

Abstract
Long‐chain acyl‐coenzyme A (CoA) synthetase 3 (ACSL3) is an androgen‐responsive gene involved in the generation of fatty acyl‐CoA esters. ACSL3 is expressed in both androgen‐sensitive and castration‐resistant prostate cancer (CRPC). However, its role in prostate cancer remains elusive. We overexpressed ACSL3 in androgen‐dependent LNCaP cells and examined the downstream effectors of ACSL3. Furthermore, we examined the role of ACSL3 in the androgen metabolism of prostate cancer. ACSL3 overexpression led to upregulation of several genes such as aldo‐keto reductase 1C3 (AKR1C3) involved in steroidogenesis, which utilizes adrenal androgen dehydroepiandrosterone sulfate (DHEAS) as substrate, and downregulated androgen‐inactivating enzyme UDP‐glucuronosyltransferase 2 (UGT2B). Exposure to DHEAS significantly increased testosterone levels and cell proliferative response in ACSL3‐overexpressing cells when compared to that in control cells. A public database showed that ACSL3 level was higher in CRPC than in hormone‐sensitive prostate cancer. CRPC cells showed an increased expression of ACSL3 and an expression pattern of AKR1C3 and UGT2B similar to ACSL3‐overexpressing cells. DHEAS stimulation significantly promoted the proliferation of CRPC cells when compared to that of LNCaP cells. These findings suggest that ACSL3 contributes to the growth of CRPC through intratumoral steroidogenesis (i.e. promoting androgen synthesis from DHEAS and preventing the catabolism of active androgens).
Keywords: ACSL3, AKR1C3, castration‐resistant prostate cancer, intratumoral steroidogenesis, UGT2B
Prostate cancer is the most hormone‐dependent malignant tumor in men. The androgen and androgen receptor (AR) signaling pathways play a pivotal role in prostate cancer biology. Androgen deprivation therapy (ADT) is the standard treatment for prostate cancer. However, most prostate cancers will invariably relapse as castration‐resistant prostate cancers (CRPC) after ADT.1 Notably, AR signaling is still active after ADT and functions as a driving force for CRPC.2
Androgens stimulate the expression of over 20 enzymes involved in lipogenesis.3, 4, 5 De novo lipogenic enzymes are overexpressed in prostate cancer by AR signaling and other oncogenic pathways. Increased lipogenesis contributes to cell survival and oncogenic transformation in prostate cancer.3, 6, 7, 8, 9, 10, 11 Long‐chain acyl‐CoA synthetases (ACSL) catalyze the conversion of free long‐chain fatty acids into acyl‐CoA esters. Physiologically, ACSL‐mediated acyl‐CoA exert diverse cellular functions, including the regulation of energy and lipid metabolism and signal transduction.12, 13, 14, 15 ACSL3 is highly expressed in human lung cancer and is required for survival and the oncogenic capacity of mutant KRAS lung cancer cells.16 ACSL3 suppression impairs tumorigenesis of lung cancer cells in vivo,16 and triacsin C, an inhibitor of ACSL1, 3 and 4, induces apoptotic death in lung, colon and brain cancer cells in vitro and in vivo.17 ACSL3 is an androgen‐responsive enzyme and is abundantly expressed in normal prostate tissue and cancer.18, 19 Although the precise mechanism remains unclear, ACSL3 contributes to CRPC progression through enhancing intratumoral steroidogenesis.20
Intratumoral steroidogenesis is one of the mechanisms underlying the development of CRPC and leads to AR signaling reactivation.2, 20, 21, 22, 23, 24, 25, 26, 27, 28 To date, three major pathways have been proposed to explain the mechanism of intratumoral steroidogenesis in CRPC: de novo androgenesis from cholesterol, dihydrotestosterone (DHT) synthesis from cholesterol by bypassing testosterone, and conversion of adrenal androgen to testosterone and DHT.21, 29, 30 The last pathway has been suggested to function as the major mechanism for the acquisition of androgens in prostate cancer after ADT.27 The circulating weak adrenal androgen, dehydroepiandrosterone sulfate (DHEAS), is incorporated by members of the solute carrier organic anion transporter family, including SLCO1B3 and SLCO2B1, and is cleaved into DHEA by steroid sulfatase (STS). DHEA is then metabolized into androgens by several steroidogenic enzymes such as 3β‐hydroxysteroid dehydrogenase (HSD) 3B, aldo‐keto reductase family member (AKR) 1C3 and steroid 5α‐reductase (SRD5A). Excess intracellular androgen is glucuronide‐conjugated and inactivated by UDP‐glucuronosyltransferases (UGT) 2B15 and UGT2B17, and released into the circulation.
In this manuscript, we show that ACSL3 contributes to intratumoral steroidogenesis by modulating the steroidogenic genes and to the growth of CRPC. Our data provides the rationale for a new therapeutic strategy for CRPC.
Materials and Methods
Cell culture and reagents
The AR‐positive LNCaP, and AR‐negative DU145 and PC3 prostate cancer cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Castration‐resistant derivatives of LNCaP cells (LTAD) were previously established after long‐term androgen deprivation in our laboratory.31, 32 Short tandem repeat (STR) analysis was performed to authenticate the cell lines used in the present study.31 We performed all studies within 30 passages. LNCaP cells and DU145 or PC3 cells were grown in Roswell Park Memorial Institute Medium 1640 or DMEM, respectively, supplemented with 10% heat‐inactivated FBS and 1% penicillin/streptomycin (both from GIBCO‐BRL, Grand Island, NY, USA) in a 5% CO2 atmosphere at 37°C. Charcoal‐stripped FBS was purchased from Invitrogen (Carlsbad, CA, USA). G418 (Geneticin) was obtained from GIBCO‐BRL (Carlsbad, CA, USA). Cells were stimulated with DHT (Wako, Osaka, Japan), DHEA or DHEAS (both from Sigma, St‐Louis, MO, USA).
Vector construction and transfection
An N‐terminal FLAG‐tagged ACSL3 construct was generated by amplifying the coding sequence of human ACSL3 by PCR. This fragment was subcloned into pcDNA3.1(−)/Myc‐His B (Invitrogen) with the FLAG epitope (DYKDDDDK) (pcDNA3.1‐FLAG‐ACSL3). Increases in ACSL3 expression were confirmed by real‐time PCR or immunoblotting. To generate LNCaP and DU145 cells stably overexpressing ACSL3 or vector‐control, LNCaP and DU145 cells were transfected with pcDNA3.1‐FLAG‐ACSL3 or pcDNA3.1‐FLAG, respectively, using the FuGENE HD transfection reagent (Roche Applied Science, Basel, Switzerland). Transfected cells were selected by their ability to grow in medium containing 400 μg/mL G418, and the two isogenic cell lines were generated.
RNA interference
ACSL3 knockdown was performed with two different Stealth siRNA (HSS103535 and HSS176683) (Invitrogen), and the data shown are from one representative experiment with ACSL3 siRNA HSS103535, unless otherwise noted. A negative control siRNA (Invitrogen) was used as a control. Transfection was performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. The cells were analyzed 72 h after transfection.
