| Dose reduction due to AE n (%) | 1 (33) | 6 (27) | 7 (28) |
| Dose omission due to AEn (%) | 2 (67) | 20 (91) | 22 (88) |
AE, adverse event.
In the total population, all patients experienced at least one AE. Table 3 shows the AEs that occurred in at least 20% of patients. The most commonly reported AEs were mucositis (88%), thrombocytopenia (68%), liver function test abnormality (64%), anemia (60%), and lymphopenia (56%). Twelve patients (48%) experienced 15 SAEs, including two cases of febrile neutropenia and two cases of Pneumocystis jiroveci pneumonia. Two patients who experienced P. jiroveci pneumonia did not receive prophylaxis against this disease. Other SAEs included one case each of sepsis, peritonitis, pneumonia, thrombocytopenia, dehydration, paraparesis, respiratory insufficiency, oral mucositis, drug eruption, back pain, and renal disability. Adverse events leading to treatment discontinuation were reported in six patients (24%), these were mostly SAEs. Dose reduction occurred in seven patients (28%) and the most common reason was oral mucositis.
Table 3.
Adverse events reported in at least 20% of Japanese patients with relapsed/refractory peripheral T‐cell lymphoma enrolled in a phase I/II study of pralatrexate
| Adverse event, n (%) | Safety population (N = 25) | |
|---|---|---|
| All grades | Grade 3/4 | |
| Mucositisa | 22 (88) | 5 (20) |
| Thrombocytopeniaa | 17 (68) | 10 (40) |
| Liver function test abnormala | 16 (64) | 3 (12) |
| Anemiaa | 15 (60) | 5 (20) |
| Lymphopeniaa | 14 (56) | 13 (52) |
| Neutropeniaa | 11 (44) | 6 (24) |
| Leukopeniaa | 11 (44) | 7 (28) |
| Fever | 11 (44) | 0 (0) |
| Malaise | 9 (36) | 0 (0) |
| Nasopharyngitis | 9 (36) | 0 (0) |
| Nausea | 7 (28) | 0 (0) |
| Rash | 7 (28) | 0 (0) |
| Vomiting | 7 (28) | 0 (0) |
| Diarrhea | 6 (24) | 0 (0) |
| Hypokalemiaa | 6 (24) | 4 (16) |
| Insomnia | 6 (24) | 0 (0) |
| Edemaa | 5 (20) | 0 (0) |
Including reclassified similar adverse events.
A single death occurred during phase II (4%) due to pneumonia in a 72‐year‐old male patient with an underlying pulmonary infiltrate by PTCL. The death was deemed unrelated to the study medication.
Efficacy
Table 4 shows treatment response for the evaluable phase II group based on CT image assessment by central review. The ORR was 45% (9/20) (90% CI, 26%–65%; P < 0.001), consisting of two CRs and seven PRs. All nine patients responded within the first treatment cycle, and four remained on therapy at the data cut‐off. In the investigator assessment, ORR was also 45%, although only one of the CRs was confirmed. Including FDG‐PET/CT image evaluation in the response criteria did not change the results significantly. Two of three patients in phase I responded to treatment (one CR, one PR, and one SD) based on CT assessment by central review.
Table 4.
Tumor response by central review in Japanese patients with relapsed/refractory peripheral T‐cell lymphoma treated with pralatrexate in phase II
| Response | Evaluable patients (n = 20) |
|---|---|
| ORR, n (%) | 9 (45) |
| CR | 2 (10) |
| PR | 7 (35) |
| SD | 4 (20) |
| PD | 7 (35) |
CR, complete response; ORR, objective response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Among the 20 evaluable phase II patients, median duration of response was not reached (range, 1–358 days; Fig. 1a). Median PFS was 150 days (95% CI, 43–183 days; Fig. 1b). Median OS was not reached (range, 41–470 days) after a median follow‐up of 181 days for censored cases, but the 12‐month OS rate was 61% (Fig. 1c).
Figure 1.
