

Abstract
A phase II study of S‐1 plus leucovorin (LV) given in a 4‐week schedule (2 weeks’ administration followed by 2 weeks’ rest) for patients with untreated metastatic colorectal cancer (mCRC) showed that the combination was effective, but grade 3 toxicities (diarrhea, stomatitis and anorexia) occurred at a relatively high rate. In this phase II study, we evaluated the efficacy and safety of a 2‐week schedule of S‐1 plus LV. Patients with mCRC received oral S‐1 (40–60 mg) and LV (25 mg) twice daily for 1 week, followed by 1 week's rest. Treatment was repeated until disease progression or unacceptable toxicity. The primary endpoint was response rate. The pharmacokinetics of S‐1 and LV in Chinese patients were evaluated on day 1 of the first cycle. Seventy‐three patients were enrolled in Japan and China. Of 71 eligible patients, the response rate was 53.5%, and the disease control rate was 83.1%. Median progression‐free survival and median overall survival were 6.5 and 24.3 months, respectively. The incidences of grade 3 toxicities were diarrhea 8.3%, stomatitis 8.3%, anorexia 2.8% and neutropenia 9.7%. There were no treatment‐related deaths. The pharmacokinetics profiles of S‐1 plus LV in Chinese patients were similar to those in Japanese patients. This 2‐week schedule of S‐1 plus LV showed good efficacy and better tolerability than the 4‐week schedule. This therapy will be the base regimen for mCRC to be added by other cytotoxic or molecular‐targeted drugs. The optimized treatment schedule for S‐1 plus LV was 1 week on and 1 week off.
Keywords: Clinical trial, colorectal neoplasms, leucovorin, pharmacokinetics, S‐1 (combination)
Colorectal cancer (CRC) is the third most common cancer and the fourth most common cause of cancer deaths worldwide, accounting for roughly 1.2 million new cases and 600 000 deaths per year.1 Chemotherapy for metastatic CRC (mCRC) has been shown to be superior to best supportive care in terms of survival.2, 3, 4
5‐Fluorouracil (5‐FU) has been the backbone of combination therapy for patients with mCRC for several decades and is often administered in combination with leucovorin (LV). Due to the advantage of being more convenient for patients, oral fluoropyrimidine monotherapy or its combination chemotherapy has been replacing infusion of 5‐FU.
S‐1 is an oral fluoropyrimidine combination product containing tegafur, a prodrug of 5‐FU, gimeracil, which enhances antitumor efficacy by inhibiting degradation of 5‐FU, and oteracil potassium, which reduces gastrointestinal toxicity by specifically inhibiting phosphorylation of 5‐FU in the digestive tract, at a molar ratio of 1.0:0.4:1.0.5, 6, 7 Phase II studies of S‐1 in patients with mCRC showed response rates of 37% and good survival times.8, 9
Leucovorin enhances the antitumor efficacy of 5‐FU by stabilizing the ternary complex with thymidylate synthase and 5‐fluoro‐2′‐deoxyuridine monophosphate. A meta‐analysis showed that 5‐FU/LV improves response rates and overall survival (OS) compared with 5‐FU alone in mCRC.10 Furthermore, when used together with oral LV, oral uracil/tegafur (UFT) was shown to be as effective as infusional 5‐FU/LV, with significantly lower rates of diarrhea, nausea, vomiting and stomatitis.11, 12
In the phase I/II study of oral S‐1 plus oral LV in patients with mCRC, the recommended treatment schedule was 2 weeks of S‐1 plus LV administration followed by 2 weeks’ rest (4‐week schedule).13, 14 This regimen showed promising efficacy, with a response rate of 57.1% and median OS of 24.3 months. The incidences of grade 3 diarrhea, stomatitis and rash were 32, 20 and 2%, respectively, and the main reasons for dose reduction and extending the rest period were diarrhea, stomatitis and rash. The median times to onset of diarrhea and stomatitis were 15 and 12 days, respectively. The median times to the worst grade of these toxicities were 20 days and 14 days, respectively, and the time to resolution of these toxicities from the worst grade was 7 and 10 days, respectively.14
Based on the findings above, we modified the treatment schedule of S‐1 plus LV to 1 week administration followed by 1 week rest (2‐week schedule), which maintains the same dose intensity as the 4‐week schedule, with the expectation of decreasing these toxicities. The main purpose of the present study was optimization of the S‐1 plus LV regimen for better tolerability. In addition, when we planned the present study, molecular targeted drugs (i.e. bevacizumab or cetuximab) had just been approved in Japan and not yet approved in China. Considering the situation of clinical practice, we selected this regimen without molecular targeted drugs. In this phase II study, we evaluated the efficacy and safety of the 2‐week schedule of S‐1 plus LV regimen for mCRC.