Real‐time PCR
Total RNA was extracted using ISOGEN reagent (Nippon Gene, Tokyo, Japan). First‐strand cDNA was generated from total RNA, using SuperScript II Reverse Transcriptase (Invitrogen). Quantitative reverse transcription‐PCR was performed for each sample, using the KAPA SYBR Fast qPCR Kit (Kapa Biosystems, Woburn, MA, USA) with gene‐specific primers on a StepOnePlus Real‐Time PCR system (Applied Biosystems, Foster city, CA, USA). The relative amounts of mRNA were calculated following the comparative CT method, after normalization to β‐actin levels. The sequences of the PCR primers are listed in Table S1. All reactions were performed at least in duplicate. Data are presented as mean ± SD.
Cell proliferation assays
Cells were suspended in charcoal‐stripped FBS with 100 μM DHEAS, as described previously33 and seeded in 96‐well microplates (5 × 102 cells/well). Cell proliferation was evaluated using Cell Count Reagent SF (Nacalai Tesque, Kyoto, Japan).
Cell lysis and immunoblotting
For total cell lysis, the cells were washed twice with ice‐cold PBS and dissolved in Nonidet P‐40 (NP‐40) buffer containing 50 mmol/L Tris‐HCl (pH 7.5), 150 mmol/L NaCl, 0.5% NP‐40, and protease and phosphatase inhibitors (Nacalai Tesque). The protein concentration of each lysate was determined using a protein assay reagent kit (BioRad, Hercules, CA, USA). After sodium dodecyl sulfate‐polyacrylamide gel electrophoresis, the proteins were transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The membranes were then blocked for 20 min in blocking buffer (Nacalai Tesque) and probed overnight with primary antibodies for UGT2B (H‐300; detection of UGT2B family members and UGT2A1), KLK3, AR (all from Santa‐Cruz, Biotechnology, Santa Cruz, CA, USA), ACSL3, AKR1C3, FLAG and β‐actin (all from Sigma). After extensive washing, bound antibodies were visualized using HRP‐conjugated secondary antibodies and the signal was enhanced by chemiluminescence (ECL; GE Healthcare, Tokyo, Japan).
Immunofluorescence and lipid staining
Cells were fixed with 4% paraformaldehyde, and lipids were stained using the BODIPY 493/503 lipid probe (Invitrogen). 4′,6′‐DAPI (Invitrogen) was used for nuclear counterstaining. For immunofluorescence, after treatment with 0.2% Triton X‐100, the cells were blocked with Protein Block (Dako Cytomation, Carpinteria, CA, USA) for 10 min at room temperature and then incubated with anti‐ACSL3, anti‐FLAG (both from Sigma), anti‐calnexin or anti‐COX 4 (3E11; Alexa Fluor 594 conjugate; both from Cell Signaling, Beverly, MA, USA) antibody at 4°C overnight. Cells were incubated with fluorescein isothiocyanate‐conjugated secondary antibodies (Dako Cytomation) followed by counterstaining with DAPI. Fluorescence images were obtained using a confocal laser‐scanning microscope, FluoView FV10i (Olympus, Tokyo, Japan).
Immunohistochemistry
Formalin‐fixed, paraffin‐embedded prostate cancer tissue microarrays (TMA) were purchased from Biochain (cat #Z7020091, lot# B507107, Hayward, CA, USA). Each TMA contains 14 prostate cancer and 8 non‐cancerous lesions in duplicate. Data regarding the tumor grades and clinical stages, which were based on the World Health Organization (WHO) recommendations and the TNM system, were obtained. Immunohistochemistry was performed on these TMA as described previously,34, 35 using primary antibodies for ACSL3, AKR1C3 and UGT2B.
Measurement of androgen levels
Cells were cultured in regular medium in the presence or absence of 100 μM DHEAS or 100 nM DHEA for 24 h, and then harvested together with the culture medium. Concentrations of testosterone and DHT were measured by liquid chromatography/electrospray tandem mass spectrometry (LC‐MS/MS; ASKA Pharma Medical, Kawasaki, Japan), as described previously.36 Briefly, a suspension composed of a mixture of cells and cultured medium was homogenized using an Ultra‐Turrax homogenizer. As internal standards, 400 pg of 13C3‐T and 100 pg of 13C3‐DHT were added to the homogenized suspensions. Steroids were then extracted using 3 mL of ethyl acetate. The concentration was standardized by sample weight (pg/g). The lower limit of quantification was 1 pg/assay for both DHT and testosterone.
Measurement of acyl‐CoA and acyl‐carnitine levels
Total lipids from cells were extracted according to a modified Folch method, using chloroform/methanol/water (1/2/0.2; v/v/v).37 Reversed‐phase LC separation was achieved using an ACQUITY UPLC HSS T3 column (2.1 × 50 mm i.d., Waters Corporation, Milford, MA, USA) at 45°C. The mobile phase was prepared by mixing solvents (A) acetonitrile/methanol/water (1/1/3; v/v/v) containing 5 mM ammonium formate and (B) isopropanol containing 5 mM ammonium formate, and the required composition was produced by mixing these solvents. The mobile phase was pumped at a flow rate of 300 μL/min. Acyl‐CoA and acyl‐carnitine measurements were performed by multiple reaction monitoring in the positive ion mode, using a 5500 QTRAP mass spectrometer (AB SCIEX, Foster City, CA, USA) with an Agilent 1290 Infinity LC system (Agilent Technologies, Loveland, CO, USA).
Statistical analysis
Two‐tailed t‐test with unequal variance was performed for continuous variables. Fold changes of mRNA expression relative to the vehicle control were analyzed by one‐way anova with Dunnett's post‐test. All statistical analyses were performed using JMP for Mac version 10.0.0 (SAS, Cary, NC, USA). For all analyses, P < 0.05 was considered statistically significant.
Results
ACSL3 is overexpressed in prostate cancer and is an androgen‐responsive gene
To examine which type of cancer predominantly expresses ACSL3, tissue distribution of ACSL3 was assessed using the Oncomine database. ACSL3 expression was significantly higher in prostate cancer (median; 4.426) than in other types of malignancies (P = 5.42E‐19, fold change = 2.711; Fig. 1a). Furthermore, ACSL3 expression was significantly higher in prostate cancer (median; 0.791) than in the prostate gland (median; 0.161) (P = 1.68E‐6, fold change = 1.502) in several studies (Fig. 1b and Fig. S1a and S1b). In our immunohistochemistry analysis, ACSL3‐positive staining pattern was cytoplasmic dot‐like (Fig. 1c, middle panel), and was commonly observed in perinuclear regions of cancer cells (Fig. 1c, lower panel), whereas normal prostatic epithelium presented very low levels (Fig. 1c, upper panel). To precisely determine the subcellular localization of ACSL3, co‐immunofluorescence staining for ACSL3 and markers for the endoplasmic reticulum (ER; calnexin) or mitochondria (COX‐4) was performed in LNCaP cells. ACSL3 co‐localized with both ER (Fig. 1d, upper panels) and mitochondrial markers (Fig. 1d, middle panels). As described in the next section, we generated FLAG‐tagged ACSL3‐overexpressing LNCaP cells, and exogenous ACSL3 protein was detected using both anti‐FLAG and anti‐ACSL3 antibodies (Fig. S1c). Lipids in ACSL3‐LNCaP cells were stained with BODIPY, and the results revealed that ACSL3 was also localized on the surface of lipid droplets (Fig. 1d, lower panels). These results were in agreement with previous studies.38
Figure 1.
