

Abstract
The anaplastic lymphoma kinase (ALK) gene was initially identified as a fusion partner of the nucleophosmin gene in anaplastic large‐cell lymphoma with t(2;5)(p23;q35) translocation, and then described with different genetic abnormalities in a number of tumors. Although ALK is known to be involved in the pathogenesis of neuroblastoma through activating mutations or gene amplification, its role in the pathogenesis of other pediatric cancers is still elusive. In addition to neuroblastoma, the high‐grade amplification of ALK has been described in a subset of rhabdomyosarcoma cases. Normal ALK protein expression is restricted to the nervous systems of adult mammals, but the aberrant expression of ALK has been observed in a variety of pediatric cancers, including glioma and Ewing sarcoma. The discovery of oncogenic activation of ALK in neuroblastoma suggests that this cancer could be potentially treated with an ALK inhibitor, as could other cancers, such as non‐small‐cell lung cancer and anaplastic large‐cell lymphoma. However, cellular responses to mutant ALK are complex when compared to rearranged ALK, and treatment remains a challenge. This review focuses on the biology of ALK in pediatric cancers and possible therapeutic strategies for ALK‐associated tumors.
Keywords: ALK cancer, anaplastic large‐cell lymphoma, inflammatory myofibroblastic tumor, neuroblastoma
In recent years, the discovery of a variety of genetic alterations in different malignancies leading to oncogenesis has provided insight into the complexity of tumorigenesis and the development for target‐specific therapies with the objective of improving clinical outcomes.1 Among them, the breakthrough of the BCR‐ABL fusion in patients with chronic myeloid leukemia has become a paradigm for personalized or precision medicine.2 In current clinical practice, personalized cancer therapy is well established for a number of gene targets, including various kinase encoded genes.1 The anaplastic lymphoma kinase (ALK) gene encodes a transmembrane receptor tyrosine kinase (RTK), which belongs to the insulin receptor superfamily.3 ALK was first identified as part of the nucleophosmin (NPM)‐ALK gene fusion transcript, which is derived from the t(2;5)(p23;q35) translocation that is involved in the pathogenesis of a subset of cases of anaplastic large‐cell lymphoma (ALCL).4, 5 ALK has been found to be rearranged, mutated, or amplified in a variety of tumors, including neuroblastoma, inflammatory myofibroblastic tumor (IMT), and non‐small‐cell lung cancer (NSCLC).6, 7, 8, 9, 10, 11 This pivotal discovery has designated the ALK protein as a potentially relevant biomarker and therapeutic target in a wide variety of solid tumors and hematological malignancies in which ALK is a critical mediator of carcinogenesis. In fact, dramatic responses to ALK inhibitors have been documented in NSCLC, ALCL, and IMT patients.12, 13 The identification of recurrent oncogenic alterations of ALK in ALCL, IMT, and neuroblastoma has highlighted the importance for ALK in histologically diverse pediatric cancers. Therefore, there is a need to better understand the role of ALK in cancer biology to optimize treatment strategies for pediatric cancers.
This review summarizes the recent discoveries of the oncogenic roles of ALK in pediatric cancers.
Structure, Function, and ALK Signaling
ALK encodes a highly conserved, 1620‐amino acid RTK, which is located on chromosome 2p23.2.3 Together with leukocyte receptor tyrosine kinase and reactive oxygen species, ALK belongs to the insulin receptor superfamily of cellular transmembrane receptors that display intrinsic tyrosine kinase activity.5 The structure of this gene product includes an extracellular domain (ECD), a single transmembrane region, and an intracellular kinase domain.3 The ALK ECD is unique among RTK family members, containing a glycine‐rich region and a low‐density lipoprotein receptor class A domain sandwiched between two meprin, A‐5 protein, and receptor protein tyrosine phosphatase mu (MAM) domains (Fig. 1).3 The ALK ECD can be divided into several regions with presumed functions of ligand binding, interactions with potential co‐receptors and secreted regulatory proteins, and dimerization, all of which may potentially relay conformational changes to initiate the activation of the intracellular kinase domain.3 The activation of endogenous ALK requires ligand‐dependent receptor dimerization and autophosphorylation. The binding site for two putative ALK ligands, pleiotrophin and midkine, has been mapped between residues 391 and 401.14, 15 More recently, augmentor α and β (FAM150) have been established as ALK ligands.16 However, the mechanism by which ALK is physiologically activated has not been completely elucidated. The ALK intracellular domains are composed of a tyrosine kinase region with three phosphorylation sites (Y1278, Y1283, and Y1283), followed by a carboxyl‐terminal lobe containing interaction sites for phospholipase C‐γ and Src homology 2 domain‐containing (SHC).17
Figure 1.