Materials and Methods
Patients
Chemotherapy‐naïve patients with histologically confirmed mCRC were eligible for inclusion in the study. Other inclusion criteria were: at least one measurable lesion according to the Response Evaluation Criteria In Solid Tumors (RECIST) version 1.0; adequate oral intake; age ≥20 years; no prior chemotherapy, radiation therapy or hormonal therapy (adjuvant chemotherapy other than S‐1, completed more than 180 days before enrollment, was allowed); ECOG performance status 0 to 1; and adequate bone marrow, liver and renal functions (serum hemoglobin concentration ≥10.0 g/dL, white blood cell count ≤12 000/mm3, neutrophil count ≥2000/mm3, platelet count ≥75 000/mm3, serum total bilirubin concentration less than 1.5 times the upper limit of the normal institutional level [ULN], serum aminotransferase [AST] and alanine aminotransferase [ALT] concentration less than 2.5 times the ULN and serum creatinine level less than the ULN).
All patients provided written informed consent. The study was approved by the institutional review boards of all participating centers and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. The present study was sponsored by Taiho Pharmaceutical (ClinicalTrials.gov: NCT00891332).
Study drug dosing and treatment
S‐1 (capsules containing 20 or 25 mg of tegafur) and LV (25‐mg tablets) were provided by Taiho Pharmaceutical (Tokyo, Japan). The dose of S‐1 was determined according to the body surface area as follows: <1.25 m2, 40 mg; 1.25 to <1.50 m2, 50 mg; and ≥1.50 m2, 60 mg. LV was given at a fixed dose of 25 mg. S‐1 and LV were given together orally twice a day after meals for 1 week, followed by a 1‐week rest period (1 week on, 1 week off).
Treatment was repeated until radiographically documented progression, unacceptable toxicity, or patient's refusal to continue treatment. The dose of S‐1 could be decreased by one level in cases of the following toxicity: grade 4 hematological toxicity or grade 3 diarrhea, stomatitis or rash. The dose of LV could not be decreased.
Physical examination and laboratory tests were done at baseline and repeated every week for 4 weeks, and every 2 weeks from 5 weeks onward. Tumors were assessed at baseline (within 30 days before enrollment), and every 5 weeks after enrollment until progression. Radiological images were evaluated according to the RECIST by the independent review committee (IRC) to evaluate the response and progression. Progression‐free survival (PFS) was counted from the date of enrollment to the date when progressive disease was first confirmed or death from any cause without disease progression. For patients whose protocol treatment was discontinued for any reason without confirmation of disease progression by the IRC, PFS was censored on the date of the last tumor assessment with non‐progression status during the treatment period. OS was calculated from the date of enrollment to the date of death or censored at the last confirmation of survival.
Efficacy and safety assessments
The primary endpoint was response rate. Secondary endpoints included disease control rate (DCR), PFS, time to treatment failure (TTF), OS, relative dose intensity (RDI) and safety. Toxicity was assessed by the National Cancer Institute Common Toxicity Criteria for Adverse Events version 3.0.
Pharmacokinetics analysis
In the Chinese patients, on day 1 of the first cycle, blood samples for pharmacokinetics (PK) analysis of S‐1 and LV were collected before administration of these drugs and 1, 2, 4, 6 and 8 h after administration. Plasma concentrations of FT, 5‐FU, CDHP, Oxo, LV and 5‐methyltetrahydrofolate (5‐MeTHF) were quantified as described previously.15, 16 Maximum drug concentrations in plasma (Cmax), area under the concentration‐time curve (AUC0–8) and elimination half‐life (T1/2) were calculated by non‐compartment model analysis, performed with the use of WinNonlin, version 6.1 (Pharsight, Cary, NC, USA).
Statistical analysis
The response rate in the previous phase II study of S‐1 alone in patients with CRC was 37.4% (95% confidence interval [CI], 27.9 to 47.7). Therefore, the threshold of response rate under the null hypothesis was set at 30%, and the expected response rate under the alternative hypothesis was 50%, which was similar response rate as that in the phase II study of S‐1 plus LV (2 weeks on, 2 weeks off) in patients with CRC, and was approximately 20% higher than the response rate for S‐1 alone. If the response rate was 50%, a sample size of 68 patients would ensure a power of at least 90% for a one‐sided significance level of 2.5% in order to reject the null hypothesis that the response rate was <30%. To evaluate the exposure to S‐1 and LV in Chinese patients of the 1 week on and 1 week off regimen, the Cmax and AUC0–8 of S‐1 components, 5‐FU, LV and 5‐MeTHF in Chinese patients were compared with the respective values in Japanese patients of a previous phase I study (2 weeks on, 2 weeks off).13 Differences in Cmax and AUC0–8 between Japanese patients and Chinese patients were evaluated by applying ANOVA to log‐transformed means for each parameter.