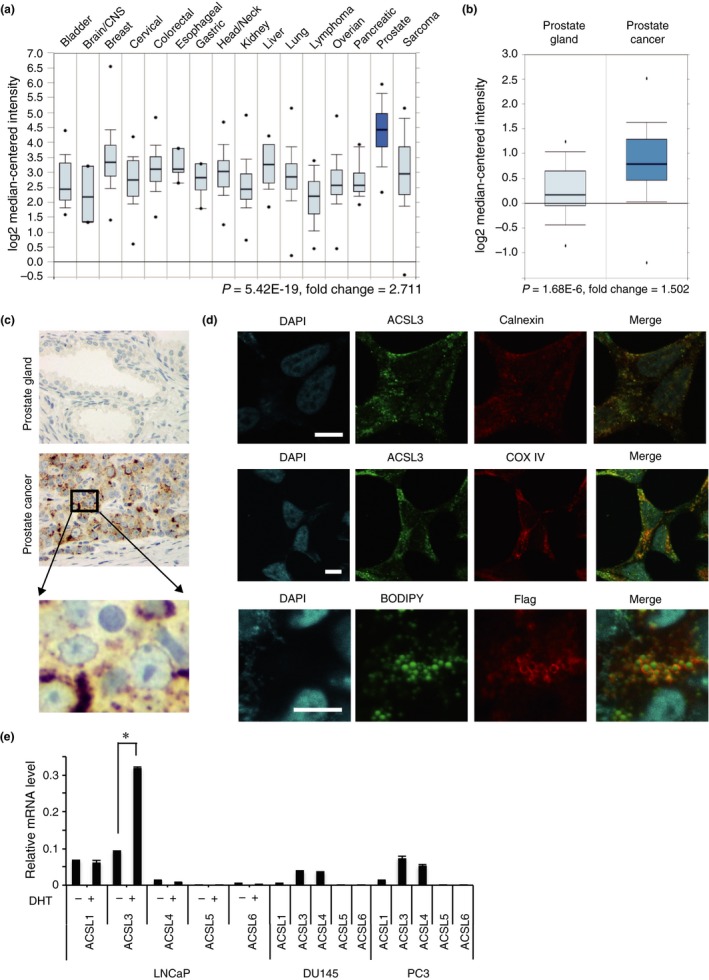
Long‐chain acyl‐coenzyme A (CoA) synthetase 3 (ACSL3) is overexpressed in prostate cancer. (a) ACSL3 mRNA expression levels in various organs were obtained from Bittner Multi‐cancer study in the Oncomine database (1911 samples). Statistical analysis was performed by t‐test (P = 5.42E‐19, fold‐change = 2.711). (b) ACSL3 mRNA levels in the prostate gland (n = 41) and in prostate cancer (n = 62) were obtained from the study of Lapointe in Oncomine. Statistical analysis was performed by t‐test (P = 1.68E‐6, fold‐change = 1.502). (c) Representative picture of ACSL3 immunostaining in the prostate gland (upper panel) and in prostate cancer (middle panel). Higher magnification of the indicated area focused on an ACSL3 immunostaining (lower panel). (d) Upper and middle panels: LNCaP cells were fixed and treated with anti‐ACSL3 and anti‐calnexin (endoplasmic reticulum marker; upper panels), or with anti‐ACSL3 and anti‐COX 4 (mitochondrial marker; middle panels). Lower panels: LNCaP‐ACSL3 cells were treated with anti‐FLAG antibody and stained with BODIPY 493/503 for lipids detection (green, BODIPY 493/503; red, FLAG; and blue, DAPI). Nuclei were stained by DAPI. Immunofluorescence staining was analyzed by confocal microscopy. Scale bar, 10 μm. (e) mRNA levels of endogenous ACSL isozymes quantified by real‐time PCR in LNCaP cells in charcoal‐stripped FBS with or without dihydrotestosterone, and in DU145 and PC3 cells in regular medium. The data were normalized to that of β‐actin, which was used as an internal control. The asterisk (*) indicates P < 0.05.
To determine the expression levels of ACSL isozymes in prostate cancer cell lines, the mRNA levels of the ACSL isozymes were measured in androgen‐dependent LNCaP cells and androgen‐independent DU145 and PC3 cells. We observed that the ACSL1 and ACSL3 isozymes were dominant in LNCaP cells, whereas ACSL3 and ACSL4 were dominant in DU145 and PC3 cells. Only the ACSL3 isozyme was responsive to DHT in LNCaP cells (Fig. 1e), which was in agreement with previous reports.3, 18, 39, 40
Collectively, these results indicate that ACSL3 is a unique androgen‐responsive ACSL isozyme overexpressed in prostate cancer and localized at the ER, mitochondria and the lipid droplet surface.
ACSL3 overexpression enhances long‐chain acyl‐CoA production
To explore the role of ACSL3 in prostate cancer, we generated ACSL3‐overexpressing LNCaP cells. ACSL3 overexpression was confirmed at the mRNA level by real‐time PCR (Fig. 2a), and at the protein level by immunoblotting using both anti‐FLAG and anti‐ACSL3 antibodies (Fig. 2b). Next, to confirm whether the exogenous ACSL3 is functional, we measured the acyl‐CoA level by mass spectrometry. Specifically, ACSL3 overexpression increased the levels of saturated or unsaturated C12‐C20 fatty acyl‐CoA (Fig. 2c). Total acyl‐CoA levels were significantly increased by ACSL3 overexpression (Fig. 2c, smaller bar graph). In addition, the levels of acyl‐carnitines, which are more stable derivatives than acyl‐CoA, were also increased by ACSL3 overexpression (data not shown). This indicates that overexpressed ACSL3 is actually functional and modulates lipid metabolism in LNCaP cells.
Figure 2.
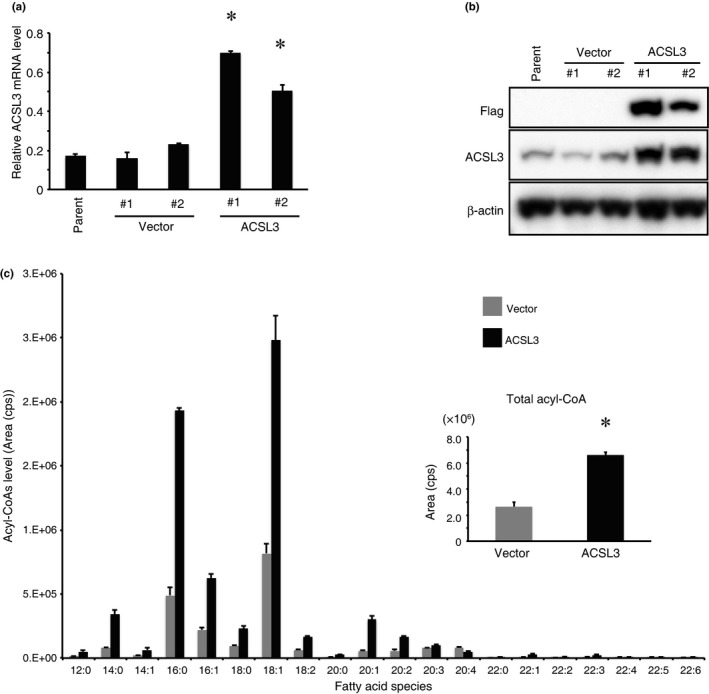
ACSL3 overexpression promotes acyl‐CoA production. (a) ACSL3 mRNA levels were quantified by real‐time PCR in parental vector control (LNCaP‐vector #1 and #2) or ACSL3‐overexpressing LNCaP (LNCaP‐ACSL3 #1 and #2) cells. The data were normalized to that of β‐actin. Differences between LNCaP‐vector and LNCaP‐ACSL3 cells were analyzed for statistical significance. *P < 0.05. (b) ACSL3 and FLAG protein levels in parental LNCaP, LNCaP‐vector, and LNCaP‐ACSL3 cells were measured by immunoblotting using antibodies against FLAG and ACSL3. β‐actin was used as a loading control. (c) Lipids were extracted from LNCaP‐vector (grey bar) and LNCaP‐ACSL3 (black bar) cells, and acyl‐CoA levels in each fatty acid species were measured by liquid chromatography tandem mass spectrometry (LC‐MS/MS). Horizontal axis: C12:0‐C22:6. Data of each fatty acid species were summed up for total levels of acyl‐CoA (small bar graph). Differences between LNCaP‐vector and LNCaP‐ACSL3 cells were analyzed for statistical significance. *P < 0.05.
