

Abstract
Polycomb repressive complex 2 (PRC2) methylates histone H3 lysine 27 and represses gene expression to regulate cell proliferation and differentiation. Enhancer of zeste homolog 2 (EZH2) or its close homolog EZH1 functions as a catalytic subunit of PRC2, so there are two PRC2 complexes containing either EZH2 or EZH1. Tumorigenic functions of EZH2 and its synthetic lethality with some subunits of SWItch/Sucrose Non‐Fermentable (SWI/SNF) chromatin remodeling complexes have been observed. However, little is known about the function of EZH1 in tumorigenesis. Herein, we developed novel, orally bioavailable EZH1/2 dual inhibitors that strongly and selectively inhibited methyltransferase activity of both EZH2 and EZH1. EZH1/2 dual inhibitors suppressed trimethylation of histone H3 lysine 27 in cells more than EZH2 selective inhibitors. They also showed greater antitumor efficacy than EZH2 selective inhibitor in vitro and in vivo against diffuse large B‐cell lymphoma cells harboring gain‐of‐function mutation in EZH2. A hematological cancer panel assay indicated that EZH1/2 dual inhibitor has efficacy against some lymphomas, multiple myeloma, and leukemia with fusion genes such as MLL‐AF9,MLL‐AF4, and AML1‐ETO. A solid cancer panel assay demonstrated that some cancer cell lines are sensitive to EZH1/2 dual inhibitor in vitro and in vivo. No clear correlation was detected between sensitivity to EZH1/2 dual inhibitor and SWI/SNF mutations, with a few exceptions. Severe toxicity was not seen in rats treated with EZH1/2 dual inhibitor for 14 days at drug levels higher than those used in the antitumor study. Our results indicate the possibility of EZH1/2 dual inhibitors for clinical applications.
Keywords: Dual inhibitor, EZH1, EZH2, H3K27me3, histone methyltransferase
Polycomb repressive complex (PRC) genes were identified by their ability to control embryogenesis in Drosophila, and they are conserved in vertebrates.1 These gene products form two protein complexes, PRC1 and PRC2; PRC2 catalyzes trimethylation of histone H3 lysine 27 (H3K27me3).2, 3 In mammals, either enhancer of zeste homolog 1 (EZH1) or EZH2 functions as a catalytic subunit of PRC2, so that there are two types of PRC2 complex (i.e. PRC2‐EZH1 and PRC2‐EZH2).4, 5, 6, 7, 8 In ES cells, Ezh1 and Ezh2 work cooperatively or complementarily, and both are important for the maintenance of ES cells.6 EZH1 and EZH2 are essential for hematopoietic stem cell maintenance and hair follicle homeostasis.9, 10, 11, 12 They also regulate the Merkel cell differentiation program in skin stem cells.13
Overexpression of EZH2 has been reported in many cancers, and correlations between EZH2 expression and poor prognosis were detected.14, 15, 16, 17, 18, 19, 20, 21, 22, 23 Somatic mutations of the EZH2 Y641, A677, and A687 residues were also found in diffuse large B‐cell lymphomas (DLBCL) and follicular lymphomas (FL).24, 25, 26, 27, 28 These were gain‐of‐function (GOF) mutations and dramatically increased the amount of H3K27me3 in the cancer cells. Recently, small molecule inhibitors of the methyltransferase activity of EZH2 have been developed, which decreased growth of cancer cells harboring EZH2 GOF mutations in vitro and in vivo.29, 30, 31, 32, 33
Loss‐of‐function mutations in EZH2 were also found in myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), and T‐cell acute lymphoblastic leukemia (T‐ALL).34, 35, 36, 37, 38 In these cases, EZH2 has been suggested to act as a tumor suppressor. EZH2 is now considered to have a context‐dependent bilateral character, acting as an oncogene or tumor suppressor gene depending on the circumstances.
Until now, a number of reports have shown a correlation between EZH2 and cancer, but not between EZH1 and cancer. However, the importance of EZH1 in cancer progression and maintenance has recently been demonstrated. EZH1 is essential for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) caused by EZH2 loss‐of‐function mutations.39 Inhibition of PRC2‐EZH2 alone is not enough to suppress MLL‐AF9‐mediated AML, but simultaneous inhibition of both PRC2‐EZH2 and PRC2‐EZH1 produces complete suppression.40, 41 These results indicate that some cancers depend on both EZH2 and EZH1, and simultaneous inhibition of EZH2 and EZH1 would be effective to target them.
In the present study, we identified orally bioavailable small molecule compounds that inhibit histone methyltransferase activity of both EZH1 and EZH2 (i.e. EZH1/2 dual inhibitors). We compared activities between EZH1/2 dual inhibitors and EZH2 selective inhibitor. Panel assays for cancer cell lines were used to find lines that were sensitive to EZH1/2 dual inhibitor. A repeated dose toxicity study in rats demonstrated that the toxicity of EZH1/2 dual inhibitor was tolerable.
Materials and Methods
Full methods and any associated references are available as Supporting Information (Doc. S1).
Results
EZH1/2 dual inhibitors suppress H3K27me3 in cells more highly than do EZH2 selective inhibitors
As EZH2 is a promising target for anticancer therapy, we first tried to acquire small molecule compounds that inhibit histone methyltransferase activity of EZH2. Daiichi Sankyo's original compound library (285 000 compounds) and the following two assay systems were used for the screening.
A cell‐free assay to detect methyltransferase activities of PRC2‐EZH2 in vitro. Recombinant PRC2‐EZH2, which consists of EZH2, EED, SUZ12, RbAp48, and AEBP2, was used as a protein source. For compounds that strongly inhibited PRC2‐EZH2, we also tested the activity to inhibit PRC2‐EZH1.
A cell‐based assay to detect H3K27me3 in HCT116 colorectal cancer cells.
Throughout the compound screening and derivatization, we found that compounds inhibiting both EZH1 and EZH2 (i.e. EZH1/2 dual inhibitors) more potently reduced H3K27me3 in cells than EZH2 selective inhibitors (Fig. 1a). From these results, we hypothesized that EZH1/2 dual inhibitors are superior to EZH2 selective inhibitors as anticancer drugs. As tools to support this hypothesis, we selected OR‐S1 and OR‐S2 as EZH1/2 dual inhibitors and OR‐S0 as an EZH2 selective inhibitor. IC50 values of OR‐S1, OR‐S2, and OR‐S0 against EZH2 were 24, 16, and 11 nM, respectively, and IC50 values against EZH1 were 21, 23, and 91 nM, respectively. These compounds inhibited H3K27me3 in HCT116 cells, with IC50 values of 0.55 (OR‐S1), 0.62 (OR‐S2), and 24 nM (OR‐S0). OR‐S1 and OR‐S2 are racemic compounds, which have a chiral center in the molecule. We identified the R‐form of these compounds, (R)‐OR‐S1 and (R)‐OR‐S2 (Fig. 1b), as having greater inhibitory activity than the racemic mixtures (Fig. 1a, 3b).
Figure 1.