Results
Patients’ characteristics
From October 2008 through June 2009, 73 patients were enrolled at 9 hospitals in Japan and 5 hospitals in China. One patient who had synchronous cancer (with gastric cancer) was excluded from all analyses, and 1 patient who had received prior chemotherapy (5‐FU intraperitoneal) was excluded from the efficacy analysis. Accordingly, 72 and 71 patients formed the safety analysis set (SAS) and the full analysis set (FAS), respectively.
The demographic data, site of metastatic tumor, and other patient characteristics are summarized in Table 1. The median age was 60 years (range 27–84 years) and 53.5% of patients had a performance status of 0.
Table 1.
Patients’ characteristics
| Number (n = 71) | % | |
|---|---|---|
| Country | ||
| Japan | 32 | 45.1 |
| China | 39 | 54.9 |
| Age (years) | ||
| Median [range] | 60 [27–84] | |
| Sex | ||
| Male | 38 | 53.5 |
| Female | 33 | 46.5 |
| Performance status | ||
| 0 | 38 | 53.5 |
| 1 | 33 | 46.5 |
| Stage | ||
| Stage IV | 44 | 62.0 |
| Recurrent | 27 | 38.0 |
| Primary tumor site | ||
| Colon | 41 | 57.7 |
| Rectum | 28 | 39.4 |
| Colon and rectum | 2 | 2.8 |
| Number of organs involved | ||
| 1 | 23 | 32.4 |
| 2 | 14 | 19.7 |
| ≥3 | 34 | 26.8 |
Efficacy
The response rate (the primary endpoint) was evaluated in 71 patients. The response rate was 53.5% (95% CI, 41.3–65.5) and the DCR was 83.1% (95% CI, 72.3–91.0) (Table 2). As a result, the null hypothesis was rejected, with the lower limit of the 95% CI exceeding the threshold response rate, demonstrating that the primary objective of the study was met.
Table 2.
Tumor response
| No. (n = 71) | % | |
|---|---|---|
| Complete response | 0 | 0 |
| Partial response | 38 | 53.5 |
| Stable disease | 21 | 29.6 |
| Progressive disease | 6 | 8.5 |
| Non‐evaluable | 6 | 8.5 |
| Response rate | 38 | 53.5 |
| 95% CI | 41.3–65.5 | |
| Disease control rate | 59 | 83.1 |
| 95% CI | 72.3–91.0 |
Assessed by independent review committee according to Response Evaluation Criteria In Solid Tumors (RECIST) version 1.0. CI, confidence interval.
Among the 71 patients in the FAS, median PFS and TTF were 6.5 months (95% CI, 5.4–7.1) and 5.5 months (95% CI, 4.6–6.7), respectively (Fig. 1). With a median follow‐up of 26.4 months, the median survival time (MST) was 24.3 months (95% CI, 18.7–XXX) with a survival rate of 77.5% at 1 year and 53.2% at 2 years (Fig. 2).
Figure 1.
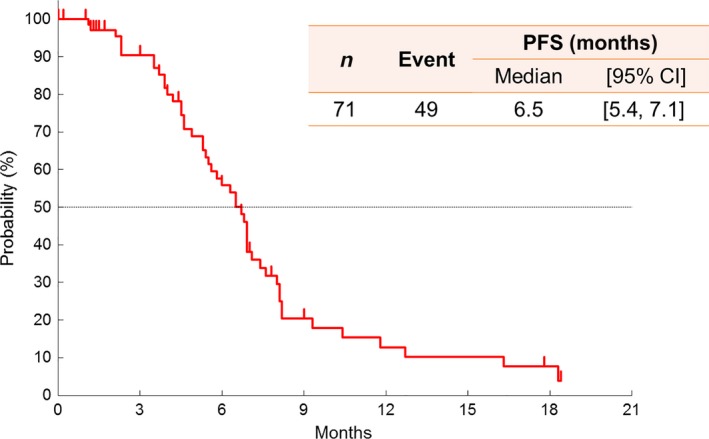
Kaplan–Meier curve of PFS assessed by the independent review committee. CI, confidence interval; PFS, progression‐free survival.
Figure 2.