ACSL3 overexpression modulates gene expression and promotes testosterone production and cell growth by adrenal androgens
Lipogenic enzymes are involved in intratumoral steroidogenesis and contribute to CRPC development.20, 23, 24, 41 Therefore, we investigated whether ACSL3 influences intratumoral steroidogenesis. Intriguingly, compared to that in LNCaP‐vector cells, ACSL3 overexpression significantly upregulated several steroidogenesis‐related genes, including SLCO1B3, which encodes an uptake transporter of DHEAS, and AKR1C3 and HSD3B1, both of which encode enzymes involved in testosterone synthesis (Fig. 3a and Fig. S2). Meanwhile, ACSL3 overexpression significantly reduced the expression of SRD5A1, UGT2B15 and UGT2B17 (Fig. 3a and Fig. S2). Upregulation of AKR1C3 and downregulation of UGT2B by ACSL3 overexpression were also confirmed by immunoblotting (Fig. 3c), and a similar relationship between ACSL3 and AKR1C3 or UGT2B was observed by immunohistochemistry (Fig. S3). The androgen‐responsive KLK3 protein level was increased without remarkable change in the AR level (Fig. 3b), suggesting that ACSL3‐mediated AR reactivation is regulated by a post‐translational mechanism. To confirm that ACSL3 regulates the expression of these genes, we knocked down ACSL3 in LNCaP cells to confirm that ACSL3 regulates the expression of these genes. AKR1C3 expression was significantly decreased by ACSL3 knockdown, whereas the expression of both SRD5A1 and UGT2B15 was significantly increased by ACSL3 knockdown (Fig. 3c). The gene involved in de novo androgenesis utilizing cholesterol, such as steroidogenic acute regulatory protein (STAR) and a cytochrome P‐450 family member (CYP11A1), was either downregulated or remained unchanged (Fig. S4). This suggests that ACSL3 drives intratumoral steroidogenesis, but not through cholesterol anabolism. In addition, ACSL3 overexpression in the AR‐negative cell line, DU145, did not affect the expression of these genes (data not shown). LNCaP cells exhibited resistance to tunicamycin, an ER stress inducer, whereas DU145 cells were sensitive to it (Fig. S5a). ACSL3 overexpression conferred resistance to tunicamycin in DU145 cells (Fig. S5b). These findings suggest that ACSL3 may enhance ER function to confer a survival advantage to cancer cells, and may drive steroidogenesis in the presence of the functional AR signaling.
Figure 3.
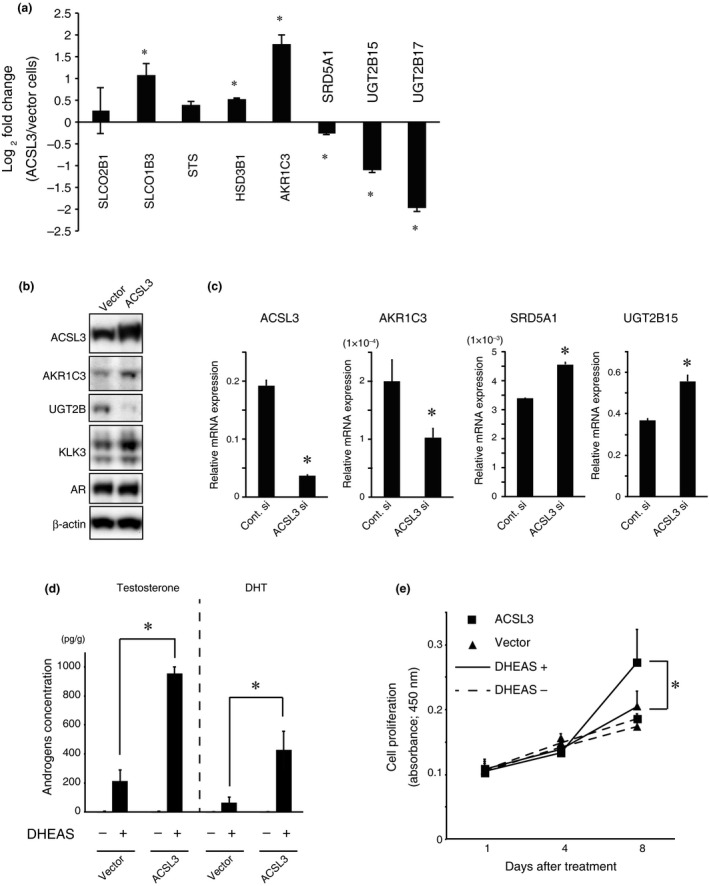
ACSL3 overexpression modulates steroidogenic gene expression and promotes androgen production and cell proliferation induced by dehydroepiandrosterone sulfate (DHEAS) stimulation. (a) Relative mRNA levels were quantified by real‐time PCR for SLCO2B1,SLCO1B3,STS,HSD3B1,AKR1C3,SRD5A1,UGT2B15 and UGT2B17 in LNCaP‐vector and LNCaP‐ACSL3 cells. The fold changes of gene expression in LNCaP‐ACSL3 cells relative to that in LNCaP‐vector cells were calculated. *P < 0.05. (b) Immunoblotting was performed for ACSL3, AKR1C3, UGT2B, KLK3, AR and β‐actin in LNCaP‐vector and LNCaP‐ACSL3 cells. (c) LNCaP cells were treated with negative control siRNA or ACSL3 siRNA, and relative mRNA levels for SLCO1B3,AKR1C3,SRD5A1 and UGT2B15 were quantified as in Figure 3b. (d) LNCaP‐vector and LNCaP‐ACSL3 cells were stimulated with 100 μM DHEAS for 24 h, and then testosterone and dihydrotestosterone levels in the mixture of cells and medium were measured by LC‐MS/MS. *P < 0.05. (e) LNCaP‐vector and LNCaP‐ACSL3 cells were cultured under androgen‐depleted conditions in the presence or absence of 100 μM DHEAS for 8 days, and cell proliferation was monitored using Cell Count Reagent SF. *P < 0.05.
To address whether ACSL3 drives androgen synthesis through DHEAS, the intracellular testosterone and DHT levels were compared between LNCaP‐vector and LNCaP‐ACSL3 cells in the presence or absence of exogenous DHEAS. Although testosterone or DHT levels were undetectable in the absence of DHEAS in both LNCaP‐vector and LNCaP‐ACSL3 cells, DHEAS stimulation significantly enhanced the production of testicular androgen, in particular testosterone, in the LNCaP‐ACSL3 cells relative to that in LNCaP‐vector cells (Fig. 3d).