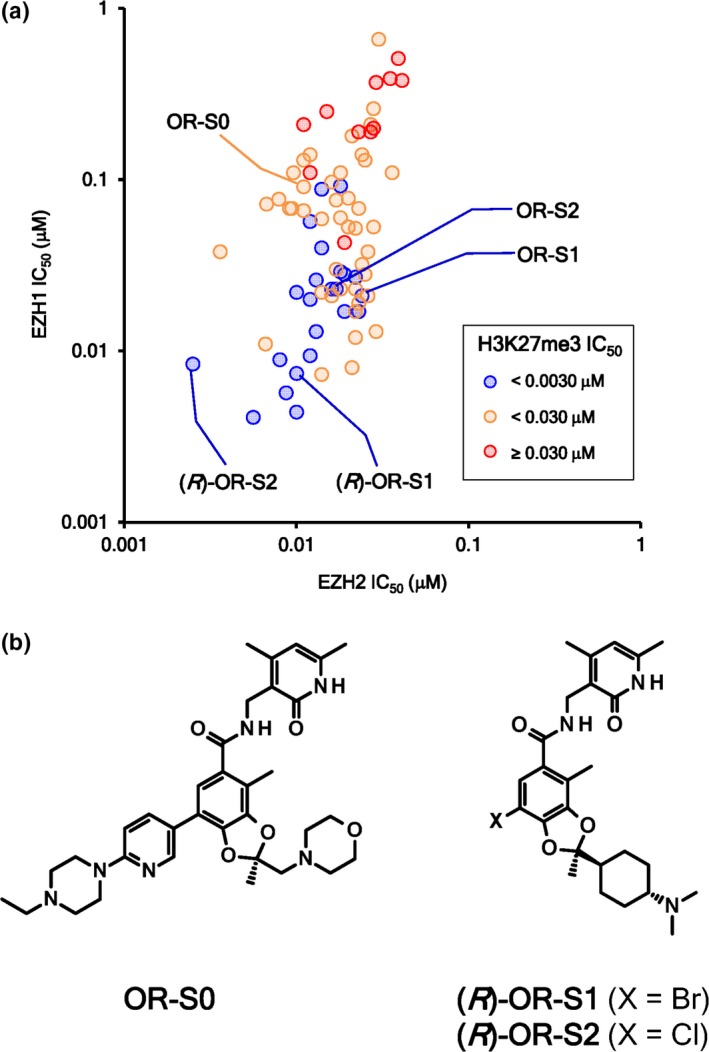
Enhancer of zeste homologs 1/2 (EZH1/2) dual inhibitors suppress trimethylation of histone H3 lysine 27 (H3K27me3) in cells more highly than do EZH2 selective inhibitors. (a) Plot of IC 50 values of 77 compounds against polycomb repressive complex 2 (PRC2)‐EZH2 (x‐axis) and PRC2‐EZH1 (y‐axis). Cell‐based H3K27me3 reduction activities in HCT116 colorectal cancer cells are plotted as colored dots. (b) Structures of EZH2 selective inhibitor, OR‐S0, and EZH1/2 dual inhibitors, (R)‐OR‐S1 and (R)‐OR‐S2.
OR‐S1 and OR‐S2 are S‐adenosylmethionine (SAM)‐competitive and highly selective EZH1/2 dual inhibitors
We conducted saturation transfer difference nuclear magnetic resonance (STD‐NMR) studies, using SAM as a cofactor, to test the binding of (R)‐OR‐S1 and (R)‐OR‐S2 to PRC2‐EZH2. Figure 2a shows the STD‐NMR and competition‐STD‐NMR spectra acquired with SAM. Decrease in the intensity of the signals for SAM acquired in the presence of (R)‐OR‐S1 or (R)‐OR‐S2 indicated that SAM and these compounds bind to the same site on PRC2‐EZH2. We confirmed SAM competitive binding of (R)‐OR‐S1 to PRC2‐EZH2 using a molecular docking study. A PRC2/(R)‐OR‐S1 model based on the PRC2 crystal structure (PDB ID: 5ij7)42 is shown in Figure 2b. The predicted binding mode of (R)‐OR‐S1 was similar to “Inhibitor 1” in PDB ID: 5ij7, or to the CPI‐1205 analog in PDB ID: 5ls6.43 The pyridone group makes hydrogen bonds (green dotted lines) with the backbone of W624 in the conserved GXG motif of the SET domain, and with the bridging water molecule (small red sphere). This pyridone binding site partially overlaps with the SAM/S‐adenosylhomocysteine (SAH) binding site as previously described.42, 44 The homocysteine moiety of SAH (from PDB ID: 5hyn, structurally aligned to our model) also overlaps the pyridone group of (R)‐OR‐S1 in our model. The partial overlap between the pyridone group of (R)‐OR‐S1 and the homocysteine moiety of SAM/SAH is also consistent with the SAM competitive binding of our NMR study. A similar result was obtained for (R)‐OR‐S2 (data not shown).
Figure 2.
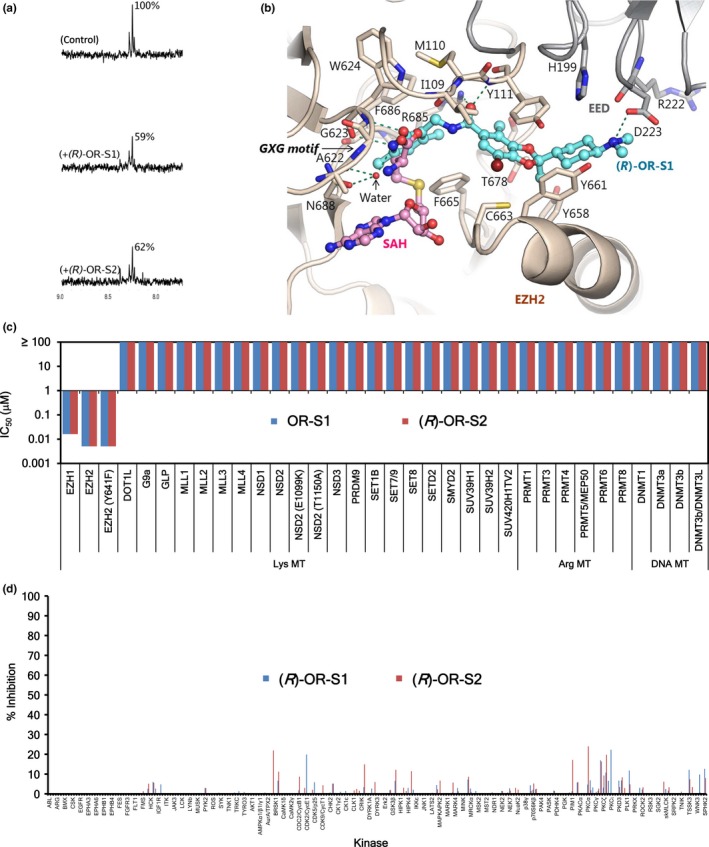
OR‐S1 and OR‐S2 are S‐adenosylmethionine (SAM)‐competitive and highly selective enhancer of zeste homologs 1/2 (EZH1/2) dual inhibitors. (a) Saturation transfer difference nuclear magnetic resonance (STD‐NMR) SAM/compound competition experiments for polycomb repressive complex 2 (PRC2). Upper spectrum shows an expansion of the aromatic region of the STD spectrum obtained for PRC2‐EZH2 in the presence of SAM. Middle and lower spectra show competition of SAM with (R)‐OR‐S1 and (R)‐OR‐S2. (b) Model of the PRC2/(R)‐OR‐S1 complex superimposed on the PRC2/S‐adenosylhomocysteine (SAH) complex (PDB ID: 5hyn44). EED, EZH2, (R)‐OR‐S1, and SAH are colored in gray, light brown, cyan, and pink, respectively. From PRC2/SAH complex, only SAH is shown. (c) In vitro inhibitory activities of OR‐S1 and (R)‐OR‐S2 against 24 histone lysine, six histone arginine, and four DNA methyltransferases. (d) In vitro inhibitory activities of (R)‐OR‐S1 and (R)‐OR‐S2 at 1 μM against 253 kinases.