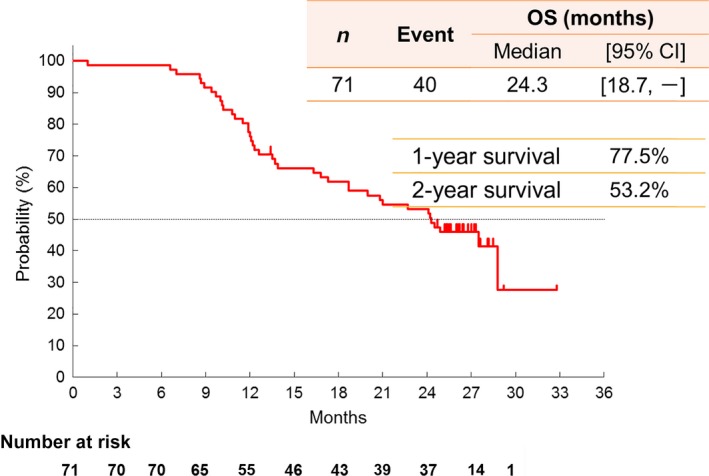
Kaplan–Meier curve of OS. CI, confidence interval; OS, overall survival.
Safety assessment
A summary of the toxicities is shown in Table 3. Grade 3/4 toxicities occurred in 31 of 72 patients (43.1%) in the SAS, and events occurring at a frequency of greater than 5% were neutropenia (9.7%), lymphopenia (9.7%), diarrhea (8.3%) and stomatitis (8.3%). Grade 4 toxicity occurred in only 1 patient (increased amylase). There were no treatment‐related deaths. The safety profile between Japanese and Chinese patients showed similar tendency (data not shown).
Table 3.
Hematological and non‐hematological adverse drug reactions
| Adverse event (n = 72a) | All (%) | Grade 2 (%) | Grade 3 (%) | Grade 4 (%) |
|---|---|---|---|---|
| Leukopenia | 47.2 | 18.1 | 2.8 | 0 |
| Anemia | 40.3 | 29.2 | 0 | 0 |
| Neutropenia | 43.1 | 13.9 | 9.7 | 0 |
| Lymphopenia | 25.0 | 12.5 | 9.7 | 0 |
| Thrombocytopenia | 11.1 | 1.4 | 0 | 0 |
| Total bilirubin increased | 31.9 | 25.0 | 0 | 0 |
| Anorexia | 63.9 | 19.4 | 2.8 | 0 |
| Nausea | 48.6 | 9.7 | 1.4 | 0 |
| Stomatitis | 58.3 | 18.1 | 8.3 | 0 |
| Diarrhea | 43.1 | 20.8 | 8.3 | 0 |
| Fatigue | 61.1 | 13.9 | 1.4 | 0 |
| Rash | 38.9 | 6.9 | 0 | 0 |
| Hyperpigmentation | 76.4 | 18.1 | 0 | 0 |
Analysis set included all treated patients.
The main reasons for dose reductions were stomatitis, rash and diarrhea. A dose reduction occurred at least once in 23 patients (32.4%), and two dose reductions occurred in 4 patients (5.6%). The incidence of treatment discontinuation during the cycle was 25.4%. The median RDI was 93% for S‐1 and 94% for LV. The median times to onset of diarrhea and stomatitis were 25 and 10.5 days, respectively. The median times to the worst grade of these toxicities were 42 and 15 days, respectively.
Reasons for treatment discontinuation and post‐treatment
Treatment was discontinued due to progressive disease in 52 patients (73.2%), adverse events in 4 (5.6%), surgery in 4 (5.6%), refusal in 7 (9.9%) and other reasons in 4 (5.6%).
Fifty‐six patients (78.9%) were given post‐treatment according to the investigators’ choice, of whom 29 (51.8%) received oxaliplatin‐based chemotherapy (FOLFOX ± bevacizumab, FOLFOX plus cetuximab or XELOX ± bevacizumab), 7 (12.5%) received 5‐FU‐based (S‐1 or capecitabine) monotherapy or combination therapy, 1 (1.8%) received irinotecan and 11 (19.6%) underwent surgery.
Pharmacokinetics
Twelve Chinese patients underwent PK analysis of S‐1 and LV. The PK parameters are summarized in Table 4 and the comparison of AUC0–8 of each compound in Chinese patients (1 week on, 1 week off) with that in Japanese patients (2 weeks on, 2 weeks off) is shown in Figure 3. No significant differences (P < 0.05) in AUC0–8 of all compounds in Chinese patients of the 1 week on and 1 week off regimen were found from those in Japanese patients undergoing the 2 weeks on and 2 weeks off regimen. There were also no significant differences in Cmax of all compounds between Chinese and Japanese patients (data not shown).
Table 4.