To assess the effect of DHEAS on cell proliferation, we compared the growth of LNCaP‐vector and LNCaP‐ACSL3 cells in the presence or absence of DHEAS. In the absence of DHEAS, there was no significant difference in proliferation between LNCaP‐vector and LNCaP‐ACSL3 cells. However, in the presence of DHEAS, proliferation of LNCaP‐ACSL3 cells was significantly promoted compared to that of LNCaP‐vector cells (Fig. 3e). Similar results were obtained using DHEA stimulation instead of DHEAS (data not shown).
These findings suggest that ACSL3 promotes intratumoral steroidogenesis, utilizing adrenal androgens, DHEAS and DHEA, and contributes to cancer cell growth.
Expression of steroidogenic genes in castration‐resistant prostate cancer cells is similar to that in ACSL3 overexpressing‐cells, and cell growth is promoted by dehydroepiandrosterone sulfate
To examine whether ACSL3 is involved in CRPC, ACSL3 mRNA level in hormone sensitive or naïve prostate cancer and CRPC was assessed using the Oncomine database. Comprehensive gene expression analysis showed that ACSL3 mRNA level was higher in CRPC than in hormone‐sensitive or naïve prostate cancer (Fig. 4a and Fig. S6).42, 43, 44 Next, we measured the endogenous ACSL levels in both LNCaP and castration‐resistant LTAD cells. ACSL3 was the most abundant ACSL isozyme in LTAD cells under conditions of androgen depletion, whereas ACSL3 expression was low in LNCaP cells under the same conditions (Fig. 4b). Similar to that in ACSL3‐overexpressing cells, the expression levels of SLCO2B1, SLCO1B3, STS, HSD3B1 and AKR1C3 were higher in LTAD cells than in parental LNCaP cells, whereas the expression levels of SRD5A1, UGT2B15 and UGT2B17 were significantly lower in LTAD cells than in parental LNCaP cells (Fig. 4c). In addition, LTAD cells grew significantly faster than LNCaP cells in the presence of DHEAS (Fig. 4d).
Figure 4.
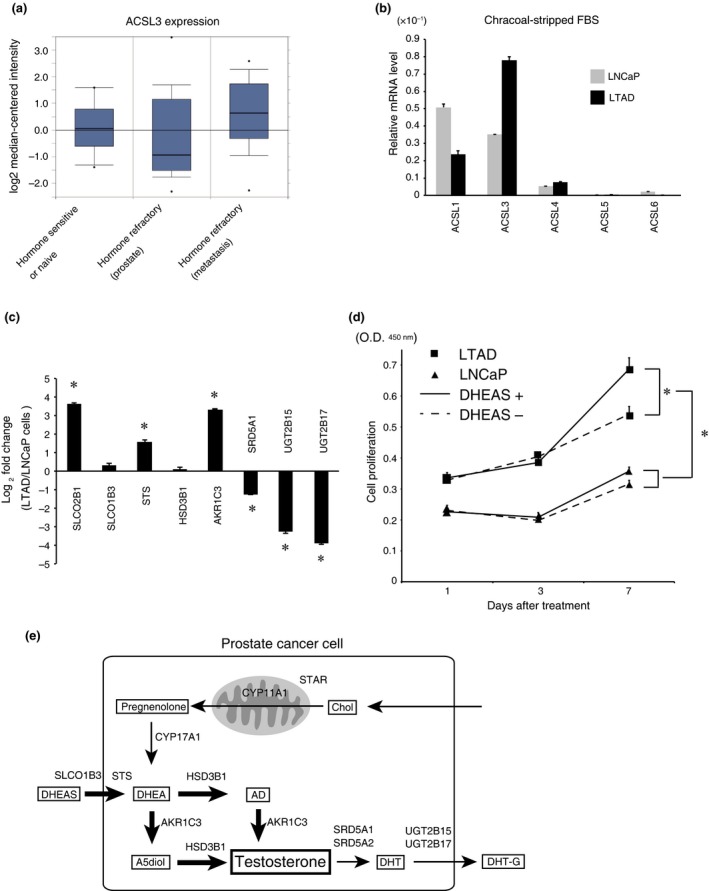
ACSL3 is overexpressed in the castration‐resistant prostate cancer (CRPC) model LTAD cells, and dehydroepiandrosterone sulfate (DHEAS) promotes the growth of LTAD cells. (a) ACSL3 expression data in hormone therapy response status were obtained from the dataset of Tamura prostate statics in Oncomine. The data includes 10 samples of hormone‐sensitive or hormone‐naïve prostate cancer, 13 samples of hormone‐refractory prostate cancer (from prostate) and 12 samples of hormone‐refractory prostate cancer (from metastasis). (b) LNCaP (grey bars) and LTAD (black bars) cells were cultured in the medium with charcoal‐stripped FBS for 48 h, and the relative mRNA levels of the ACSL isozymes were quantified by real‐time PCR. The data were normalized to β‐actin. (c) LNCaP and LTAD cells were cultured in the medium with charcoal‐stripped FBS for 48 h, and the relative mRNA levels were quantified by real‐time PCR for SLCO1B3,HSD3B1,AKR1C3 and UGT2B17. The fold changes of gene expression in LTAD cells relative to those in LNCaP cells were calculated. *P < 0.05. (d) LNCaP and LTAD cells were cultured as described in Figure 3d, and cell proliferation was assayed. *P < 0.05. (e) Schematic model of the potential mechanism of ACSL3‐mediated intratumoral steroidogenesis in prostate cancer. Bold arrows indicate an increased pathway. AD, androstenedione; A5diol, androstenediol; Chol, cholesterol; DHEA, dehydroepiandrosterone; DHEAS, dehydroepiandrosterone sulfate; DHT, dihydrotestosterone; DHT‐G, DHT‐glucuronide.
Collectively, these data suggest that ACSL3 contributes to intratumoral steroidogenesis by stimulating androgen synthesis, utilizing DHEAS and suppressing androgen inactivation (Fig. 4e).
Discussion
ACSL3 is overexpressed in both androgen sensitive prostate cancer and castration‐resistant prostate cancer
A previous study demonstrated that the ACSL3 level is lower in prostate cancer than in normal tissue.45 However, a recent meta‐analysis revealed that ACSL3 mRNA levels as well as those of other lipogenic enzymes were higher in prostate cancer than in normal prostate tissue.46 ACSL3 expression is decreased after ADT in prostate cancer.20 However, ACSL3 expression increases again during CRPC progression.20 This phenomenon is also observed in other lipogenic enzymes and their transcriptional factors, such as fatty acid synthase (FASN) and sterol regulatory element‐binding protein (SREBP), respectively.6, 47 The expression of these molecules is associated with prostate cancer aggressiveness.11, 47, 48 Thus, ACSL3 could be a useful biological marker of prostate cancer progression in a clinical setting. ACSL3 immunohistochemical detection on biopsy specimens could provide information on the efficacy of ADT. Circulating tumor cells (CTC) are rare cancer cells that have been shed from primary or metastatic tumors, and CTC burden is a useful biomarker of treatment response and overall survival in patients with CRPC.49 Because AR expression in CTC is associated with a worse outcome in patients with CRPC,50 ACSL3 expression in CTC could also predict and monitor the progression to CRPC.