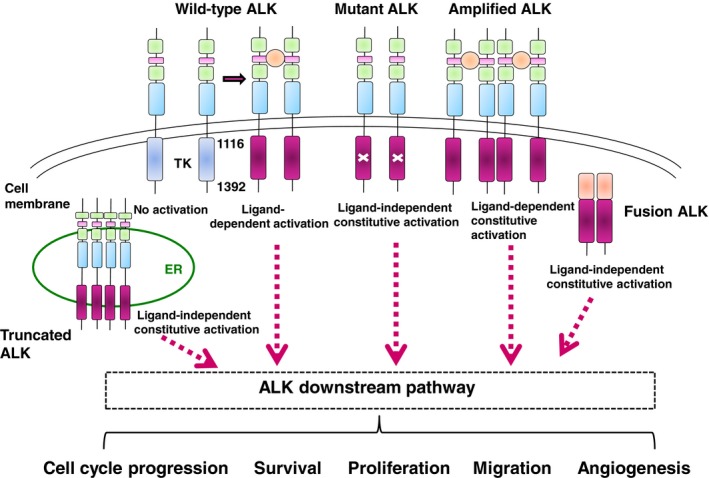
Anaplastic lymphoma kinase (ALK) signaling in normal and cancer cells. Normal activation of ALK through ligand binding is shown (the ligand is indicated in orange). The full‐length ALK receptor is a classical receptor tyrosine kinase, composed of an amino‐terminal extracellular domain and intracellular tyrosine kinase domain (inactive in blue and active in red), and connected by a single transmembrane domain. The ALK extracellular domain contains two MAM domains (in green), one LDL domain (in pink), and a glycine‐rich region (in light blue). ALK mutations result in ligand‐independent constitutive activation of the downstream ALK pathway, whereas ALK amplification results in ligand‐dependent constitutive activation of ALK signaling. In expressed ALK fusion proteins, ligand‐independent dimerization with the oligomerization domains of partner genes leads to the constitutive activation of the ALK pathway. An aberrant form of ALK that lacks exons 2 and 3 was amplified, leading to the high‐level expression of an N‐terminal truncated kinase. This short form of ALK is mainly located at endoplasmic reticulum and aberrantly activates the STAT3 pathway from ER. TK, tyrosine kinase.
Expression of ALK in humans is limited to rare neural cells and scattered pericytes, as well as to endothelial cells after birth, implying a role in neural development and differentiation.3 However, the native function of ALK in humans is still unclear. At present, the distribution and function of the ALK gene have been investigated in a number of model systems. A single ALK family member has been found in worms (SCD‐2) and flies (Alk).18, 19 In Caenorhabditis elegans, SCD‐2 signaling is required for the integration of sensory input but is not essential for development.18 In Drosophila melanogaster, Alk is activated by its ligand Jelly belly (Jeb) and regulates the development of the gut musculature and neuronal circuitry within the visual system.20 Flies lacking Alk die due to a lack of founder cell specification in the embryonic visceral muscle.19 Mice with a homozygous deletion of the Alk tyrosine kinase domain have a normal appearance and no obvious tissue abnormalities, but preliminary observations have revealed an increase in the number of progenitor cells in the hippocampus and modifications of adult brain function.21 Additionally, results in ALK gain‐of‐function, knock‐in mice have revealed a role for ALK in neurogenesis and neuroblastoma progression combined with MYCN overexpression.22
The ALK enzyme can activate other signaling pathways, including the PI3K‐AKT, JAK‐STAT, CRKL‐C3G, MEKK2/3‐MEK5‐ERK, and MAPK pathways, which in turn transmit a signal downstream through the signal pathway to the nucleus where the expression of different genes are controlled (Fig. 1).23 The activation of adaptor proteins and other cellular proteins, such as protein tyrosine phosphatase, non‐receptor type 11, SRC, fibroblast growth factor receptor substrate 2, and SHC, has been observed downstream of ALK, implicating roles for these alternative pathways.17 Other reported ALK downstream targets are Bcl‐2‐like protein 11, p27, Cyclin D2, nuclear interacting partner of ALK, Ras‐related C3 botulinum toxin substrate 1, cell division cycle 42, p130CAS, Src homology region 2 domain‐containing phosphatase‐1, and PIKFYVE.23, 24, 25, 26
ALK Rearrangements in Pediatric Cancers
Various chromosomal rearrangements have been reported at the ALK locus on chromosome 2q23.2, leading to the generation of fusion genes that encode the entire intracellular domain of ALK at the 3′‐end fused to various 5′‐end partner genes. Recent genome‐wide studies have revealed the presence of nearly 30 different ALK fusion partner genes in multiple types of cancer (Table 1). Even though a large number of different N‐terminal partners have been identified, they all share common oncogenic features. The subsequent expression of fusion proteins is regulated by the promoter of the N‐terminal partner, which is generally expressed in various normal tissues and can lead to the ectopic expression of ALK kinase.40 As in other RTK‐related, oncogenic fusions, the N‐termini of the partner genes are characterized by the presence of oligomerization domains, which are essential for the oncogenic potential of the fusion protein. The role of ALK fusion proteins as oncogenic drivers has been well established in preclinical models, including transgenic mouse models.7
Table 1.