PK analysis
| Cmax (ng/mL) | AUC0–8 (ng·hr/mL) | T1/2 a (hr) | |
|---|---|---|---|
| FT | 1940 ± 441 | 9801 ± 2194 | 5.8 ± 2.1 |
| 5‐FU | 172.9 ± 53.7 | 774.5 ± 275.9 | 1.5 ± 0.2 |
| CDHP | 351.1 ± 118.7 | 1172 ± 295 | 2.3 ± 0.2 |
| Oxo | 55.6 ± 58.5 | 201.4 ± 215.9 | 2.2 ± 0.8 |
| LV | 434.5 ± 374.0 | 2336 ± 1938 | 7.0 ± 1.2 |
| 5‐MeTHF | 430.7 ± 121.7 | 1900 ± 632 | 2.3 ± 0.3 |
T1/2 of FT, 5‐FU, CDHP, Oxo, LV and 5‐MeTHF were calculated from 10, 8, 10, 9, 7 and 5 patients, respectively. PK, pharmacokinetics; FT, tegafur; 5‐FU, 5‐fluorouracil; CDHP, 5‐chloro‐2,4‐dihydroxypyridine; Oxo, potassium oxonate; LV, leucovorin; 5‐MeTHF, 5‐methyltetrahydrofolate.
Figure 3.
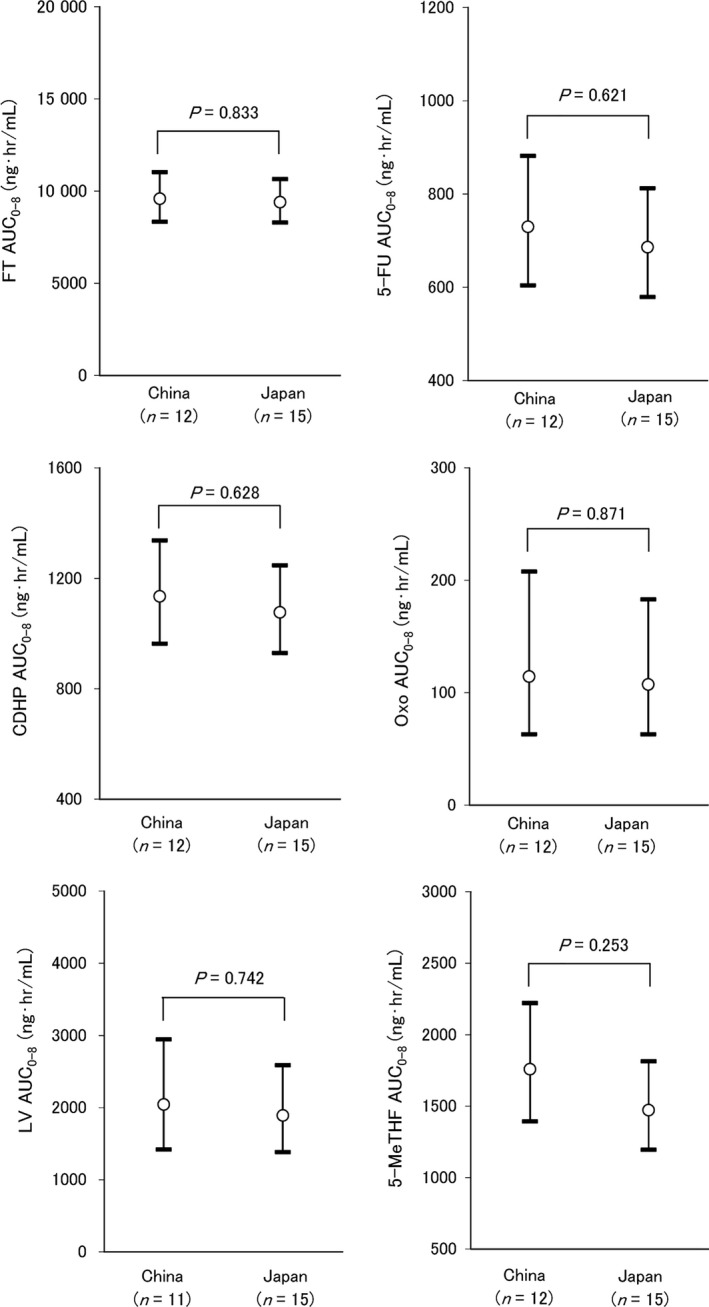
Comparison of AUC 0–8 of FT, 5‐FU, CDHP, Oxo, LV and 5‐MeTHF in S‐1 plus LV combination chemotherapy between Chinese and Japanese on day 1. China, S‐1 plus LV combination therapy in Chinese (this study); Japan, S‐1 plus LV combination chemotherapy in Japanese (reference 13). AUC, area under the concentration time curve; FT tegafur, 5‐FU 5‐fluorouracil; CDHP, 5‐chloro‐2,4‐dihydroxypyridine, Oxo, potassium oxonate; LV, leucovorin; 5‐MeTHF, 5‐methyltetrahydrofolate
Discussion
As shown by many studies, when combined with 5‐FU, LV improves response rates and OS compared with 5‐FU alone in mCRC. However, a randomized phase II study reported response rates for combination therapy with LV plus capecitabine of 23%, and for capecitabine alone of 21 and 24% in CRC.17 The addition of LV to capecitabine did not result in a marked increase in response rate due to its severe toxicities. It appears necessary to optimize the dose and treatment schedule of combination chemotherapy with oral fluoropyrimidine and LV.