Collectively, ACSL3 expression is increased in both hormone‐sensitive and refractory prostate cancer and may be utilized as a biomarker for the detection of CRPC.
ACSL3 regulates genes are involved in intratumoral steroidogenesis and contributes to cell growth of castration‐resistant prostate cancer
Our study showed that ACSL3 is involved in intratumoral steroidogenesis in LNCaP cells, but not in DU145 cells. Because DU145 cells express neither AR nor HSD3B1,51 the latter of which is the essential enzyme to catalyze both DHEA to androstenedione and androstenediol to testosterone (Fig. 4e), DU145 cells per se cannot generate steroids. ACSL3 overexpression upregulated the expression of SLCO1B3, HSD3B1 and AKR1C3, whereas it downregulated the expression of SRD5A1, UGT2B15 and UGT2B17. A number of studies support this alteration of gene expression in aggressive prostate cancer. The levels of DHEAS transporters, SLCO2B1 and SLCO1B3, are increased in metastatic sites of prostate cancer, and their polymorphism is associated with clinical outcome.52 Testosterone‐synthesizing enzymes, HSD3B1 and AKR1C3, are highly expressed in CRPC.25, 26, 28 The activity of the DHT‐synthesizing enzyme, SRD5A, is consistently depressed in lymph node metastasis and primary prostate cancer.53 Androgen‐inactivating enzymes, UGT2B15 and UGT2B17, are major determinants of AR response in LNCaP cells, and both reduce the intracellular active androgen.54, 55 Because ACSL3 overexpression induced the upregulation of AKR1C3 and downregulation of SRD5A1, ACSL3 overexpression enhances testosterone production rather than DHT. Intratumoral testosterone levels are higher at metastatic sites than at primary sites, whereas DHT levels are markedly decreased in metastatic sites.2, 26, 56, 57 Moreover, it has been suggested that testosterone is more crucial than DHT in CRPC.27, 58 Our data highlighted the significance of testosterone in metastatic CRPC.
New hormonal agents, abiraterone and enzalutamide, demonstrated a survival advantage in patients with CRPC. However, prolonged treatment with abiraterone or enzalutamide induces intratumoral steroidogenesis due to several mechanisms like CYP17A1 upregulation,59, 60 the induction of AR and AR splicing variants,60 and AKR1C3 activation.61 Thus, the use of such new agents against prostate cancer with ACSL3 overexpression warrants further investigation. We recently reported that ACSL3 is the major target of the complex between AR and the AR‐interacting partner, Oct1, and that Oct1 inhibition downregulated ACSL3 expression and impaired CRPC cell growth.62 Thus, ACSL3 could be a therapeutic target of CRPC.
Our data suggest that ACSL3 stimulates intratumoral steroidogenesis; it preferably utilizes adrenal androgen rather than cholesterol for androgen synthesis (Fig. 4e). ACSL3 expression may be upregulated in CRPC cells, allowing them to grow under low‐androgen conditions.
Possible mechanisms of the regulation of steroidogenic genes by ACSL3
The most significant change in expression in ACSL3‐overexpressing LNCaP cells and CRPC cells was the upregulation of AKR1C3 and downregulation of UGT2B. The mechanism underlying this change remains elusive. However, two molecules may be involved in this change: protein kinase C (PKC) and hepatocyte nuclear factor‐4α (HNF4A). ACSL3‐mediated acyl‐CoA bind to acyl‐CoA binding protein (ACBP), which directly regulates a metabolic transcriptional factor, HNF4A.12, 13 Saturated acyl‐CoAs stimulate the transcriptional activity of HNF4A, whereas unsaturated acyl‐CoAs inhibit it.14, 15, 63, 64 Thus, HNF4A negatively and positively regulates target genes, including AKR1C4,65 UGT2B family,66 and other steroidogenic genes such as the SLCO and HSD family.67 We observed that HNF4A overexpression upregulated AKR1C3 expression and downregulated UGT2B17 expression in LNCaP cells (data not shown), suggesting that HNF4A can be a downstream target of ACSL3‐mediated acyl‐CoA. PKC is directly or indirectly activated by acyl‐CoA13 and can phosphorylate AR at serine 578, leading to AR reactivation.68, 69 Activation of AR downregulates UGT2B15 and 17 expression.70, 71 PKC also phosphorylates nuclear factor erythroid‐2 related factor 2 (Nrf2), which drives AKR1C3 expression through binding to an anti‐oxidant responsive element.72, 73 In addition to the gene regulation mediated by acyl‐CoA, intracellular acyl‐CoA function as detergents in microsomes, and the high level of acyl‐CoA inhibits UGT2B activity.13, 74, 75 Altogether, ACSL3‐mediated alteration of these steroidogenic genes may contribute to intratumoral steroidogenesis. However, the current study was performed using a limited number of cell lines and the mode of action of acyl‐CoA remains unclear. Confirmation of this hypothesis awaits further study.
In conclusion, our studies reveal a novel function of androgen‐responsive ACSL3 in prostate cancer. The results provide new insights into the molecular link between increased de novo lipogenesis and intratumoral steroidogenesis in prostate cancer. Taking into account this positive feedback loop, ACSL3 might be a novel target for the development of new treatment strategies for CRPC.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Expression levels of ACSL3 mRNA in prostate cancer and its subcellular localization.
Fig. S2. Effect of DHT supplementation and ACSL3 overexpression on the expression levels of SLCO1B3, AKR1C3, SRD5A1 and UGT2B15.
Fig. S3. Representative pictures of immunohistochemistry of ACSL3, AKR1C3 and UGT2B in prostate cancer.
Fig. S4. Effect of ACSL3 overexpression on steroidogenic genes utilizing cholesterol.
Fig. S5. Effect of ACSL3 overexpression on the sensitivity to tunicamycin in DU145 cells.
Fig. S6. Expression levels of ACSL3 mRNA in hormone sensitive prostate cancer and castration‐resistant prostate cancer (CRPC).
Table S1. Oligonucleotide sequences of PCR primers.
Acknowledgments
The authors thank Noriko Sasaki and Keisuke Kodama (both from the Department of Anti‐Aging Medicine, Graduate School of Medicine, The University of Tokyo), Hidehiko Sasamoto (ASKA Pharma Medical), and Haruka Takahashi and Haruka Sato (both from the Institute for Advanced Biosciences, Keio University) for technical assistance. This work was supported by Grants of the Cell Innovation Program (S.I.), P‐DIRECT and P‐CREATE from the MEXT, Japan; by Grants (T.M., K.T., T.U., S.T. and S.I.) from the JSPS, Japan; by AstraZeneca VRI Research Grant 2011 (T.M.); by Grants‐in‐Aid (S.I.) from the MHLW, Japan; by the 2010 Research Grant of the 60th Anniversary Memorial Fund (D.O.) from Nihon University Medical Alumni Association; by the Young Researcher Promotion Grant (D.O.) from The Japanese Urological Association; and by the Advanced Research for Medical Products Mining Program (S.I.), NIBIO, Japan.
Cancer Sci 108 (2017) 2011–2021
Funding Information
Japan Society for the Promotion of Science.
Contributor Information
Toshiro Migita, Email: toshiro.migita@gmail.com.