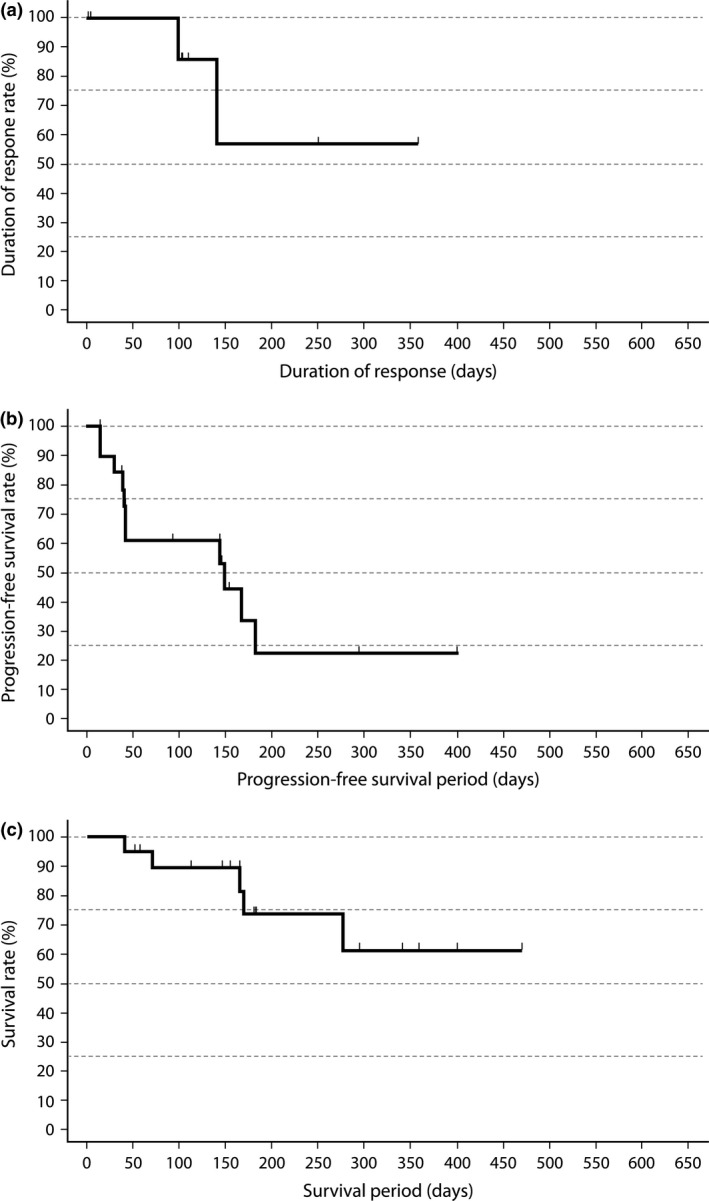
(a) Duration of response in evaluable Japanese patients with relapsed/refractory peripheral T‐cell lymphoma (PTCL) treated with pralatrexate in phase II (n = 9). (b) Progression‐free survival in evaluable Japanese patients with relapsed/refractory PTCL treated with pralatrexate in phase II (n = 20). (c) Overall survival in evaluable Japanese patients with relapsed/refractory PTCL treated with pralatrexate in phase II (n = 20).
There were no apparent differences in ORR within clinically relevant subgroups in an exploratory analysis across both treatment phases (n = 23). Responses were seen regardless of age, number of prior treatments, response to and time from last therapy, histology, stage, or LDH level (Table 5).
Table 5.