In a previous phase I/II study, the recommended treatment schedule for S‐1 plus oral LV was 2 weeks of administration followed by a 2‐week rest period.13, 14 The response rate was 57.1%, median time to tumor progression was 6.7 months, and median OS was 24.3 months,14 while response rates for S‐1 alone in two phase II studies in mCRC conducted in Japan were 37%.8, 9 It is suggested that LV enhances the antitumor efficacy of S‐1.
In this phase II study in China and Japan, we evaluated the efficacy and safety of a 2‐week schedule for S‐1 plus oral LV as first‐line treatment for mCRC. The response rate, the primary endpoint of this study, was 53.5%. With a median follow‐up of 26.4 months, the median PFS was 6.5 months, MST was 24.3 months, and survival rates were 77.5% at 1 year and 53.2% at 2 years. The modified treatment schedule of S‐1 plus LV (1 week on, 1 week off) showed similar efficacy to the previous treatment schedule (2 weeks on, 2 weeks off).
As for the toxicities for S‐1 plus LV (2 weeks on, 2 weeks off), grade 3/4 toxicities occurred in 35 of 56 patients (55%), and the incidences of grade 3 diarrhea and stomatitis were 32 and 20%, respectively. In this study, the incidences of grade 3 diarrhea and stomatitis were remarkably decreased. Time to onset and recovery of diarrhea and stomatitis in the previous study with a 4‐week schedule14 and in this study with a 2‐week schedule are shown in Figure 4. Grade 3/4 toxicities occurred in 31 of 72 patients (43.1%), and the incidences of grade 3 or higher diarrhea and stomatitis were as low as 8.3%. The incidence of dose reduction of S‐1 decreased from 58.9% in the previous study to 32.4% in this study, and that of treatment discontinuation during the cycle decreased from 62.5 to 25.4%, respectively. The median RDI of S‐1 was 81% in the previous study and 93% in this study, and the median RDI of LV was 93 and 94%, respectively. Therefore, this regimen can maintain a high RDI during treatment. It is considered that a short duration of administration could succeed in reducing toxicities of S‐1 plus LV (Fig. 4).
Figure 4.
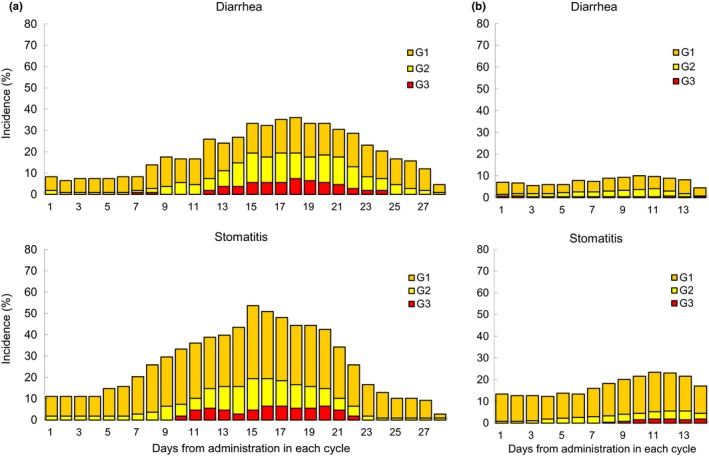
Time to onset and recovery of diarrhea and stomatitis in (a) the S‐1/LV 2 weeks on, 2 weeks off regimen (n = 108; Cycle 1 = 56, Cycle 2 = 52) and (b) the S‐1/LV 1 week on, 1 week off regimen in this study (n = 269; Cycle 1 = 72, Cycle 2 = 69, Cycle 3 = 66, Cycle 4 = 62). G, grade; LV, leucovorin.
As for the PK, while it was reported that PK comparisons showed similar 5‐FU exposure despite higher FT metabolism in Caucasians than Asian,18 there were no significant differences in the PK parameters of S‐1 and LV between Chinese patients and Japanese patients, indicating that no obvious ethnic differences existed between Chinese and Japanese patients. Furthermore, this result suggests that the daily exposures to 5‐FU, which is an active component of S‐1 and LV, in the 1 week on and 1 week off regimen were comparable to those in the 2 week on and 2 week off regimen, but total exposures during the one cycle to these compounds during repeated administrations for the 1 week on and 1 week off regimen are lower than those in the 2 week on and 2 week off regimen.