Satoshi Inoue, Email: sinoue@tmig.or.jp.
References
- 1. Wang Q, Li W, Zhang Y et al Androgen receptor regulates a distinct transcription program in androgen‐independent prostate cancer. Cell 2009; 138: 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mostaghel EA, Montgomery B, Nelson PS. Castration‐resistant prostate cancer: targeting androgen metabolic pathways in recurrent disease. Urol Oncol 2009; 27: 251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Swinnen JV, Heemers H, van de Sande T et al Androgens, lipogenesis and prostate cancer. J Steroid Biochem Mol Biol 2004; 92: 273–9. [DOI] [PubMed] [Google Scholar]
- 4. Barfeld SJ, Itkonen HM, Urbanucci A, Mills IG. Androgen‐regulated metabolism and biosynthesis in prostate cancer. Endocr Relat Cancer 2014; 21: T57–66. [DOI] [PubMed] [Google Scholar]
- 5. Swinnen JV, Ulrix W, Heyns W, Verhoeven G. Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc Natl Acad Sci USA 1997; 94: 12975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ettinger SL, Sobel R, Whitmore TG, et al Dysregulation of sterol response element‐binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res 2004; 64: 2212–21. [DOI] [PubMed] [Google Scholar]
- 7. Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta 2013; 1831: 1518–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? J Cell Biochem 2004; 91: 47–53. [DOI] [PubMed] [Google Scholar]
- 9. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7: 763–77. [DOI] [PubMed] [Google Scholar]
- 10. Migita T, Ruiz S, Fornari A et al Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst 2009; 101: 519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care 2006; 9: 358–65. [DOI] [PubMed] [Google Scholar]
- 12. Mashek DG, Li LO, Coleman RA. Long‐chain acyl‐CoA synthetases and fatty acid channeling. Future Lipidol 2007; 2: 465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Faergeman NJ, Knudsen J. Role of long‐chain fatty acyl‐CoA esters in the regulation of metabolism and in cell signalling. Biochem J 1997; 323(Pt 1): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schroeder F, Petrescu AD, Huang H et al Role of fatty acid binding proteins and long chain fatty acids in modulating nuclear receptors and gene transcription. Lipids 2008; 43: 1–17. [DOI] [PubMed] [Google Scholar]
- 15. Pegorier JP, Le May C, Girard J. Control of gene expression by fatty acids. J Nutr 2004; 134: 2444S–9S. [DOI] [PubMed] [Google Scholar]
- 16. Padanad MS, Konstantinidou G, Venkateswaran N et al Fatty acid oxidation mediated by Acyl‐CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep 2016; 16: 1614–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mashima T, Oh‐hara T, Sato S et al p53‐defective tumors with a functional apoptosome‐mediated pathway: a new therapeutic target. J Natl Cancer Inst 2005; 97: 765–77. [DOI] [PubMed] [Google Scholar]
- 18. Nelson PS, Clegg N, Arnold H et al The program of androgen‐responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci USA 2002; 99: 11890–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minekura H, Kang MJ, Inagaki Y et al Genomic organization and transcription units of the human acyl‐CoA synthetase 3 gene. Gene 2001; 278: 185–92. [DOI] [PubMed] [Google Scholar]
- 20. Locke JA, Guns ES, Lehman ML et al Arachidonic acid activation of intratumoral steroid synthesis during prostate cancer progression to castration resistance. Prostate 2010; 70: 239–51. [DOI] [PubMed] [Google Scholar]
- 21. Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer 2011; 18: R175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hofland J, van Weerden WM, Dits NF et al Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res 2010; 70: 1256–64. [DOI] [PubMed] [Google Scholar]
- 23. Leon CG, Locke JA, Adomat HH et al Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration‐resistant prostate cancer in a mouse xenograft model. Prostate 2010; 70: 390–400. [DOI] [PubMed] [Google Scholar]
- 24. Locke JA, Guns ES, Lubik AA et al Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration‐resistant prostate cancer. Cancer Res 2008; 68: 6407–15. [DOI] [PubMed] [Google Scholar]
- 25. Mitsiades N, Sung CC, Schultz N et al Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res 2012; 72: 6142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montgomery RB, Mostaghel EA, Vessella R et al Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration‐resistant tumor growth. Cancer Res 2008; 68: 4447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharifi N. Minireview: androgen metabolism in castration‐resistant prostate cancer. Mol Endocrinol 2013; 27: 708–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stanbrough M, Bubley GJ, Ross K et al Increased expression of genes converting adrenal androgens to testosterone in androgen‐independent prostate cancer. Cancer Res 2006; 66: 2815–25. [DOI] [PubMed] [Google Scholar]
- 29. Chang KH, Li R, Papari‐Zareei M et al Dihydrotestosterone synthesis bypasses testosterone to drive castration‐resistant prostate cancer. Proc Natl Acad Sci USA 2011; 108: 13728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Powell K, Semaan L, Conley‐LaComb MK et al ERG/AKR1C3/AR constitutes a feed‐forward loop for AR signaling in prostate cancer cells. Clin Cancer Res 2015; 21: 2569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takayama K, Misawa A, Suzuki T et al TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat Commun 2015; 6: 8219. [DOI] [PubMed] [Google Scholar]
- 32. Takayama K, Horie‐Inoue K, Katayama S et al Androgen‐responsive long noncoding RNA CTBP1‐AS promotes prostate cancer. EMBO J 2013; 32: 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang M, Xie W, Mostaghel E et al SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J Clin Oncol 2011; 29: 2565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Migita T, Narita T, Nomura K et al ATP citrate lyase: activation and therapeutic implications in non‐small cell lung cancer. Cancer Res 2008; 68: 8547–54. [DOI] [PubMed] [Google Scholar]
- 35. Migita T, Okabe S, Ikeda K et al Inhibition of ATP citrate lyase induces an anticancer effect via reactive oxygen species: AMPK as a predictive biomarker for therapeutic impact. Am J Pathol 2013; 182: 1800–10. [DOI] [PubMed] [Google Scholar]
- 36. Yamashita K, Miyashiro Y, Maekubo H et al Development of highly sensitive quantification method for testosterone and dihydrotestosterone in human serum and prostate tissue by liquid chromatography‐electrospray ionization tandem mass spectrometry. Steroids 2009; 74: 920–6. [DOI] [PubMed] [Google Scholar]
- 37. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957; 226: 497–509. [PubMed] [Google Scholar]
- 38. Perez‐Chacon G, Astudillo AM, Balgoma D, Balboa MA, Balsinde J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim Biophys Acta 2009; 1791: 1103–13. [DOI] [PubMed] [Google Scholar]
- 39. Takayama K, Tsutsumi S, Katayama S et al Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome‐wide androgen receptor signaling in prostate cancer cells. Oncogene 2011; 30: 619–30. [DOI] [PubMed] [Google Scholar]
- 40. Attard G, Clark J, Ambroisine L et al Heterogeneity and clinical significance of ETV1 translocations in human prostate cancer. Br J Cancer 2008; 99: 314–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tamura K, Makino A, Hullin‐Matsuda F et al Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long‐chain fatty acid metabolism. Cancer Res 2009; 69: 8133–40. [DOI] [PubMed] [Google Scholar]