Exploratory analysis of objective response rate (central review) in a phase I/II study of pralatrexate in Japanese patients with relapsed or refractory peripheral T‐cell lymphoma (PTCL), according to baseline characteristics (n = 23)
| Characteristic | Number of cases | Number with response | % (90% CI) |
|---|---|---|---|
| Age, years | |||
| <65 | 7 | 5 | 71 (34–95) |
| ≥65 | 16 | 6 | 38 (18–61) |
| Sex | |||
| Male | 16 | 8 | 50 (28–72) |
| Female | 7 | 3 | 43 (13–77) |
| ECOG PS | |||
| 0 | 11 | 8 | 73 (44–92) |
| 1 | 12 | 3 | 25 (7–53) |
| Histology by central review | |||
| PTCL‐NOS | 12 | 6 | 50 (25–75) |
| AITL | 9 | 4 | 44 (17–75) |
| ALCL, ALK‐negative | 2 | 1 | 50 (3–97) |
| Ann Arbor Staging | |||
| I | 2 | 1 | 50 (3–97) |
| II | 0 | – | – |
| III | 11 | 7 | 64 (35–86) |
| IV | 10 | 3 | 30 (9–61) |
| Prior systemic therapiesa | |||
| 1 | 4 | 3 | 75 (25–99) |
| 2 | 7 | 4 | 57 (23–87) |
| 3 | 2 | 1 | 50 (3–97) |
| 4 or more | 10 | 3 | 30 (9–61) |
| Response to most recent therapy | |||
| CR/PR | 10 | 6 | 60 (30–85) |
| SD/PD | 7 | 3 | 43 (13–77) |
| UE/NE | 6 | 2 | 33 (6–73) |
| Time from most recent therapy | |||
| <3 months | 12 | 4 | 33 (12–61) |
| ≥3 months | 11 | 7 | 64 (35–86) |
| LDH level at base line | |||
| ≤ULN | 12 | 3 | 25 (7–53) |
| >ULN | 11 | 8 | 73 (44–92) |
Not including systemic corticosteroid monotherapy. AITL, angioimmunoblastic T‐cell lymphoma; ALCL, anaplastic large‐cell lymphoma; ALK, anaplastic lymphoma kinase; CI, confidence interval; CR, complete response; LDH, lactate dehydrogenase; NOS, not otherwise specified; PD, progressive disease; PR, partial response; PS, performance status; SD, stable disease; UE/NE, unestimable/not evaluable; ULN, upper limit of normal; –, not applicable.
Pharmacokinetics
Results for the pharmacokinetic analysis are shown in Table 6. For both stereoisomers, the plasma pralatrexate concentration reached its maximum directly after administration in cycle 1, visit 1, then promptly declined. The distribution half‐life ranged from 10 to 20 min. Similar pharmacokinetic results were obtained in cycle 1, visit 6, indicating a lack of drug accumulation with repeat dosing. Urinary excretion data revealed that 20%–30% of the dose is excreted in the urine within 24 h.
Table 6.
Pharmacokinetics of pralatrexate in Japanese patients with relapsed/refractory peripheral T‐cell lymphoma enrolled in phase I/II study
| C max, ng/mL | t max, min | AUCinf, ng·min/mL | CLtot, mL/min | Vdss, L | t 1/2 term, min | t 1/2,a min | |
|---|---|---|---|---|---|---|---|
| Plasma pralatrexate‐10a pharmacokinetic parameters (n = 6), cycle 1, visit 1 | |||||||
| Mean (SD) | 2945 (2035) | 6 (2) | 94 500 (68 300) | 367 (170) | 102 (138) | 1198 (954) | 12 (4) |
| Median (range) | 2335 (628.3–6531) | 7 (4–9) | 67 600 (47 700–228 000) | 375 (110–587) | 51.0 (24.4–382) | 1071 (175–2410) | 11 (7–19) |
| CV% | 69.1 | 31.5 | 72.3 | 46.4 | 136.2 | 79.6 | 34.4 |
| Plasma pralatrexate‐10a pharmacokinetic parameters (n = 5), cycle 1, visit 6 | |||||||
| Mean (SD) | 2729 (1093) | 6 (2) | 75 600 (27 500) | 379 (115) | 83.1 (58.2) | 1398 (855) | 10 (2) |
| Median (range) | 2285 (1731–4144) | 5 (5–9) | 60 600 (48 000–108 000) | 413 (260–521) | 88.6 (25.8–164) | 1744 (184–2355) | 11 (7–13) |
| CV% | 40.0 | 30.5 | 36.4 | 30.2 | 70.0 | 61.2 | 25.1 |
| Plasma pralatrexate‐10b pharmacokinetic parameters (n = 6), cycle 1, visit 1 | |||||||
| Mean (SD) | 3888 (2094) | 11 (11) | 178 000 (78 100) | 166 (59.6) | 30.0 (20.1) | 718 (435) | 20 (15) |
| Median (range) | 3485 (868.6–7190) | 7 (4–33) | 155 000 (115 000–324 000) | 175 (77.2–244) | 22.9 (18.0–70.8) | 690 (284–1221) | 14 (11–50) |
| CV% | 53.8 | 99.1 | 43.9 | 35.8 | 67.1 | 60.6 | 75.0 |
| Plasma pralatrexate‐10b pharmacokinetic parameters (n = 5), cycle 1, visit 6 | |||||||
| Mean (SD) | 3754 (985.9) | 6 (2) | 156 000 (30 600) | 173 (32.6) | 34.6 (18.4) | 1006 (503) | 14 (1) |
| Median (range) | 3628 (2552–5213) | 5 (5–9) | 162 000 (111 000–186 000) | 155 (150–226) | 24.2 (17.5–60.9) | 912 (409–1550) | 14 (13–16) |
| CV% | 26.3 | 30.5 | 19.7 | 18.9 | 53.1 | 50.0 | 8.2 |
t 1/2 estimated from plasma concentration–time profiles by two–compartment method (0–720 min).