After the end of this study, 56 patients (78.9%) were given post‐treatment according to the investigators’ preference, 29 of whom received oxaliplatin‐based chemotherapy, 1 received irinotecan‐based chemotherapy, and 4 underwent curative surgery. The median OS in Japan and China were 28.8 and 20.8 months, respectively, but there were no significant differences in PK between Japanese and Chinese. The reasons for the differences in OS may include the content of subsequent therapy. The proportions of patients receiving subsequent chemotherapy were 78% in Japan and 46% in China. In addition, all patients who received oxaliplatin‐based plus bevacizumab chemotherapy were Japanese because bevacizumab was not approved in China during the time of the study. Several studies have shown that the addition of bevacizumab to oxaliplatin‐based chemotherapy improves survival duration or PFS for patients with previously treated mCRC and for those receiving first‐line treatment.19, 20 The response rate (59% in Japan, 49% in China), the median PFS (6.3 months in Japan, 6.8 months in China), the median RDI of S‐1 (85% in Japan, 94% in China), safety profile, and PK profile were similar in both countries, suggesting that the difference in OS was mainly affected by post‐treatment.
In conclusion, the S‐1 plus LV (1 week on, 1 week off) treatment regimen showed promising activity and safety in patients with untreated mCRC. Based on these findings, phase II trials of S‐1 plus LV‐based combination therapy with oxaliplatin and bevacizumab were conducted in Japan, and showed promising efficacy with feasible toxicity for mCRC.21, 22 In addition, S‐1 plus LV therapy was evaluated in patients with gemcitabine‐refractory advanced pancreatic cancer23 and as first‐line treatment for advanced gastric cancer 24 in phase II studies. For the patients to be treated with more intensive chemotherapy, this 2‐week regimen will be the base regimen to be added by other cytotoxic drugs or molecular‐targeted drugs. Further study of this combination chemotherapy is warranted.
Disclosure Statement
Ken Shimada, Takeshi Kato and Hideo Baba received honoraria from Taiho Pharmaceutical. Tadamichi Denda, Yasushi Toh and Hideo Baba received research funding from Taiho Pharmaceutical. All remaining authors have no conflicts of interest to declare. The study was designed under the responsibility of Taiho Pharmaceutical, in conjunction with the investigators. The study was funded by Taiho Pharmaceutical. The study drug was provided by Taiho Pharmaceutical. Taiho Pharmaceutical. collected and analyzed the data and participated in the interpretation of the study. The corresponding author had final responsibility for the decision to submit the paper for publication.
Acknowledgments
We appreciate the contribution of all patients, their families, the investigators and the medical staff. We are grateful to Yoshihiko Maehara, Keisuke Aiba, Atsushi Ohtsu and Maolin Jin for their kind advice as members of the independent data and safety monitoring committee and to Kohki Yoshikawa, Junji Tanaka and Takayuki Yoshino as members of the independent review committee. We also thank Narikazu Boku for his advice as the medical advisor. In addition, we thank Taiho Pharmaceutical for providing data from a previous S‐1 plus LV study. This study was sponsored by Taiho Pharmaceutical.
Cancer Sci 108 (2017) 2045–2051
Clinical trial registration: NCT00891332
Funding Information
Taiho Pharmaceutical Co., Ltd.
References
- 1. International Agency for Research on Cancer, World Health Organization . GLOBOCAN 2012: Estimated cancer incidence, mortality and prevalence worldwide in 2012. [Cited 01 Mar 2017.] Available from URL: http://globocan.iarc.fr/
- 2. Scheithauer W, Rosen H, Kornek GV et al Randomised comparison of combination chemotherapy plus supportive care with supportive care alone in patients with metastatic colorectal cancer. BMJ 1993; 306: 752–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simmonds PC. Palliative chemotherapy for advanced colorectal cancer: systematic review and meta‐analysis. Colorectal Cancer Collaborative Group. BMJ 2000; 321: 531–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cunningham D, Pyrhönen S, James RD et al Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet 1998; 352: 1413–18. [DOI] [PubMed] [Google Scholar]
- 5. Shirasaka T, Shimamato Y, Ohshimo H et al Development of a novel form of an oral 5‐fluorouracil derivative (S‐1) directed to the potentiation of the tumor selective cytotoxicity of 5‐fluorouracil by two biochemical modulators. Anticancer Drugs 1996; 7: 548–57. [DOI] [PubMed] [Google Scholar]