Next, we investigated the selectivity of our EZH1/2 dual inhibitors across a panel of methyltransferases and kinases. In the methyltransferase panel assay, we tested histone lysine, histone arginine, and DNA methyltransferases. OR‐S1 and (R)‐OR‐S2 strongly inhibited both EZH1 and EZH2, but not other methyltransferases (Fig. 2c; Table S1). OR‐S1 and (R)‐OR‐S2 also inhibited EZH2 (Y641F), which is a GOF mutant found in DLBCL patients (Fig. 2c).24 In the kinase panel assay, we surveyed 253 kinases and found none to be strongly inhibited by (R)‐OR‐S1 and (R)‐OR‐S2 at 1 μM (Fig. 2d; Table S2). These results demonstrated that OR‐S1 and OR‐S2 are highly selective methyltransferase inhibitors against EZH1 and EZH2.
EZH1/2 dual inhibitors show greater antitumor efficacy than EZH2 selective inhibitor against KARPAS‐422 cells harboring a GOF mutation in EZH2
KARPAS‐422 is a DLBCL cell line harboring a Y641N GOF mutation in EZH2, which has been shown to be sensitive to EZH2 selective inhibitors.29, 30 Using this cell line, we compared the antitumor efficacy between EZH1/2 dual inhibitors and EZH2 selective inhibitor. First, we treated KARPAS‐422 cells with (R)‐OR‐S1, (R)‐OR‐S2 or OR‐S0 for 10 days to evaluate their antiproliferative effects (Fig. 3a). All three compounds inhibited the growth of KARPAS‐422 cells in a dose‐dependent manner. (R)‐OR‐S1 and (R)‐OR‐S2 showed greater antiproliferative efficacy than OR‐S0, although all had similar inhibitory activities against EZH2 in vitro (Fig. 3b). Second, we compared in vivo antitumor efficacy between (R)‐OR‐S1 and OR‐S0, using the KARPAS‐422 xenograft mice model. All in vivo studies in this report were approved by the Institutional Animal Care and Use Committee of Daiichi Sankyo Co. Ltd. Orally given (R)‐OR‐S1 inhibited tumor growth dose‐dependently and caused tumor regression at a dose of 50 mg/kg (Fig. 3e). This effect continued after cessation of the 50 mg/kg dose until the tumors were completely eliminated (Fig. S1). OR‐S0 also showed dose‐dependent tumor growth inhibition, and tumor regression was detected at a dose of 160 mg/kg (Fig. 3d). We carried out pharmacokinetic (PK) analysis of these two compounds at the doses that afforded a 50% reduction in tumor volumes (Fig. 3c). Values of area under the curve (AUC0‐24) indicate that the antitumor activity of (R)‐OR‐S1 is 28‐fold more potent than OR‐S0. Subsets of mice from each group in Figures 3d and 3e were killed 1 day after cessation of the dosing, and tumors were harvested to quantify the amount of H3K27me3. Dose‐dependent reductions of H3K27me3 were detected, and they correlated with the antitumor activities of the compounds (Fig. 3f,g).
Figure 3.
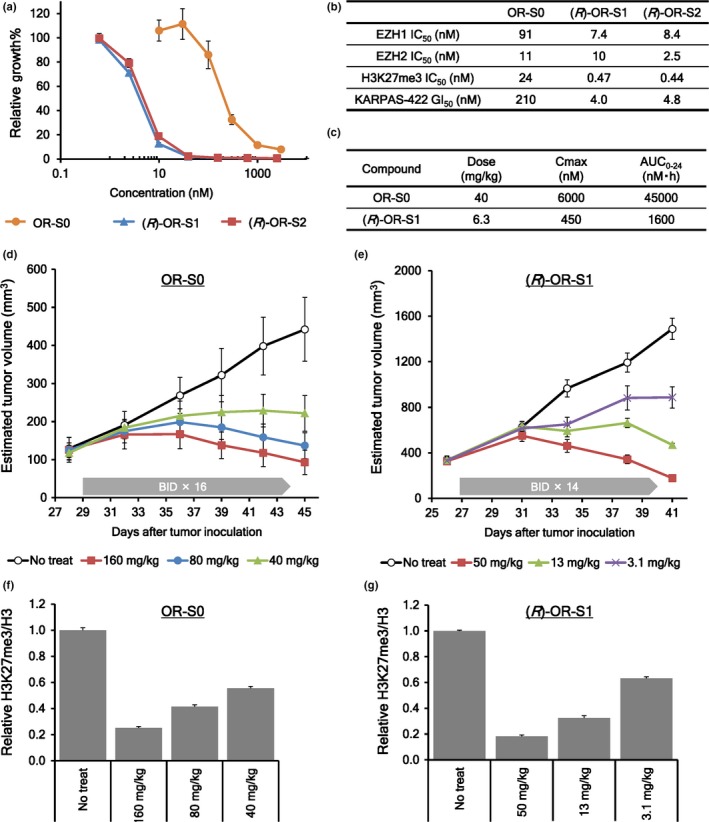
Enhancer of zeste homologs 1/2 (EZH1/2) dual inhibitors show greater antitumor activity than an EZH2 selective inhibitor. (a) Dose‐dependent in vitro growth inhibition of KARPAS‐422 cells by OR‐S0, (R)‐OR‐S1, and (R)‐OR‐S2. (b) Summary of activities of OR‐S0, (R)‐OR‐S1, and (R)‐OR‐S2. Cell‐free polycomb repressive complex 2 (PRC2)‐EZH1 and PRC2‐EZH2 inhibition activities, cell‐based trimethylation of histone H3 lysine 27 (H3K27me3) inhibition activities in HCT116 cells, and in vitro growth inhibition activities against KARPAS‐422 cells of OR‐S0, (R)‐OR‐S1, and (R)‐OR‐S2 are shown. (c) Summary of pharmacokinetic parameters of single‐dose OR‐S0 and (R)‐OR‐S1 in BALB/c mice. (d–g) Antitumor activities and H3K27me3 reduction activities of (d,f) OR‐S0 and (e,g) (R)‐OR‐S1 in a KARPAS‐422 xenograft model. Mean estimated tumor volumes ± standard errors (n = 5, d,e), and mean relative H3K27me3/H3 values ± standard errors (n = 3, f,g) are plotted on the graphs.
Collectively, EZH1/2 dual inhibitors showed greater antitumor efficacy than an EZH2 selective inhibitor in a model of DLBCL harboring Y641N oncogenic mutation in EZH2.