Chromosomal rearrangements involving ALK locus in pediatric cancers
| Cancer type | ALK fusion gene | Chromosomal abnormalities | Reference no. |
|---|---|---|---|
| ALCL | TPM3‐ALK | t(1;2) (q25;p23) | 23 |
| ATIC‐ALK | inv(2) (p23;q35) | 23 | |
| TFG‐ALK | t(2;3) (p23;q21) | 27 | |
| NPM1‐ALK | t(2;5) (p23;q35) | 4, 5 | |
| TRAF1‐ALK | t(2;9) (p23;q32.2) | 28 | |
| CLTC‐ALK | t(2;17) (p23;q23.1) | 23 | |
| RNF213‐ALK | t(2;17) (p23;q25.3) | 29 | |
| TPM4‐ALK | t(2;19) (p23;q13.1) | 23 | |
| MYH9‐ALK | t(2;22) (p23;q12.3) | 23 | |
| MSN‐ALK | t(2;X) (p23;q12) | 23 | |
| IMT | TPM3‐ALK | t(1;2) (q25;p23) | 30 |
| RANBP2‐ALK | t(2;2) (p23;q13) | 31 | |
| ATIC‐ALK | inv(2) (p23;q35) | 32 | |
| SEC31A‐ALK | t(2;4) (p23;q21.22) | 33 | |
| CARS‐ALK | t(2;11) (p23;p15.4) | 23 | |
| PPFIBP1‐ALK | t(2;12) (p23;p11) | 34 | |
| CLTC‐ALK | t(2;17) (p23;q23.1) | 23 | |
| TPM4‐ALK | t(2;19) (p23;q13.1) | 30 | |
| RCC | TPM3‐ALK | t(1;2) (q25;p23) | 36, 37 |
| EML4‐ALK a | inv(2) (p21;p23) | 35 | |
| STRN‐ALK | t(2;2) (p23;p22) | 37 | |
| VCL‐ALK | t(2;10) (p23;q22) | 36 | |
| PTC | EML4‐ALK | inv(2) (p21;p23) | 39 |
| STRN‐ALK | t(2;2) (p23;p22) | 38, 39 | |
| GTF21RD1‐ALK | t(2;7) (p23;q11.23) | 39 |
Reported only in adult cases. ALCL, anaplastic large cell lymphoma; IMT, inflammatory myofibroblastic tumor; PTC, papillary thyroid cancer; RCC, renal cell carcinoma.
Anaplastic large‐cell lymphoma
Anaplastic large‐cell lymphoma is a rare type of T‐cell non‐Hodgkin's lymphoma that is characterized by large cells with variable shapes (anaplastic patterns) expressing the membrane receptor CD30, which is a member of the nerve growth factor/tumor necrosis factor receptor family.41 Anaplastic large‐cell lymphoma accounts for 2–5% of all non‐Hodgkin's lymphoma, although its prevalence is higher in children and young adults with a frequency of 10–15%.41 Up to 90% of all pediatric ALCL cases have ALK rearrangements that lead to the ectopic overexpression of ALK kinase, whereas only approximately 30% of adult cases harbor these same genetic abnormalities.42 The most recurrent rearrangements of ALK in ALCL is the NPM‐ALK fusion, accounting for 75–80% of all ALK‐positive ALCLs, followed by the tropomyosin 3 (TPM3)‐ALK fusion, with a frequency of 12–18%.4, 5, 43 NPM, located on chromosome 5q35, is an abundant, nucleolar phosphoprotein that shuttles between the nucleus and cytoplasm;44 it is involved in numerous cellular processes, including ribonucleoprotein transport, centrosome duplication, and control of genomic stability.44 Tropomyosin 3 is a member of the tropomyosin family of actin‐binding proteins, located on chromosome 1q25.45 Tropomyosins are components of cytoskeletal microfilaments, providing actin filament stability and regulating interactions with other actin‐binding proteins.45 Other fusions have been found at a much lower frequency (<2%) and include TRK‐fused gene (TFG)‐ALK, clathrin heavy chain (CLTC)‐ALK, and TPM4‐ALK.23, 27, 28, 29 These fusions possess subtly different characteristics when transfected into NIH3T3 fibroblasts and implanted as xenografts. The effects of the different ALK N‐terminal partners were assessed by expressing five ALK fusion variants in NIH3T3 cells:40 NPM‐, TFG‐, CLTC‐, and 5‐aminoimidazole‐4‐carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase (ATIC) ‐ALK were found to increase proliferation and soft agar colony formation, whereas TPM3‐ALK had a stronger effect on invasion. The expressed TPM3‐ALK protein was subsequently shown to co‐immunoprecipitate endogenous tropomyosin, further supporting an effect on cytoskeleton organization with a concomitant decrease in cell adhesion.30, 46 Transfection of the different ALK fusions in NIH3T3 cells led to the development of tumors in nude mice, but NPM‐ALK‐ and TFG‐ALK‐transfected cells gave rise to more rapidly growing tumors.27 Notably, several ALK fusions, such as TPM3/4‐ALK, CLTC‐ALK, and Ran‐binding protein 2 (RANBP2)‐ALK, have been detected in IMTs and diffuse B‐cell large cell lymphoma, implicating a preferential choice for the ALK rearrangement partner, regardless of the affected organs or tissues. Importantly, clinical outcomes for ALK‐positive ALCL patients approach the 70–80% cure rate, compared to 15–45% for ALK‐negative ALCL patients.41, 42