Discussion
Clinical outcomes for Japanese patients with PTCL remain poor, and there are few therapeutic options in the relapsed/refractory disease setting. The current phase I/II study is the first study of pralatrexate specifically in Japanese patients with relapsed or refractory PTCL. It was carried out at 12 centers in Japan and included patients with PTCL‐NOS, AITL, and ALK‐negative ALCL. In phase I, pralatrexate was generally well tolerated, and none of three patients experienced a DLT at the FDA‐approved dose of 30 mg/m2 given weekly for 6 weeks of a 7‐week treatment cycle. Based on these results, the trial continued to phase II to further evaluate safety and determine efficacy based on ORR.
In phase II, nine of 20 evaluable patients (45%) responded to treatment, all within the first cycle of pralatrexate, suggesting its rapid antitumor effects. This ORR is higher than expected based on a large phase II study among patients in North America and Europe (PROPEL), which found an ORR of 29%. The difference in ORR between these two studies may be caused not by the patients’ racial background but by different sample sizes, because an earlier dose‐ranging study undertaken in the USA found a 54% ORR in relapsed or refractory TCL.17 The median duration of response could not be determined in the current study at the data cut‐off; however, median PFS was approximately 5 months, which compares favorably to published reports that show a median PFS less than 4 months with traditional therapy given at first relapse or time of first progression.4 Median OS was not reached in this study, but the 12‐month OS rate of 61% also compares favorably to historical data that show a median OS less than 6 months in this setting.4
In the exploratory analysis, the treatment benefit appeared to be maintained across all clinically relevant subgroups, although this finding should be interpreted cautiously given the small numbers in each subgroup. Nonetheless, the data are encouraging. Responses were observed even in patients with stage IV disease, a history of more than three prior therapies, disease that was refractory to the last therapy, or a high LDH level at baseline. These were consistent with a subgroup analysis of a previous phase II study (PROPEL), which showed durable responses regardless of number of prior treatments or response to most recent prior therapy.8
Although prognosis and response to treatment have been reported to vary by PTCL subtype in some studies, this study found similar ORR for patients with PTCL‐NOS (6/12; 50%), AITL (4/9; 44%), and ALK‐negative ALCL (1/2; 50%). In the PROPEL trial, the ORR was ≥25% for all PTCL subtypes except AITL.8 In the AITL subgroup, only 1 of 13 patients (8%) responded to pralatrexate. The current data are therefore encouraging, although due to small numbers in both studies, more data are needed to draw firm conclusions about the relative effectiveness of pralatrexate in AITL.
Pralatrexate was generally well tolerated, and the safety profile and pharmacokinetics were consistent with prior studies.8, 15, 17 The most commonly reported AEs included mucositis, thrombocytopenia, liver function test abnormality, anemia, lymphopenia, and neutropenia. Unlike the PROPEL study, there were no cases of grade 4 mucositis in the current trial. In addition to concurrent vitamin B12 and folic acid supplementation, all patients in the current trial used cryotherapy prophylactically, and many used sodium azulene sulfonate‐containing mouthwashes and/or commercially available oral care mouthwashes and oral moisturizers, in addition to regular dental care that included tartar and plaque removal. These preventative measures may be responsible for the reduction in high‐grade mucositis seen in this trial relative to prior studies. Although no patient received leucovorin in the current study, leucovorin might be another option to reduce the risk of severe mucositis by pralatrexate, as reported in the USA.18
In summary, although the total number of patients was relatively small, the results of this phase I/II study show that pralatrexate is a promising treatment option with a manageable safety profile in Japanese patients with relapsed/refractory PTCL. Clinical benefit, as indicated by a relatively higher ORR than that of several conventional chemotherapies that was achieved within one treatment cycle, was greater than expected in a patient population with limited therapeutic options. Further evaluation of pralatrexate is warranted in this setting.