Search for hematological cancer cell lines sensitive to EZH1/2 dual inhibitor
As dysregulation of PRC2 is linked to hematological malignancies,45, 46, 47, 48, 49 and we found that EZH1/2 dual inhibitors are more potent than EZH2 selective inhibitors, we evaluated the growth inhibition efficacy of EZH1/2 dual inhibitor ((R)‐OR‐S2) in a panel of hematological cancer cell lines (Fig. 4a). In all cell lines, no correlation was detected between sensitivity to (R)‐OR‐S2 and mRNA expression levels of EZH1 and/or EZH2. Among the DLBCL cell lines, those harboring a GOF mutation in EZH2 (green bars in Fig. 4a) showed higher sensitivity to (R)‐OR‐S2 than those that did not have a mutation in EZH2, with one exception. This result is consistent with previous reports that these cell lines are sensitive to EZH2 selective inhibitors.29, 30 RL cell line has a Y641N mutation in EZH2 but did not show sensitivity to (R)‐OR‐S2, which also corroborated a previous report that this line is not sensitive to GSK126, an EZH2 selective inhibitor.29 As half maximal growth inhibition (GI50) values of (R)‐OR‐S2 against these higher sensitive cells were ≤100 nM, we decided to label cell lines with GI50 values of ≤100 nM as hypersensitive ones. Among lymphoma subgroups, most peripheral T‐cell lymphoma (PTCL) cell lines also showed hypersensitivity to (R)‐OR‐S2. As for myelomas, six out of eight multiple myeloma (MM) cell lines showed hypersensitivity to (R)‐OR‐S2. Compared to lymphomas and MM, the percentage of hypersensitive cell lines was low with leukemia. However, all hypersensitive cell lines of AML; namely MOLM‐14, THP‐1, MV‐4‐11, and Kasumi‐1 (orange bars in Fig. 4a; Table S3), had the fusion genes, MLL‐AF9 (MOLM‐14 and THP‐1), MLL‐AF4 (MV‐4‐11), or AML1‐ETO (Kasumi‐1). Among cell lines of acute lymphoblastic leukemia (ALL), RS4;11 (red bar in Fig. 4a) has an MLL‐AF4 fusion gene like MV‐4‐11, and RS4;11 also showed hypersensitivity to (R)‐OR‐S2. Collectively, these results suggest that lymphomas (e.g. DLBCL with EZH2 mutations and PTCL), MM, and leukemia with fusion genes (e.g. MLL‐AF9, MLL‐AF4, and AML1‐ETO) are good clinical targets of EZH1/2 dual inhibitors.
Figure 4.
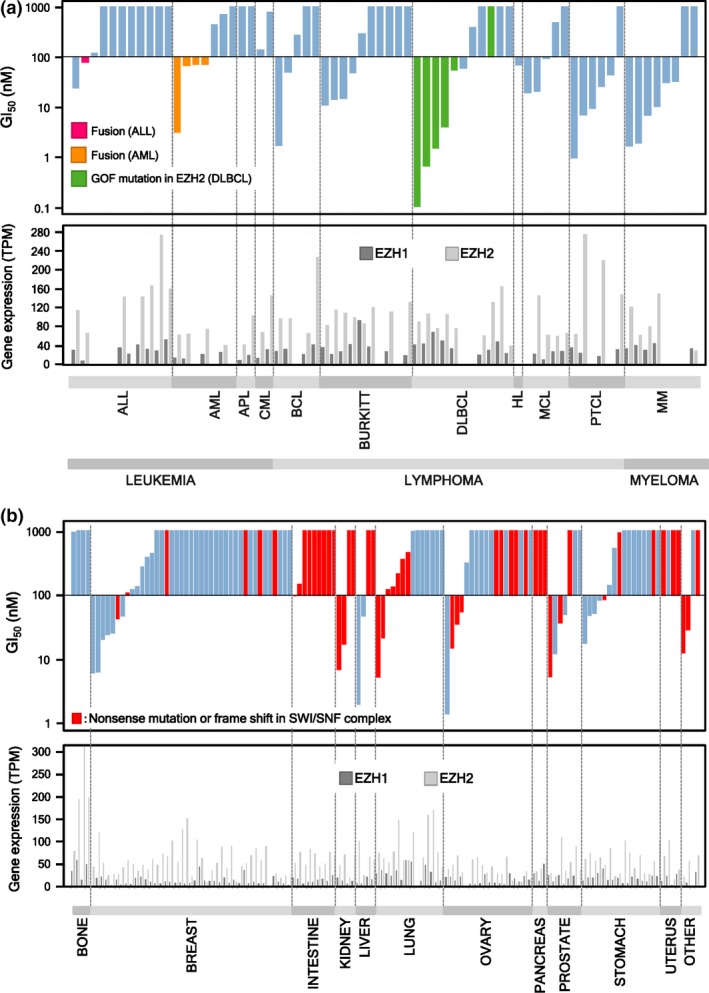
In vitro growth inhibition activities of (R)‐OR‐S2 against various cancer cell lines. Fifty percent growth inhibitory activities against (a) hematological cancer cell lines and (b) solid cancer cell lines, with the cellular gene expression profiles of enhancer of zeste homolog 1 (EZH1) and EZH2. Gene expression profiles of EZH1 and EZH2 were obtained from the Cancer Cell Line Encyclopedia (CCLE).60 Information of the mutations of SWItch/Sucrose Non‐Fermentable (SWI/SNF) subunits in each cell line was obtained from CCLE, the Catalogue Of Somatic Mutations In Cancer (COSMIC),61 and the cBioPortal for Cancer Genomics.62, 63 ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; BCL, B‐cell lymphoma; BURKITT, Burkitt lymphoma; DLBCL, diffuse large B‐cell lymphoma; HL, Hodgkin's lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma; PTCL, peripheral T‐cell lymphoma.
Search for solid cancer cell lines sensitive to EZH1/2 dual inhibitor
In addition to hematological cancer, an oncogenic dependency on EZH2 has been suggested for solid cancers.14, 15, 16, 17, 18, 19, 20, 21, 22, 23 SWItch/Sucrose Non‐Fermentable (SWI/SNF) is a chromatin remodeling complex that has 12–15 subunits, and mutations in some of these subunits are found in nearly 20% of human cancers.50, 51 Recently, it has been suggested that loss‐of‐function mutations in some subunits of the SWI/SNF complex become synthetic lethal with EZH2 inhibition.52, 53, 54 For example, EZH2 knockdown or inhibition of its methyltransferase activity with a small molecule compound suppressed the progression of tumors harboring mutations of SWI/SNF subunits, such as SMARCB1 in malignant rhabdoid tumor (MRT)31, 52 or ARID1A in ovarian clear cell carcinoma (OCCC).54 However, Kim et al. suggested that cancer cells harboring mutations in SWI/SNF subunits, such as ARID1A, SMARCA4, and PBRM1, are primarily dependent on a non‐catalytic role of EZH2 and partially dependent on its methyltransferase activity, but the Ras‐pathway mutation abrogates this EZH2 dependency.53 To find solid cancer cells which are sensitive to EZH1/2 dual inhibitor and to clarify their dependency on SWI/SNF mutations, we screened a panel of solid cancer cells (Fig. 4b). In this experiment, we also labeled cell lines for which (R)‐OR‐S2 showed GI50 values of ≤100 nM as hypersensitive. No correlation was detected between sensitivity to (R)‐OR‐S2 and mRNA expression levels of EZH1 and/or EZH2 (Fig. 4b). Clear correlations were generally not detected between sensitivity to EZH1/2 dual inhibitor, and mutations in SWI/SNF subunits and/or Ras‐pathway (Fig. 4b; Table S3). As for lung cancer cell lines, however, good correlation was detected between them. Interestingly, when we divided lung cancer cell lines into two groups (i.e. non‐small cell lung cancer [NSCLC] and small cell lung cancer [SCLC]), a more refined correlation was obtained among SCLC cell lines (Fig. S2).