Inflammatory myofibroblastic tumors
Inflammatory myofibroblastic tumors are rare benign or locally aggressive soft‐tissue mesenchymal neoplasms.47 They occur primarily in children and young adults, but can develop at any age. Histologically, IMTs are characterized by the presence of a dense, inflammatory infiltrate amid spindle cells in a myxoid to collagenous stroma.47 Surgical resection is usually the first line of treatment, but many cases develop a more aggressive phenotype with occurrence of metastases. Approximately 50% of IMTs possess chromosomal rearrangements involving the 2q23.2 region, resulting in TPM3‐4‐ALK fusion transcripts.30 Currently, several other ALK fusions, including cysteinyl‐TRNA synthetase (CARS)‐ALK, CLTC‐ALK, ATIC‐ALK, RANBP2‐ALK, and SEC31L1‐ALK, have been identified at frequencies <5%.6, 31, 32, 33, 34 With the exception of RANBP2‐ALK, which is localized to the nuclear membrane when expressed, the other expressed fusion proteins display a typical cytoplasmic staining. Anaplastic lymphoma kinase expression is generally seen in younger patients and correlates with local recurrence rather than with distant metastasis formation; thus, ALK fusions are thought to be associated with better prognosis in cases of IMT.47 Published studies suggest that the identification of the ALK gene rearrangements is useful to differentiate IMTs from other spindle cell neoplasms of soft tissues and viscera.
Other pediatric and adolescent cancers with ALK fusions
Renal cell carcinoma (RCC) is rare in children; it accounts for approximately 2–4% of all pediatric renal tumors.35 Initially, ALK rearrangements were described in pediatric RCC cases, but further investigations showed that a small proportion of adult RCCs belong to this ALK‐rearranged category.36 ALK‐rearranged RCC is a distinct type of disease included in the so‐called class of emerging/provisional RCCs.36, 48 Several gene partners, including VCL, TPM3, striatin (STRN), echinoderm microtubule‐associated protein like 4 (EML4), and HOOK1, have been reported in cases of RCC to date.37, 48, 49, 50 Histologically, ALK‐rearranged RCCs most commonly feature a solid architecture with occasional trabecular and tubular features, and are composed of sheets of epithelioid cells with abundant pale eosinophilic cytoplasm and frequent intracytoplasmic lumina.48 The clinical course for ALK‐rearranged RCCs is frequently indolent; however, in adult patients, progression is frequently more aggressive.48
Additionally, in adult cases of thyroid cancer, various ALK fusions, including EML4‐ALK, glutamine‐fructose‐6‐phosphate transaminase 1 (GFPT1)‐ALK, TFG‐ALK, and STRN‐ALK, have been detected. These ALK fusions have been found in approximately 2% of patients with papillary thyroid cancer and medullary thyroid carcinoma.38, 51 Although rare, ALK rearrangements, such as STRN‐ALK and EML4‐ALK, have been observed in pediatric papillary thyroid cancer cases as well.39
Furthermore, ALK rearrangements occur in approximately 10–20% of spitzoid tumors (Spitz nevi, atypical Spitz tumors, and spitzoid melanomas), which are melanocytic neoplasms with distinctive histopathological features such as increased cell size and an epithelioid or spindle morphology.52 Spitzoid tumors are more common in children and adolescents, but can occur at all ages. Those with ALK fusions show unique histopathologic features, which promise to improve the classification of these diagnostically challenging tumors.