Disclosure Statement
The study was funded by Mundipharma and designed under the company's responsibility, in conjunction with the steering committee. D. Maruyama has received research funding from Amgen Astellas Biopharma, Pfizer, Otsuka, GlaxoSmithKline, Celgene, ONO, Chugai, Janssen, Takeda, Eisai, Mundipharma K.K., Novartis, Abbvie, Bayer, MSD, Solasia, Daiichi Sankyo, Quintiles Transnational, Zenyaku Kogyo, and CMIC and honoraria from Takeda, Janssen, Eisai, and Biomedis International. H. Nagai has received research funding from Janssen, Mundipharma, Celgene, Bayer, Abbvie, Takeda, Chugai, Kyowa Hakko Kirin, and Eisai and honoraria from Chugai, Mundipharma, Eisai, Sanofi, and Janssen. K. Izutsu has received research funding from Celgene and Eisai and honoraria from Takeda, Eisai, and Kyowa Hakko Kirin. R. Ueda has received research funding from Kyowa Hakko Kirin, Chugai, Rikaken, and Medical Laboratories, and served in an advisory role for Mundipharma and Termo. K. Tobinai has received research funding from Kyowa Hakko Kirin, Celgene, Eisai, Mundipharma, and Takeda and honoraria from Eisai, Takeda, Mundipharma, and HUYA Bioscience International. The other authors have no conflict of interest.
Abbreviations
- AE
adverse event
- AITL
angioimmunoblastic T‐cell lymphoma
- ALCL
anaplastic large‐cell lymphoma
- ALK
anaplastic lymphoma kinase
- CCR4
CC chemokine receptor 4
- CI
confidence interval
- CR
complete response
- CT
computed tomography
- DLT
dose‐limiting toxicity
- FDG‐PET/CT
fluorine‐18‐2‐fluoro‐2‐deoxy‐D‐glucose positron emission tomography/computed tomography
- HBV
hepatitis B virus
- LDH
lactate dehydrogenase
- NKTCL
natural killer/T‐cell lymphoma
- ORR
objective response rate
- OS
overall survival
- PFS
progression‐free survival
- PR
partial response
- PS
performance status
- PTCL‐NOS
peripheral T‐cell lymphoma, not otherwise specified
- PTCL
peripheral T‐cell lymphoma
- SAE
serious adverse event
- TCL
T‐cell lymphoma
Supporting information
Table S1. Protocol‐mandated pralatrexate dose modifications for adverse events.
Acknowledgments
We sincerely thank all the patients who participated in this study and their families, as well as the investigators, medical staff, and operation staff at the investigational sites, including Dr. Tatsuro Jo (Japanese Red Cross Nagasaki Genbaku Hospital), Dr. Hikaru Kobayashi (Nagano Red Cross Hospital) and Dr. Nobuyuki Aotsuka (Japanese Red Cross Narita Hospital). We give special thanks to Dr. Yoshihiro Matsuno (Hokkaido University Hospital), Dr. Shigeo Nakamura (Nagoya University Hospital) and Dr. Koichi Ohshima (Kurume University School of Medicine) for their contributions to central pathology review; Dr. Takashi Terauchi (Cancer Institute Hospital of Japanese Foundation for Cancer Research), Dr. Ukihide Tateishi (Tokyo Medical and Dental University), and Dr. Mitsuaki Tatsumi (Osaka University Hospital) for their contributions to central CT and FDG‐PET/CT review; and Dr. Noboru Horikoshi (Juntendo University School of Medicine) and Dr. Noriko Usui (Jikei University School of Medicine) for their contributions as members of the Efficacy and Safety Evaluation Committee. Medical writing support was provided by Phillips Gilmore Oncology Communications, funded by Mundipharma.
Cancer Sci 108 (2017) 2061–2068
Funding information
Mundipharma K.K.