G401 is a SMARCB1‐negative MRT cell line, and previous studies demonstrated that G401 is sensitive to EZH2 selective inhibitors in vitro and in vivo.31 In the solid cancer panel assay, the GI50 value of EZH1/2 dual inhibitor against G401 cells was 17.7 nM, classifying this cell as hypersensitive (Table S3). In a G401 xenograft model, once daily administration of 50 mg/kg OR‐S1 for 12 days completely suppressed tumor growth (Fig. 5a). NCI‐N87 is a gastric carcinoma cell line, which was not classified as hypersensitive (GI50 = 536 nM). As the growth of NCI‐N87 cells is slower than other cell lines, we hypothesized that a longer exposure to EZH1/2 dual inhibitor could improve its efficacy. To investigate this hypothesis, we carried out an in vivo antitumor test, using s.c. xenografts of NCI‐N87 cells (Fig. 5b). During a 28‐day administration of OR‐S1 (once‐daily), tumor regression was not detected. However, after dose cessation, tumor growth stopped, and this effect continued for at least 40 days. These results indicate that long compound exposure time in vivo is necessary to observe antitumor effects of EZH1/2 dual inhibitor against NCI‐N87 cells. Similar result was obtained in an in vitro study (data not shown).
Figure 5.
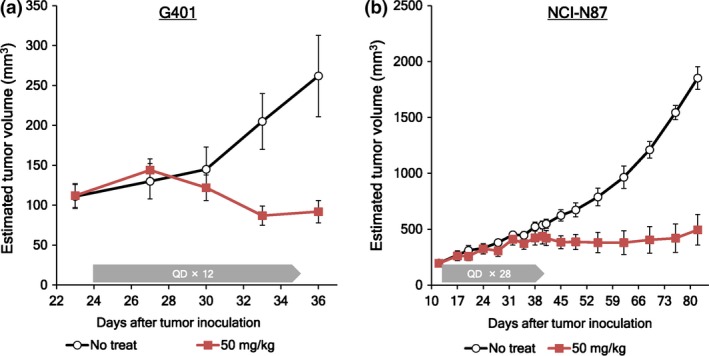
Antitumor activities of OR‐S1 against solid cancer cell lines. Antitumor activities in a xenograft model of OR‐S1 against (a) rhabdoid tumor cell line, G401 and (b) gastric cancer cell line, NCI‐N87. Mean estimated tumor volumes ± standard errors are plotted on the graphs (n = 5).
Repeated dose toxicity study of EZH1/2 dual inhibitor in rats
To investigate potential toxicity of EZH1/2 dual inhibitor, we carried out a 14‐day, repeated dose toxicity study of (R)‐OR‐S1 in rats. We used 100 mg/kg per day as the dose of (R)‐OR‐S1. This dose was expected to produce an exposure that was dozens of times higher than the pharmacologically active dose in the KARPAS‐422 xenograft mouse model. No abnormal clinical signs were observed in the animals throughout the study period. Food consumption and bodyweight in the (R)‐OR‐S1 group tended to be lower than those in the control group during the dosing period (Fig. S3). In hematology, white blood cell, neutrophil, and lymphocyte counts were significantly lower in the (R)‐OR‐S1 group than those in the control group (Table 1). In blood chemistry, statistically significant differences between the (R)‐OR‐S1 and control groups were detected in alanine aminotransferase and total bilirubin levels, although the differences were slight (Table 2). Histopathological changes in rats treated with (R)‐OR‐S1 are summarized in Table 3. A small thymus, with moderately decreased numbers of lymphocytes in the cortex, was observed in all animals in the (R)‐OR‐S1 group. Vacuolation of histiocytes in the thymus was also observed in two of the (R)‐OR‐S1‐treated animals. Additionally, one rat showed slight atrophy of the spleen, which consisted of decreased cellularity of white pulp lymphocytes and decreased hematopoiesis. Slight atrophy of hepatocytes and slight hypertrophy of zona fasciculata and reticularis cells in the adrenal were noted in one rat treated with (R)‐OR‐S1, although these changes were considered secondary to undernutrition or stress. No treatment‐related changes were observed in any other organ examined. In toxicokinetics analysis, no marked change with systemic exposure of (R)‐OR‐S1 was observed after repeated dosing. Mean AUC0‐24 h and Cmax of (R)‐OR‐S1 on the last day of dosing were 27 900 nM·h and 2440 nM, respectively (Table S4).
Table 1.
Effect of (R)‐OR‐S1 on hematological parameters in rats
| Parameter | Group | |
|---|---|---|
| Control | (R)‐OR‐S1 | |
| Red blood cell count (106/μL) | 6.71 ± 0.386 | 6.59 ± 0.160 |
| Hemoglobin (g/dL) | 13.5 ± 0.386 | 13.0 ± 0.377 |
| Hematocrit (%) | 41.2 ± 1.26 | 40.1 ± 0.594 |
| Mean corpuscular volume (fL) | 61.5 ± 1.92 | 60.9 ± 1.34 |
| MCHC (%) | 32.7 ± 0.050 | 32.5 ± 0.750 |
| Reticulocyte percentile (%) | 4.68 ± 0.802 | 4.48 ± 0.785 |
| White blood cell count (103/μL) | 9.55 ± 1.72 | 4.56 ± 1.02* |
| Neutrophil count (103/μL) | 1.73 ± 0.276 | 0.583 ± 0.0359** |
| Basophil count (103/μL) | 0.023 ± 0.0189 | 0.010 ± 0.0000 |
| Eosinophil count (103/μL) | 0.085 ± 0.0404 | 0.168 ± 0.0613 |
| Lymphocyte count (103/μL) | 7.37 ± 1.60 | 3.67 ± 0.928* |
| Monocyte count (103/μL) | 0.203 ± 0.0907 | 0.105 ± 0.0465 |
| Platelet count (103/μL) | 1160 ± 65.1 | 1120 ± 56.8 |
(R)‐OR‐S1 at a dose of 100 mg/kg per day or vehicle was orally given to rats for 14 days. Data are expressed as the mean ± SD of four animals. Statistical difference was determined by Student's t‐test (*P < 0.01) or Aspin‐Welch's t‐test (**P < 0.01). MCHC, mean corpuscular hemoglobin concentration.
Table 2.