ALK mutations in pediatric cancers
Gain‐of‐function mutations in the full‐length ALK were first described in sporadic and familial neuroblastomas, common childhood solid tumors.8, 9, 10, 11 Subsequently, activating ALK mutations were reported in adult cases of anaplastic thyroid cancer.53 In addition, next‐generation sequencing revealed the presence of several mutations in the ALK kinase domain in pediatric cases with rhabdomyosarcoma, primitive neuroectodermal tumor, and osteosarcoma (Fig. 2). Most of these mutations are located in the kinase domain and can be classified into three groups: ligand‐independent mutations (F1174I, F1174S, F1174L, and R1275Q), ligand‐dependent mutations (D1091N, T1151M, and A1234T), and a kinase‐dead mutation (I1250T) (Fig. 2). ALK mutations are occasionally acquired in ALK fusion genes as a result of resistance to ALK inhibitors in cases of NSCLC as well as ALCL.54, 55
Figure 2.
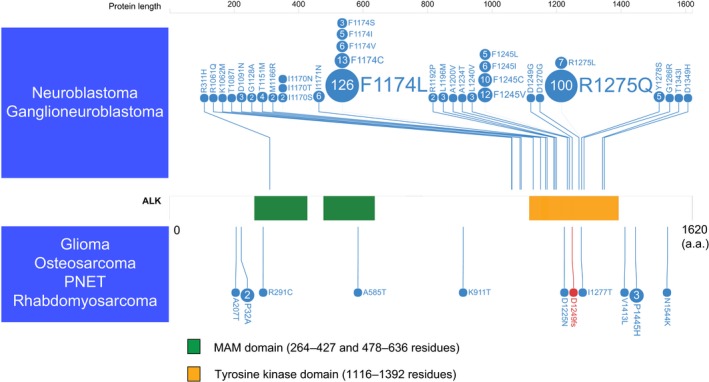
Anaplastic lymphoma kinase (ALK) mutations detected in pediatric cancers. The nucleotide changes detected in neuroblastoma, ganglioneuroblastoma, glioma, osteosarcoma, primitive neuroectodermal tumor, and rhabdomyosarcoma are shown. COSMIC frequencies of ALK mutations from published reports with functional and/or therapeutic significance are shown. A missense mutation is indicated by a blue circle, and a frameshift mutation is indicated by a red circle. The numbers in the circles are the reported mutation numbers. The green box indicates MAM domains, and the orange box indicates the tyrosine kinase domain. a.a., amino acids; PNET, primitive neuroectodermal tumor.
Neuroblastoma
Neuroblastoma is the most common pediatric extracranial solid tumor, a disease of the sympathoadrenal lineage of the neural crest, accounting for approximately 15% of all pediatric oncology deaths.56, 57 This disease has a broad spectrum of clinical behaviors, ranging from localized disease with spontaneous regression to aggressive clinical course and death from progressive disease.56, 57 Although mostly sporadic, neuroblastomas may occur in familial or syndromic contexts.57 The best characterized genetic alteration associated with a poor prognosis of neuroblastoma is the amplification of the MYCN oncogene.56 A loss of heterozygosity on chromosome 11q was also found to correlate with poor prognosis in neuroblastoma.58
Previously, we and other groups discovered ALK to be a major oncogene target in sporadic and familial neuroblastoma cases.8, 9, 10, 11 In familial cases, germline ALK mutations were observed in 50% of pedigree, whereas approximately 10% of sporadic cases showed ALK oncogenic mutations. Greater than 90% of all ALK mutations occurred within the kinase domain, which clearly showed two mutation hot spots at positions F1174 and R1275.59 Mutations of the F1245 residue were also frequently observed in neuroblastoma, after the more common F1174 and the R1275 mutations. The R1275 mutation is more frequently observed in familial cases compared to the F1174 mutation, which is a common mutation in sporadic cases.59 Intriguingly, the R1275 mutation is adjacent to the corresponding L858R mutation in the epidermal growth factor receptor, which is the most common epidermal growth factor receptor mutation in lung cancer.60 According to insulin RTK homology models, F1174 is located at the end of the Cα1 helix, whereas the other two common mutations are located in the two β sheets: before the catalytic loop (β6) (F1245) and within the activation loop (β9) (R1275).60 The R1192P mutation, which has only been reported in familial cases, is found in the β4 strand of the kinase domain.60 Expression of the F1174L and K1062M mutants in NIH3T3 cells induces transformation capacity; cells transduced with mutant proteins display increased colony formation in soft agar and tumor generation in nude mice, and mutant kinases show increased autophosphorylation and in vitro kinase activity compared to wild‐type.8 In accordance with these findings, molecules located downstream of ALK, including AKT, STAT3, and ERK, were found to be activated in cells expressing mutant ALK (Fig. 3).8, 9, 10, 11 Furthermore, both F1174L and R1275Q mutants promote cytokine‐dependent growth of BaF3 cells, an immortalized murine bone marrow‐derived pro‐B‐cell line whose growth and proliferation depend on the presence of interleukin‐3.11 Importantly, RNAi‐mediated ALK knockdown resulted in reduced cell proliferation in a cell line harboring the F1174L mutation, but these effects were less clear in wild‐type ALK‐expressing neuroblastoma cells.8, 9 ALK mutations were found to highly correlate with MYCN amplification: approximately 50% of ALK mutations coexist with MYCN amplification.8, 61 Transgenic and knock‐in ALK mutation models highlight the synergy between ALK and MYCN in the pathogenesis of neuroblastoma.22, 62
Figure 3.