- 6. Shirasaka T, Shimamoto Y, Fukushima M. Inhibition by oxonic acid of gastrointestinal toxicity of 5‐fluorouracil without loss of its antitumor activity in rats. Cancer Res 1993; 53: 4004–9. [PubMed] [Google Scholar]
- 7. Takechi T, Nakano K, Uchida J et al Antitumor activity and low intestinal toxicity of S‐1, a new formulation of oral tegafur, in experimental tumor models in rats. Cancer Chemother Pharmacol 1997; 39: 205–11. [DOI] [PubMed] [Google Scholar]
- 8. Ohtsu A, Baba H, Sakata Y et al Phase II study of S‐1, a novel oral fluoropyrimidine derivative, in patients with metastatic colorectal carcinoma. Br J Cancer 2000; 83: 141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shirao K, Ohtsu A, Takada H et al Phase II study of oral S‐1 for treatment of metastatic colorectal carcinoma. Cancer 2004; 100: 2355–61. [DOI] [PubMed] [Google Scholar]
- 10. Thirion P, Michiels S, Pignon JP et al Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: an updated meta‐analysis. J Clin Oncol 2004; 22: 3766–75. [DOI] [PubMed] [Google Scholar]
- 11. Douillard JY, Hoff PM, Skillings JR et al Multicenter phase III study of uracil/tegafur and oral leucovorin versus fluorouracil and leucovorin in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2002; 20: 3605–16. [DOI] [PubMed] [Google Scholar]
- 12. Carmichael J, Popiela T, Radstone D et al Randomized comparative study of tegafur/uracil and oral leucovorin versus parenteral fluorouracil and leucovorin in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2002; 20: 3617–27. [DOI] [PubMed] [Google Scholar]
- 13. Yoshino T, Hyodo I, Nishina T et al Phase I clinical and pharmacokinetic study of S‐1 plus oral leucovorin in patients with metastatic colorectal cancer. Cancer Chemother Pharmacol 2017; 79: 107–16. [DOI] [PubMed] [Google Scholar]
- 14. Koizumi W, Boku N, Yamaguchi K et al Phase II study of S‐1 plus leucovorin in patients with metastatic colorectal cancer. Ann Oncol 2010; 21: 766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsushima E, Yoshida K, Kitamura R, Yoshida K. Determination of S‐1 (combined drug of tegafur, 5‐chloro‐2,4‐dihydroxypyridine and potassium oxonate) and 5‐fluorouracil in human plasma and urine using high‐performance liquid chromatography and gas chromatography‐negative ion chemical ionization mass spectrometry. J Chromatogr B Biomed Sci Appl 1997; 691: 95–104. [DOI] [PubMed] [Google Scholar]
- 16. Shirao K, Hoff PM, Ohtsu A et al Comparison of the efficacy, toxicity, and pharmacokinetics of a uracil/tegafur (UFT) plus oral leucovorin (LV) regimen between Japanese and American patients with advanced colorectal cancer: Joint United States and Japan study of UFT/LV. J Clin Oncol 2004; 22: 3466–74. [DOI] [PubMed] [Google Scholar]
- 17. Van Cutsem E, Findlay M, Osterwalder B et al Capecitabine, an oral fluoropyrimidine carbamate with substantial activity in advanced colorectal cancer: Results of a randomized phase II study. J Clin Oncol 2000; 18: 1337–45. [DOI] [PubMed] [Google Scholar]
- 18. Chuah B, Goh BC, Lee SC et al Comparison of the pharmacokinetics and pharmacodynamics of S‐1 between Caucasian and East Asian patients. Cancer Sci 2011; 102: 478–83. [DOI] [PubMed] [Google Scholar]
- 19. Giantonio BJ, Catalano PJ, Meropol NJ et al Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 2007; 25: 1539–44. [DOI] [PubMed] [Google Scholar]
- 20. Saltz LB, Clarke S, Díaz‐Rubio E et al Bevacizumab in combination with oxaliplatin‐based chemotherapy as first‐line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008; 26: 2013–19. [DOI] [PubMed] [Google Scholar]
- 21. Yamazaki K, Kuwano H, Ojima H et al A randomized phase II study of combination therapy with S‐1, oral leucovorin, and oxaliplatin (SOL) and mFOLFOX6 in patients with previously untreated metastatic colorectal cancer. Cancer Chemother Pharmacol 2015; 75: 569–77. [DOI] [PubMed] [Google Scholar]
- 22. Nishina T, Kato T, Yamazaki K et al A phase II study of S‐1, oxaliplatin, oral leucovorin, and bevacizumab combination therapy (SOLA) in patients with unresectable metastatic colorectal cancer. Cancer Chemother Pharmacol 2015; 76: 547–53. [DOI] [PubMed] [Google Scholar]
- 23. Ueno M, Okusaka T, Omuro Y et al A randomized phase II study of S‐1 plus oral leucovorin versus S‐1 monotherapy in patients with gemcitabine‐refractory advanced pancreatic cancer. Ann Oncol 2016; 27: 502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hironaka S, Sugimoto N, Yamaguchi K et al S‐1 plus leucovorin versus S‐1 plus leucovorin and oxaliplatin versus S‐1 plus cisplatin in patients with advanced gastric cancer: a randomised, multicentre, open‐label, phase 2 trial. Lancet Oncol 2016; 17: 99–108. [DOI] [PubMed] [Google Scholar]