Effect of (R)‐OR‐S1 on blood chemical parameters in rats
| Parameter | Group | |
|---|---|---|
| Control | (R)‐OR‐S1 | |
| Aspartate aminotransferase (U/L) | 72.0 ± 5.03 | 71.8 ± 8.85 |
| Alanine aminotransferase (U/L) | 35.5 ± 1.91 | 27.5 ± 4.73* |
| Alkaline phosphatase (U/L) | 1340 ± 199 | 1120 ± 169 |
| Lactase dehydrogenase (U/L) | 103.0 ± 16.1 | 137 ± 57.6 |
| Creatine kinase (U/L) | 164 ± 5.00 | 186 ± 38.2 |
| Total bilirubin (mg/dL) | 0.003 ± 0.0050 | 0.028 ± 0.0096** |
| Total cholesterol (mg/dL) | 63.5 ± 5.20 | 64.0 ± 6.16 |
| Triglyceride (mg/dL) | 132 ± 30.0 | 90.0 ± 58.5 |
| Glucose (mg/dL) | 193 ± 10.4 | 216 ± 19.0 |
| Albumin (g/dL) | 3.85 ± 0.129 | 3.75 ± 0.058 |
| Globulin (g/dL) | 1.60 ± 0.082 | 1.55 ± 0.100 |
| Urea nitrogen (mg/dL) | 16.7 ± 2.39 | 16.8 ± 1.85 |
| Creatinine (mg/dL) | 0.223 ± 0.0126 | 0.238 ± 0.0096 |
| Calcium (mg/dL) | 10.1 ± 0.096 | 9.88 ± 0.189 |
| Inorganic phosphorus (mg/dL) | 6.02 ± 0.628 | 5.97 ± 0.487 |
| Sodium (mEq/L) | 141 ± 0.82 | 142 ± 1.26 |
| Potassium (mEq/L) | 4.18 ± 0.189 | 4.20 ± 0.216 |
| Chloride (mEq/L) | 104 ± 1.50 | 105 ± 1.41 |
(R)‐OR‐S1 at a dose of 100 mg/kg per day or vehicle was orally given to rats for 14 days. Data are expressed as the mean ± SD of four animals. Statistical difference was determined by Student's t‐test (*P < 0.05, **P < 0.01).
Table 3.
Histopathological changes in (R)‐OR‐S1‐treated rats
| Organ | Group No. animals Grade | Control | (R)‐OR‐S1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 | 4 | ||||||||
| – | 1 | 2 | 3 | – | 1 | 2 | 3 | ||
| Thymus | |||||||||
| Decreased number of lymphocytes in cortex | 4 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | |
| Vacuolation of histiocytes | 4 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | |
| Spleen | |||||||||
| Atrophy of spleen | 4 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | |
| Adrenal | |||||||||
| Hypertrophy of zona fasciculata/reticularis cells | 4 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | |
| Liver | |||||||||
| Atrophy of hepatocytes | 4 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | |
(R)‐OR‐S1 at a dose of 100 mg/kg per day or vehicle was orally given to rats for 14 days. All animals were necropsied on the day after the last dosing. Number of animals was indicated according to the abnormal grades. No abnormalities were observed in organs other than those in the table when examined microscopically. Grade: –, within normal limits; 1, slight; 2, moderate; 3, marked.
Discussion
The present study demonstrated that EZH1/2 dual inhibitors have greater activity than EZH2 selective inhibitors to reduce cellular H3K27me3 in HCT116 cells (Figs 1a, 3b). We confirmed this finding using other cell lines with different EZH1 and EZH2 expression pattern. We used LS180 and OCI‐LY7 cell lines whose EZH1 and EZH2 expression patterns were different from that of HCT116 (Fig. S4a). In these cells, EZH1/2 dual inhibitors, OR‐S1 and (R)‐OR‐S2, also showed greater H3K27me3 inhibition than the EZH2 selective inhibitor, OR‐S0 (Fig. S4e, S4g). In HCT116 cells, higher concentration of EZH1/2 dual inhibitors reduced the amount of PRC2 components such as EZH2 and SUZ12 (Fig. S4c); however, this effect was not detected in LS180 and OCI‐LY7 cells (Fig. S4e, S4g). The effect of EZH2 selective inhibitor on growth of HCT116 was weak, and EZH1/2 dual inhibitors also showed weak growth inhibition on this cell line (Fig. S4b). In contrast, EZH2 selective inhibitor completely inhibited the growth of LS180 and OCI‐LY7 and, for these cells, EZH1/2 dual inhibitors showed growth inhibition greater than the EZH2 selective inhibitor (Fig. S4d, S4f). These results indicate that dual inhibition of the methyltransferase activity of EZH1 and EZH2 shows greater growth inhibition for cells sensitive to EZH2 inhibition. EZH2, but not EZH1, has been reported to be a key factor for maintaining global H3K27me3 levels. However, it has been shown that EZH1 compensates for the function of EZH2 in cells depleted of EZH2.6, 47 This might be the reason why EZH1/2 dual inhibitor has greater activity than EZH2 selective inhibitor. A hematological cancer panel assay indicated that EZH1/2 dual inhibitor would be applicable at least for DLBCL with EZH2 mutation, PTCL, MM, and leukemia harboring fusion genes, such as MLL‐AF9, MLL‐AF4, or AML1‐ETO. The involvement of EZH2 in these cancers has been reported,( 45, 46, 47, 55 ) but little is currently known about the role of EZH1 in these cancers, except for MLL‐rearranged leukemia. As for the importance of EZH1 to MLL‐rearranged leukemia, it was suggested that simultaneous inhibition of PRC2‐EZH1 and PRC2‐EZH2 is needed to suppress leukemogenecity of MLL‐AF9‐induced AML cells.40, 41, 56 UNC1999, a recently reported EZH1/2 dual inhibitor, also suppressed growth of MLL‐rearranged leukemia cells in vitro and in vivo.57 However, in vitro growth inhibition efficacy of UNC1999 against MLL‐rearranged leukemia cells is not so strong compared with (R)‐OR‐S2. For example, EC50 (half maximal effective concentration) values of UNC1999 against MV‐4‐11 and RS4;11 are 524.8 and 1956 nM, respectively,57 whereas those of (R)‐OR‐S2 are 3.19 and 78.3 nM, respectively (Table S3). UNC1999 inhibits enzymatic activity of EZH2 at IC50 of <10 nM,57 and this value is almost the same as that of (R)‐OR‐S2 (2.5 nM). In contrast, the IC50 value of UNC1999 against enzymatic activity of EZH1 is 45 nM,57 which is weaker than that of (R)‐OR‐S2 (8.4 nM). This difference may be one of the reasons why (R)‐OR‐S2 showed better growth inhibitory efficacy than UNC1999. Additional studies are needed to elucidate the role of EZH1 in hematological cancers, especially when EZH2 is impaired. This would assist the development of predictive biomarker(s) to select patients suitable for treatment with EZH1/2 dual inhibitor.