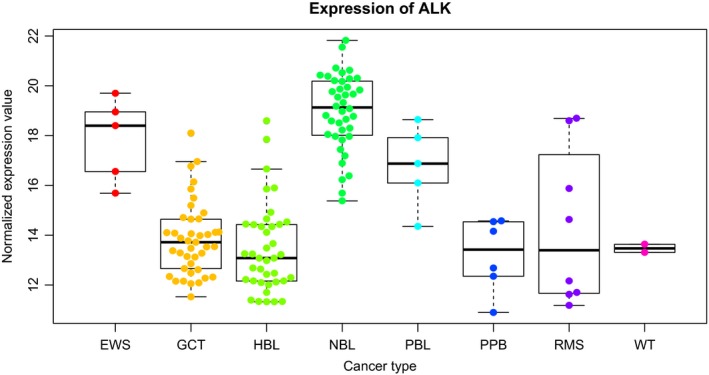
Anaplastic lymphoma kinase (ALK) expression in pediatric cancers detected by RNA sequencing. Box plots of the ALK mRNA expression in pediatric cancers are shown. ALK expression, detected by RNA sequencing, was normalized and analyzed using variance‐stabilizing transformations (VST). EWS, Ewing sarcoma; GCT, germ cell tumor; HBL, hepatoblastoma; NBL, neuroblastoma; PBL, pancreatoblastoma; PPB, pleuropulmonary blastoma; RMS, rhabdomyosarcoma; WT, Wilms' tumor.
ALK Amplification, Copy Number Gain, and Rare Structural Variants
ALK gene amplification occurs in a variety of cancers and is recognized as a therapeutic target. In neuroblastoma, high‐grade amplification of ALK has been found in cell lines and primary tissues, and the subsequent overexpression of ALK protein correlates with tumorigenesis.23 Constitutive activation of ALK results in the phosphorylation of downstream molecules, including SHC and MAPK pathway‐related proteins, in neuroblastoma cell lines.17 As with ALK mutation, amplification of ALK also strongly correlates with MYCN amplification.62 In addition, recurrent copy number gain of chromosome 2p23.2 involving the ALK locus has been reported in neuroblastoma with a frequency of 35–40%.8, 62 High ALK expression has been shown to be tightly associated with ALK amplification/mutation, as well as copy number gain of the ALK locus, suggesting the upregulation of ALK expression by increasing DNA dose.(62)
Amplification or copy number gain of the ALK locus, as reported in a subset of rhabdomyosarcoma cases, is also intriguing from a therapeutic perspective. Rhabdomyosarcoma is the most common soft‐tissue sarcoma in children and can be divided into two histological subtypes, embryonal (ERMS) and alveolar (ARMS).63 Alveolar rhabdomyosarcoma shows an aggressive phenotype, whereas embryonal has a more favorable prognosis.63 We previously found that ALK inhibitors were effective in select rhabdomyosarcoma cell lines with high ALK expression levels, suggesting a possible therapeutic target for ALK inhibitors in rhabdomyosarcoma.64 Compared with TAE684 and 2,4‐pyrimidinediamine derivative (2,4‐PDD), the different pharmacological behavior of crizotinib may be related to MET inhibition, the specific activity of crizotinib, because the rhabdomyosarcoma cell lines examined also express MET to varying degrees.64 Given that ALK amplification or copy number gain is predominantly detected in ARMS (6–17%),64, 65 rhabdomyosarcoma cases with a poor prognosis may benefit from ALK inhibitors.
We found another mechanism of ALK activation observed in a neuroblastoma‐derived cell line (NB‐1): A short form of the ALK protein (ALKdel2‐3, with a truncated extracellular domain) is overexpressed, because of the amplification of an abnormal ALK gene that lacks exons 2 and 3.66 Functional analysis revealed that ALKdel2‐3 underwent autophosphorylation in NB‐1 and NIH3T3 cells, demonstrating enhanced kinase activity and promoting downstream signaling pathways, such as the STAT3 pathway. These results confirm the oncogenic activity of ALKdel2‐3.66 Interestingly, two separate studies described aberrant ALK proteins with truncated extracellular domains, Del 1‐5 and Del 4‐11 variants, leading to constitutive kinase activity in four neuroblastoma specimens, including three primary cases. These truncated proteins resulted from chromosomal rearrangements involving the ALK locus.67, 68 Our data, together with these results, indicate that, although rare, truncated ALK proteins are involved in the pathogenesis of neuroblastoma, and patients with these rare types of ALK alterations may benefit from targeted treatments using ALK inhibitors.