- 42. Tamura K, Furihata M, Tsunoda T et al Molecular features of hormone‐refractory prostate cancer cells by genome‐wide gene expression profiles. Cancer Res 2007; 67: 5117–25. [DOI] [PubMed] [Google Scholar]
- 43. Tomlins SA, Mehra R, Rhodes DR et al Integrative molecular concept modeling of prostate cancer progression. Nat Genet 2007; 39: 41–51. [DOI] [PubMed] [Google Scholar]
- 44. Holzbeierlein J, Lal P, LaTulippe E et al Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen‐responsive genes and mechanisms of therapy resistance. Am J Pathol 2004; 164: 217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marques RB, Dits NF, Erkens‐Schulze S, van Ijcken WF, van Weerden WM, Jenster G. Modulation of androgen receptor signaling in hormonal therapy‐resistant prostate cancer cell lines. PLoS One 2011; 6: e23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol 2014; 2: 111–20. [PMC free article] [PubMed] [Google Scholar]
- 47. Rossi S, Graner E, Febbo P et al Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res 2003; 1: 707–15. [PubMed] [Google Scholar]
- 48. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP‐1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res 2012; 10: 133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyamoto DT, Sequist LV, Lee RJ. Circulating tumour cells‐monitoring treatment response in prostate cancer. Nat Rev Clin Oncol 2014; 11: 401–12. [DOI] [PubMed] [Google Scholar]
- 50. Crespo M, van Dalum G, Ferraldeschi R et al Androgen receptor expression in circulating tumour cells from castration‐resistant prostate cancer patients treated with novel endocrine agents. Br J Cancer 2015; 112: 1166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chang KH, Li R, Kuri B et al A gain‐of‐function mutation in DHT synthesis in castration‐resistant prostate cancer. Cell 2013; 154: 1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wright JL, Kwon EM, Ostrander EA et al Expression of SLCO transport genes in castration‐resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev 2011; 20: 619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Klein H, Bressel M, Kastendieck H, Voigt KD. Androgens, adrenal androgen precursors, and their metabolism in untreated primary tumors and lymph node metastases of human prostatic cancer. Am J Clin Oncol 1988; 11(Suppl 2): S30–6. [DOI] [PubMed] [Google Scholar]
- 54. Chouinard S, Barbier O, Belanger A. UDP‐glucuronosyltransferase 2B15 (UGT2B15) and UGT2B17 enzymes are major determinants of the androgen response in prostate cancer LNCaP cells. J Biol Chem 2007; 282: 33466–74. [DOI] [PubMed] [Google Scholar]
- 55. Barbier O, Belanger A. Inactivation of androgens by UDP‐glucuronosyltransferases in the human prostate. Best Pract Res Clin Endocrinol Metab 2008; 22: 259–70. [DOI] [PubMed] [Google Scholar]
- 56. Mohler JL, Gregory CW, Ford OH III et al The androgen axis in recurrent prostate cancer. Clin Cancer Res 2004; 10: 440–8. [DOI] [PubMed] [Google Scholar]
- 57. Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res 2005; 11: 4653–7. [DOI] [PubMed] [Google Scholar]
- 58. Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 2010; 21: 315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cai C, Chen S, Ng P et al Intratumoral de novo steroid synthesis activates androgen receptor in castration‐resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011; 71: 6503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mostaghel EA, Marck BT, Plymate SR et al Resistance to CYP17A1 inhibition with abiraterone in castration‐resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res 2011; 17: 5913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu C, Lou W, Zhu Y et al Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res 2015; 75: 1413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Obinata D, Takayama K, Fujiwara K et al Targeting Oct1 genomic function inhibits androgen receptor signaling and castration‐resistant prostate cancer growth. Oncogene 2016; 35: 6350–8. [DOI] [PubMed] [Google Scholar]
- 63. Hertz R, Magenheim J, Berman I, Bar‐Tana J. Fatty acyl‐CoA thioesters are ligands of hepatic nuclear factor‐4alpha. Nature 1998; 392: 512–6. [DOI] [PubMed] [Google Scholar]
- 64. Hertz R, Sheena V, Kalderon B, Berman I, Bar‐Tana J. Suppression of hepatocyte nuclear factor‐4alpha by acyl‐CoA thioesters of hypolipidemic peroxisome proliferators. Biochem Pharmacol 2001; 61: 1057–62. [DOI] [PubMed] [Google Scholar]
- 65. Bolotin E, Schnabl J, Sladek F. HNF4A. Transcription factor encyclopedia, 2010. [Cited 26 Aug 2010] Available from URL: http://www.cisreg.ca/cgi-bin/tfe/articles.pl?tfid=140.
- 66. Hwang‐Verslues WW, Sladek FM. HNF4alpha–Role in drug metabolism and potential drug target? Curr Opin Pharmacol 2010; 10: 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bolotin E, Liao H, Ta TC et al Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology 2010; 51: 642–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome‐wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 2003; 31: 3635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Patek SCW, Wilder JM, Heng J et al Androgen receptor phosphorylation status at serine 578 predicts poor outcome in prostate cancer patients. Oncotarget 2017; 8: 4875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bao BY, Chuang BF, Wang Q et al Androgen receptor mediates the expression of UDP‐glucuronosyltransferase 2 B15 and B17 genes. Prostate 2008; 68: 839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chouinard S, Pelletier G, Belanger A, Barbier O. Isoform‐specific regulation of uridine diphosphate‐glucuronosyltransferase 2B enzymes in the human prostate: differential consequences for androgen and bioactive lipid inactivation. Endocrinology 2006; 147: 5431–42. [DOI] [PubMed] [Google Scholar]
- 72. Takahashi K, Funakoshi‐Tago M, Takaoka M, Kakio S, Kobata K, Tamura H. Roasted coffee induction of aldo‐keto reductase 1C3 expression in LNCaP human prostate cancer cells is associated with Nrf2 activation. Oncol Lett 2016; 12: 5321–6. [Google Scholar]
- 73. Ebert B, Kisiela M, Wsol V, Maser E. Proteasome inhibitors MG‐132 and bortezomib induce AKR1C1, AKR1C3, AKR1B1, and AKR1B10 in human colon cancer cell lines SW‐480 and HT‐29. Chem Biol Interact 2011; 191: 239–49. [DOI] [PubMed] [Google Scholar]
- 74. Csala M, Banhegyi G, Kardon T et al Inhibition of glucuronidation by an acyl‐CoA‐mediated indirect mechanism. Biochem Pharmacol 1996; 52: 1127–31. [DOI] [PubMed] [Google Scholar]
- 75. Ishii Y, Nurrochmad A, Yamada H. Modulation of UDP‐glucuronosyltransferase activity by endogenous compounds. Drug Metab Pharmacokinet 2010; 25: 134–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression levels of ACSL3 mRNA in prostate cancer and its subcellular localization.
Fig. S2. Effect of DHT supplementation and ACSL3 overexpression on the expression levels of SLCO1B3, AKR1C3, SRD5A1 and UGT2B15.
Fig. S3. Representative pictures of immunohistochemistry of ACSL3, AKR1C3 and UGT2B in prostate cancer.
Fig. S4. Effect of ACSL3 overexpression on steroidogenic genes utilizing cholesterol.
Fig. S5. Effect of ACSL3 overexpression on the sensitivity to tunicamycin in DU145 cells.
Fig. S6. Expression levels of ACSL3 mRNA in hormone sensitive prostate cancer and castration‐resistant prostate cancer (CRPC).
Table S1. Oligonucleotide sequences of PCR primers.