Regarding SWI/SNF mutations, G401, a SMARCB1‐negative MRT cell line, showed sensitivity to EZH1/2 dual inhibitor in vitro and in vivo, which corroborated the previous findings that this cell line is sensitive to EZH2 selective inhibitors.31 However, regarding the other SWI/SNF subunits, clear correlations were generally not detected between sensitivity to EZH1/2 dual inhibitor and mutations in SWI/SNF subunits, even in ovarian cancer cells harboring ARID1A mutation. Bitler et al. has reported that inhibition of methyltransferase activity of EZH2 acts in a synthetic lethal manner in ARID1A‐mutated ovarian cancer cells.54 Our solid cancer panel included six ARID1A mutated ovarian cancer cell lines (A2780, OAW42, OVISE, OVMANA, SKOV3, and TOV‐21G), and only two of them (A2780 and TOV‐21G) were sensitive to EZH1/2 dual inhibitor (Table S3). Bitler et al. used 5 μM GSK126 as an EZH2 selective inhibitor, and difference in sensitivity thresholds might be the reason for the discrepancy between our results and those of Bitler et al. Although we could not find general correlations between sensitivity to EZH1/2 dual inhibitor and SWI/SNF mutations, there seemed to be good correlations between them among SCLC cell lines (Figs 4b, S2). We tested eight SCLC cell lines, and five of them (DMS114, SBC‐5, NCI‐H446, NCI‐H841, and NCI‐H1436) harbor loss‐of‐function mutations of SMARCA4 or ARID1A. Interestingly, all these cell lines showed higher sensitivity to EZH1/2 dual inhibitor than the other three cell lines that do not have SWI/SNF mutation (Table S3). As the number of tested SCLC cell lines is only eight, further investigation is necessary to ensure the accuracy of this correlation. Considering the results of a solid cancer panel assay so far, cell context might be important regarding the correlation between sensitivity to EZH1/2 dual inhibitor and SWI/SNF mutations.
As it has been reported that EZH2 and EZH1 are required for hematopoietic stem cell maintenance and EZH2 plays a critical role in B‐ and T‐cell development,9, 10, 11, 58, 59 we were concerned that long‐term EZH1/2 dual inhibition in vivo would cause serious on‐target toxicity in the lympho‐hematopoietic system. Repeated dose toxicity study of (R)‐OR‐S1, however, showed EZH1/2 dual inhibition in rats for 14 days induced no critical or severe toxicity up to systemic (R)‐OR‐S1 exposure (AUC0‐24 h) of 27 900 nM·h, which was approximately 17‐fold higher than exposure at pharmacological active dose in mice (Fig. 3c). Even at this high exposure, reduction of peripheral blood cell counts was only detected in leukocytes such as lymphocytes, which was consistent with the moderately decreased number of lymphocytes in the thymic cortex and spleen observed in histopathology. These results suggest that EZH1/2 dual inhibition causes no critical lympho‐hematopoietic toxicity.
In the present study, we showed that EZH1/2 dual inhibitors have greater antitumor efficacy than EZH2 selective inhibitor, and subtypes of hematological cancers sensitive to EZH1/2 dual inhibitor were suggested. Furthermore, long‐term EZH1/2 dual inhibition induced no critical toxicity in rats. These results indicate the possible use of EZH1/2 dual inhibitor for clinical applications.
Disclosure Statement
All authors, except for OK, TT, IY, and ASY, are employees of Daiichi Sankyo Co., Ltd. OK is an employee of Asubio Pharma Co., Ltd. TT and IY are employees of Daiichi Sankyo RD Novare Co., Ltd. ASY is an employee of Daiichi Sankyo India Pharma Pvt. Ltd.
Supporting information
Fig. S1. Antitumor activity of (R)‐OR‐S1 against a diffuse large B‐cell lymphoma KARPAS‐422 xenograft model.
Fig. S2. In vitro growth inhibition activities of (R)‐OR‐S2 against lung cancer cell lines.
Fig. S3. Effect of (R)‐OR‐S1 on food consumption and bodyweight in rats.
Fig. S4. Characterization of OR‐S0, OR‐S1 and (R)‐OR‐S2 using cancer cell lines.
Table S1. Assay conditions and IC50 values of OR‐S1 and (R)‐OR‐S2 for each methyltransferase.
Table S2. Selectivity of (R)‐OR‐S1 and (R)‐OR‐S2 against 253 kinases.
Table S3. Cell lines used in this study and the in vitro growth inhibitory activity of (R)‐OR‐S2 against them.
Table S4. Plasma concentrations and TK parameters of (R)‐OR‐S1 after oral dosing in rats.
Doc. S1. Materials and methods, and associated references.
Acknowledgments
The authors thank Tadashi Toki, Kentaro Ito, Mayumi Hayashi, Hiroyuki Hanzawa, and Kazusi Araki for valuable suggestions; Sachiko Takaishi and Takayuki Nagasawa for technical support; Toshihiro Kiho, Sho Takechi, and Tomoaki Hamada for compound synthesis; and Ryota Kawai and Toshihiko Makino for discussions of toxicology on the manuscript.
Cancer Sci 108 (2017) 2069–2078
Funding information
Daiichi Sankyo Co., Ltd.
References
- 1. Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 2007; 8: 9–22. [DOI] [PubMed] [Google Scholar]
- 2. Blackledge NP, Rose NR, Klose RJ. Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat Rev Mol Cell Biol 2015; 16: 643–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv 2015; 1: e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuzmichev A, Nishioka K, Erdjument‐Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 2002; 16: 2893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao R, Wang L, Wang H et al Role of histone H3 lysine 27 methylation in Polycomb‐group silencing. Science 2002; 298: 1039–43. [DOI] [PubMed] [Google Scholar]
- 6. Shen X, Liu Y, Hsu YJ et al EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008; 32: 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Margueron R, Li G, Sarma K et al Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008; 32: 503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ho L, Crabtree GR. An EZ mark to miss. Cell Stem Cell 2008; 3: 577–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mochizuki‐Kashio M, Mishima Y, Miyagi S et al Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 2011; 118: 6553–61. [DOI] [PubMed] [Google Scholar]
- 10. Hidalgo I, Herrera‐Merchan A, Ligos JM et al Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence‐like cell cycle arrest. Cell Stem Cell 2012; 11: 649–62. [DOI] [PubMed] [Google Scholar]
- 11. Xie H, Xu J, Hsu JH et al Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental‐stage‐specific manner. Cell Stem Cell 2014; 14: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev 2011; 25: 485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bardot ES, Valdes VJ, Zhang J et al Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J 2013; 32: 1990–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varambally S, Dhanasekaran SM, Zhou M et al The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419: 624–9. [DOI] [PubMed] [Google Scholar]
- 15. Kleer CG, Cao Q, Varambally S et al EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 2003; 100: 11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He LJ, Cai MY, Xu GL et al Prognostic significance of overexpression of EZH2 and H3k27me3 proteins in gastric cancer. Asian Pac J Cancer Prev 2012; 13: 3173–8. [DOI] [PubMed] [Google Scholar]
- 17. Behrens C, Solis LM, Lin H et al EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non‐small cell lung carcinoma. Clin Cancer Res 2013; 19: 6556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu CH, Han HD, Mangala LS et al Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010; 18: 185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toll AD, Dasgupta A, Potoczek M et al Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum Pathol 2010; 41: 1205–9. [DOI] [PubMed] [Google Scholar]
- 20. Wagener N, Macher‐Goeppinger S, Pritsch M et al Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer 2010; 10: 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao W, Feng Z, Cui Z et al Up‐regulation of enhancer of zeste homolog 2 is associated positively with cyclin D1 overexpression and poor clinical outcome in head and neck squamous cell carcinoma. Cancer 2012; 118: 2858–71. [DOI] [PubMed] [Google Scholar]