Expression of Full‐length ALK in Pediatric Cancers
Expression of full‐length ALK protein has been detected in a variety of human cancer cell lines and tumor specimens, such as rhabdomyosarcoma, glioblastoma, and Wilms' tumor.65, 69, 70 In our cohort of 146 cases with pediatric cancers, expression of full‐length ALK was observed across various pediatric solid tumors, such as rhabdomyosarcoma, Ewing sarcoma, and pancreatoblastoma (Fig. 3). Previously, a genome‐wide study revealed ALK as a target gene of PAX3‐FOXO1, the recurrent gene fusion in ARMS.71 Quantitative assessment of ALK mRNA expression in rhabdomyosarcoma revealed that high ALK expression correlated with a negative prognosis, independent of fusion status.72 The wide distribution of ALK expression in pediatric solid tumors suggests that ALK inhibitor‐based therapies may benefit patients with these particular tumors.
Targeted Anticancer Therapies
Recently, several ALK inhibitors were developed and examined in preclinical and clinical trials.73 Initial studies were carried out using less potent ALK inhibitors, such as WHI‐P154, pyridines, and heat shock protein 90.74, 75 Subsequently, more potent and specific ALK inhibitors, such as diaminopyrimidines or aminopyrimidines including NVP‐TAE684, 2,4‐PDD, and crizotinib (PF‐2341066), have been developed.7, 73 NVP‐TAE684 is a highly potent and specific NPM‐ALK inhibitor but is not currently being developed clinically. Such selective inhibitors can lead to the potent suppression of cell growth in tumors expressing ALK fusion proteins. Crizotinib is an inhibitor of both MET and ALK, and is the first ALK inhibitor to be tested in phase I–II clinical trials of pediatric patients including relapsed solid tumors and ALCL.13 Crizotinib was generally well tolerated in pediatric patients and showed excellent antitumor activity in patients with ALCL and IMT.9 Although neuroblastoma cell lines harboring ALK mutations or amplification were highly vulnerable to ALK inhibitors, such as NVP‐TAE684, such a robust response to crizotinib has not been observed in neuroblastoma patients with ALK mutations.8, 9 Thus, it will be important to study neuroblastoma cells expressing mutant ALK to determine whether an ALK inhibitor alone, or in combination with other kinase inhibitors, is sufficient for growth inhibition. Further development of ALK inhibitors, alone and in combination with conventional chemotherapies, is likely to be used in the near future. The development of mAbs against ALK should be investigated, as they may be particularly useful for tumors that have acquired resistance to ALK inhibitors.
Conclusion
This review provides an understanding of the role of ALK in pediatric cancers and the preclinical proof‐of‐principle for the use of ALK as a therapeutic target. There is a strong rationale and significant enthusiasm for the use of ALK inhibitors as targeted therapies against tumors harboring oncogenic fusions or activating mutations. The results of the first clinical trial with crizotinib have shown promise in highly refractory patients with tumors harboring ALK fusion proteins; these results support the ongoing efforts to select patients for subsequent trials based on ALK translocation status. Additional clinical trials of other ALK inhibitors, such as alectinib and loratinib, in neuroblastoma patients will be crucial to overcoming neuroblastomas harboring ALK mutations.76
Disclosure Statement
The author has no conflict interest.
Acknowledgments
I am grateful to Ms. Matsumura, Ms. Hoshino, Ms. Yin, Ms. Saito, Ms. Mori, Ms. Mizota, and Ms. Nakamura for their excellent technical assistance. I also wish to express my appreciation to Dr. Seishi Ogawa (Kyoto University, Kyoto, Japan), Dr. Yasuhide Hayashi (Gunma Children's Medical Center, Gunma, Japan), Dr. Satoru Miyano and Dr. Masahiro Sekiguchi (The University of Tokyo, Tokyo, Japan), and Dr. Yuyan Chen (The University of Sydney, Sydney, Australia) for their continuing support. This work was supported by the Japan Society of Promotion of Science (KAKENHI 17H04224), Research on Measures for Intractable Diseases, Health, and Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare of Japan, Research on Health Sciences focusing on Drug Innovation by the Japan Health Sciences Foundation, Core Research for Evolutional Science and Technology from the Japan Science and Technology Agency, and by P‐CREATE.