References
- 1. Vose J, Armitage J, Weisenburger D; International T‐Cell Lymphoma Project . International peripheral T‐cell and natural killer/T‐cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 2008; 26: 4124–30. [DOI] [PubMed] [Google Scholar]
- 2. Park S, Ko YH. Peripheral T cell lymphoma in Asia. Int J Hematol 2014; 99: 227–39. [DOI] [PubMed] [Google Scholar]
- 3. Kitahara H, Maruyama D, Maeshima AM et al Prognosis of patients with peripheral T cell lymphoma who achieve complete response after CHOP/CHOP‐like chemotherapy without autologous stem cell transplantation as an initial treatment. Ann Hematol 2017; 96: 411–20. [DOI] [PubMed] [Google Scholar]
- 4. Mak V, Hamm J, Chhanabhai M et al Survival of patients with peripheral T‐cell lymphoma after first relapse or progression: spectrum of disease and rare long‐term survivors. J Clin Oncol 2013; 31: 1970–6. [DOI] [PubMed] [Google Scholar]
- 5. National Comprehensive Cancer Network . NCCN clinical practice guidelines in oncology (NCCN Guidelines®): T‐cell lymphomas, version 2.2017 – February 21, 2017. [Cited 10 Mar 2017.] Available from URL: https://www.nccn.org/professionals/physician_gls/pdf/t-cell.pdf.
- 6. Tsukasaki K, Tobinai K, Uchida T et al Phase 1/2 study of forodesine in patients with relapsed peripheral t‐cell lymphoma (PTCL). J Clin Oncol 2016; 34(suppl): 7542. abstr 7542 [Google Scholar]
- 7. Malik SM, Liu K, Qiang X et al Folotyn (pralatrexate injection) for the treatment of patients with relapsed or refractory peripheral T‐cell lymphoma: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res 2010; 16: 4921–7. [DOI] [PubMed] [Google Scholar]
- 8. O'Connor OA, Pro B, Pinter‐Brown L et al Pralatrexate in patients with relapsed or refractory peripheral T‐cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 2011; 29: 1182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marchi E, Mangone M, Zullo K, O'Connor OA. Pralatrexate pharmacology and clinical development. Clin Cancer Res 2013; 19: 6657–61. [DOI] [PubMed] [Google Scholar]
- 10. Marchi E, Paoluzzi L, Scotto L et al Pralatrexate (PDX) is synergistic with the proteasome inhibitor bortezomib in in vitro and in vivo models of T‐cell lymphoid malignancies. Clin Cancer Res 2010; 16: 3648–58. [DOI] [PubMed] [Google Scholar]
- 11. Wang ES, O'Connor O, She Y et al Activity of a novel anti‐folate (PDX, 10‐propargyl 10‐deazaaminopterin) against human lymphoma is superior to methotrexate and correlates with tumor RFC‐1 gene expression. Leuk Lymphoma 2003; 44: 1027–35. [DOI] [PubMed] [Google Scholar]
- 12. Toner LE, Vrhovac R, Smith EA et al The schedule‐dependent effects of the novel antifolate pralatrexate and gemcitabine are superior to methotrexate and cytarabine in models of human non‐Hodgkin's lymphoma. Clin Cancer Res 2006; 12: 924–32. [DOI] [PubMed] [Google Scholar]
- 13. Swerdlow SH, Campo E, Harris NL et al WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th edn Lyon, France: IARC Press, 2008. [Google Scholar]
- 14. Cheson BD, Horning SJ, Coiffier B et al Report of an international workshop to standardize response criteria for non‐Hodgkin's lymphomas: NCI sponsored International Working Group. J Clin Oncol 1999; 17: 1244–53. [DOI] [PubMed] [Google Scholar]
- 15. Folotyn® [USA package insert] . Spectrum Pharmaceuticals; 2016. [Google Scholar]
- 16. Cheson BD, Pfistner B, Juweid ME et al Revised response criteria for malignant lymphoma. J Clin Oncol 2007; 25: 579–86. [DOI] [PubMed] [Google Scholar]
- 17. O'Connor OA, Horwitz S, Hamlin P et al Phase II‐I‐II study of two different doses and schedules of pralatrexate, a high‐affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T‐cell malignancies. J Clin Oncol 2009; 27: 4357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koch E, Story SK, Geskin LJ. Preemptive leucovorin administration minimizes pralatrexate toxicity without sacrificing efficacy. Leuk Lymphoma 2013; 54: 2448–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Protocol‐mandated pralatrexate dose modifications for adverse events.