- 22. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med 2016; 22: 128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gonzalez ME, Li X, Toy K et al Downregulation of EZH2 decreases growth of estrogen receptor‐negative invasive breast carcinoma and requires BRCA1. Oncogene 2009; 28: 843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morin RD, Johnson NA, Severson TM et al Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet 2010; 42: 181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wigle TJ, Knutson SK, Jin L et al The Y641C mutation of EZH2 alters substrate specificity for histone H3 lysine 27 methylation states. FEBS Lett 2011; 585: 3011–4. [DOI] [PubMed] [Google Scholar]
- 26. McCabe MT, Graves AP, Ganji G et al Mutation of A677 in histone methyltransferase EZH2 in human B‐cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA 2012; 109: 2989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Majer CR, Jin L, Scott MP et al A687V EZH2 is a gain‐of‐function mutation found in lymphoma patients. FEBS Lett 2012; 586: 3448–51. [DOI] [PubMed] [Google Scholar]
- 28. Sneeringer CJ, Scott MP, Kuntz KW et al Coordinated activities of wild‐type plus mutant EZH2 drive tumor‐associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B‐cell lymphomas. Proc Natl Acad Sci USA 2010; 107: 20980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCabe MT, Ott HM, Ganji G et al EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature 2012; 492: 108–12. [DOI] [PubMed] [Google Scholar]
- 30. Knutson SK, Wigle TJ, Warholic NM et al A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012; 8: 890–6. [DOI] [PubMed] [Google Scholar]
- 31. Knutson SK, Warholic NM, Wigle TJ et al Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA 2013; 110: 7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qi W, Chan H, Teng L et al Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA 2012; 109: 21360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gehling VS, Vaswani RG, Nasveschuk CG et al Discovery, design, and synthesis of indole‐based EZH2 inhibitors. Bioorg Med Chem Lett 2015; 25: 3644–9. [DOI] [PubMed] [Google Scholar]
- 34. Ernst T, Chase AJ, Score J et al Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42: 722–6. [DOI] [PubMed] [Google Scholar]
- 35. Nikoloski G, Langemeijer SM, Kuiper RP. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010; 42: 665–7. [DOI] [PubMed] [Google Scholar]
- 36. Ntziachristos P, Tsirigos A, Van Vlierberghe P et al Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 2012; 18: 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simon C, Chagraoui J, Krosl J et al A key role for EZH2 and associated genes in mouse and human adult T‐cell acute leukemia. Genes Dev 2012; 26: 651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sashida G, Harada H, Matsui H et al Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun 2014; 5: 4177. [DOI] [PubMed] [Google Scholar]
- 39. Mochizuki‐Kashio M, Aoyama K, Sashida G et al Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1‐dependent manner. Blood 2015; 126: 1172–83. [DOI] [PubMed] [Google Scholar]
- 40. Neff T, Sinha AU, Kluk MJ et al Polycomb repressive complex 2 is required for MLL‐AF9 leukemia. Proc Natl Acad Sci USA 2012; 109: 5028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim W, Bird GH, Neff T et al Targeted disruption of the EZH2‐EED complex inhibits EZH2‐dependent cancer. Nat Chem Biol 2013; 9: 643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brooun A, Gajiwala KS, Deng YL et al Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun 2016; 7: 11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vaswani RG, Gehling VS, Dakin LA et al Identification of (R)‐N‐((4‐Methoxy‐6‐methyl‐2‐oxo‐1,2‐dihydropyridin‐3‐yl)methyl)‐2‐methyl‐1‐(1‐(1‐(2,2,2‐trifluoroethyl)piperidin‐4‐yl)ethyl)‐1H‐indole‐3‐carboxamide (CPI‐1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B‐Cell Lymphomas. J Med Chem 2016; 59: 9928–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Justin N, Zhang Y, Tarricone C et al Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 2016; 7: 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herviou L, Cavalli G, Cartron G, Klein B, Moreaux J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2016; 7: 2284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sashida G, Iwama A. Multifaceted role of the polycomb‐group gene EZH2 in hematological malignancies. Int J Hematol 2017; 105: 23–30. [DOI] [PubMed] [Google Scholar]
- 47. Takamatsu‐Ichihara E, Kitabayashi I. The roles of Polycomb group proteins in hematopoietic stem cells and hematological malignancies. Int J Hematol 2016; 103: 634–42. [DOI] [PubMed] [Google Scholar]
- 48. Herrera‐Merchan A, Arranz L, Ligos JM, de Molina A, Dominguez O, Gonzalez S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun 2012; 3: 623. [DOI] [PubMed] [Google Scholar]
- 49. Souroullas GP, Jeck WR, Parker JS et al An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med 2016; 22: 632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kadoch C, Hargreaves DC, Hodges C et al Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013; 45: 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013; 8: e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson BG, Wang X, Shen X et al Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010; 18: 316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim KH, Kim W, Howard TP et al SWI/SNF‐mutant cancers depend on catalytic and non‐catalytic activity of EZH2. Nat Med 2015; 21: 1491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bitler BG, Aird KM, Garipov A et al Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A‐mutated cancers. Nat Med 2015; 21: 231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Giulino‐Roth L. EZH2: a potential new target in T‐cell lymphoma? Leuk Lymphoma 2015; 56: 1924–5. [DOI] [PubMed] [Google Scholar]
- 56. Shi J, Wang E, Zuber J et al The Polycomb complex PRC2 supports aberrant self‐renewal in a mouse model of MLL‐AF9;Nras(G12D) acute myeloid leukemia. Oncogene 2013; 32: 930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu B, On DM, Ma A et al Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL‐rearranged leukemia. Blood 2015; 125: 346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Su IH, Basavaraj A, Krutchinsky AN et al Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 2003; 4: 124–31. [DOI] [PubMed] [Google Scholar]
- 59. Karantanos T, Chistofides A, Barhdan K, Li L, Boussiotis VA. Regulation of T Cell differentiation and function by EZH2. Front Immunol 2016; 7: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barretina J, Caponigro G, Stransky N et al The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483: 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Forbes SA, Beare D, Gunasekaran P et al COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43: D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao J, Aksoy BA, Dogrusoz U et al Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cerami E, Gao J, Dogrusoz U et al The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Antitumor activity of (R)‐OR‐S1 against a diffuse large B‐cell lymphoma KARPAS‐422 xenograft model.
Fig. S2. In vitro growth inhibition activities of (R)‐OR‐S2 against lung cancer cell lines.
Fig. S3. Effect of (R)‐OR‐S1 on food consumption and bodyweight in rats.
Fig. S4. Characterization of OR‐S0, OR‐S1 and (R)‐OR‐S2 using cancer cell lines.
Table S1. Assay conditions and IC50 values of OR‐S1 and (R)‐OR‐S2 for each methyltransferase.
Table S2. Selectivity of (R)‐OR‐S1 and (R)‐OR‐S2 against 253 kinases.
Table S3. Cell lines used in this study and the in vitro growth inhibitory activity of (R)‐OR‐S2 against them.
Table S4. Plasma concentrations and TK parameters of (R)‐OR‐S1 after oral dosing in rats.
Doc. S1. Materials and methods, and associated references.