Cancer Sci 108 (2017) 1913–1920
Funding information
Japan Society for the Promotion of Science (26293242); Japan Health Sciences Foundation; Japan Science and Technology Agency; P‐CREATE Japan Agency for Medical Research and Development (AMED) (16cm0106509h001) (Japan); KAKENHI (17H04224).
References
- 1. McDermott U, Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol 2009; 27: 5650–9. [DOI] [PubMed] [Google Scholar]
- 2. Gambacorti‐Passerini C, Aroldi A, Cordani N, Piazza R. Chronic myeloid leukemia: Second‐line drugs of choice. Am J Hematol 2016; 91: 67–75. [DOI] [PubMed] [Google Scholar]
- 3. Iwahara T, Fujimoto J, Wen D et al Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997; 14: 439–49. [DOI] [PubMed] [Google Scholar]
- 4. Morris SW, Kirstein MN, Valentine MB et al Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non‐Hodgkin's lymphoma. Science 1994; 263: 1281–4. [DOI] [PubMed] [Google Scholar]
- 5. Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein‐tyrosine kinase similar to Ltk in a human Ki‐1 lymphoma cell line, AMS3. Oncogene 1994; 9: 1567–74. [PubMed] [Google Scholar]
- 6. Bridge JA, Kanamori M, Ma Z et al Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol 2001; 159: 411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soda M, Takada S, Takeuchi K et al A mouse model for EML4‐ALK‐positive lung cancer. Proc Natl Acad Sci U S A 2008; 105: 19893–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y, Takita J, Choi YL et al Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008; 455: 971–4. [DOI] [PubMed] [Google Scholar]
- 9. Mossé YP, Laudenslager M, Longo L et al Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008; 455: 930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janoueix‐Lerosey I, Lequin D, Brugières L et al Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008; 455: 967–70. [DOI] [PubMed] [Google Scholar]
- 11. George RE, Sanda T, Hanna M et al Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008; 455: 975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kwak EL, Bang YJ, Camidge DR et al Anaplastic lymphoma kinase inhibition in non‐small‐cell lung cancer. N Engl J Med 2010; 363: 1693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mossé YP, Lim MS, Voss SD et al Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large‐cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 2013; 14: 472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stoica GE, Kuo A, Aigner A et al Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem 2001; 276: 16772–9. [DOI] [PubMed] [Google Scholar]
- 15. Stoica GE, Kuo A, Powers C et al Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem 2002; 277: 35990–8. [DOI] [PubMed] [Google Scholar]
- 16. Reshetnyak AV, Murray PB, Shi X et al Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: hierarchy and specificity of ligand‐receptor interactions. Proc Natl Acad Sci U S A 2015; 112: 15862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyake I, Hakomori Y, Shinohara A et al Activation of anaplastic lymphoma kinase is responsible for hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene 2002; 21: 5823–34. [DOI] [PubMed] [Google Scholar]
- 18. Reiner DJ, Ailion M, Thomas JH, Meyer BJC. elegans anaplastic lymphoma kinase ortholog SCD‐2 controls dauer formation by modulating TGF‐beta signaling. Curr Biol 2008; 18: 1101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lorén CE, Englund C, Grabbe C, Hallberg B, Hunter T, Palmer RH. A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep 2003; 4: 781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bazigou E, Apitz H, Johansson J et al Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell 2007; 128: 961–75. [DOI] [PubMed] [Google Scholar]
- 21. Englund C, Lorén CE, Grabbe C et al Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 2003; 425: 512–6. [DOI] [PubMed] [Google Scholar]
- 22. Ueda T, Nakata Y, Yamasaki N et al ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene 2016; 35: 4447–58. [DOI] [PubMed] [Google Scholar]
- 23. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008; 8: 11–23. [DOI] [PubMed] [Google Scholar]
- 24. Dupuis‐Coronas S, Lagarrigue F, Ramel D et al The nucleophosmin‐anaplastic lymphoma kinase oncogene interacts, activates, and uses the kinase PIKfyve to increase invasiveness. J Biol Chem 2011; 286: 32105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Piazza R, Magistroni V, Mogavero A et al Epigenetic silencing of the proapoptotic gene BIM in anaplastic large cell lymphoma through an MeCP2/SIN3a deacetylating complex. Neoplasia 2013; 15: 511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kramer M, Ribeiro D, Arsenian‐Henriksson M, Deller T, Rohrer H. Proliferation and Survival of Embryonic Sympathetic Neuroblasts by MYCN and Activated ALK Signaling. J Neurosci 2016; 36: 10425–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hernández L, Pinyol M, Hernández S et al TRK‐fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG‐ALK translocations. Blood 1999; 94: 3265–8. [PubMed] [Google Scholar]
- 28. Feldman AL, Vasmatzis G, Asmann YW et al Novel TRAF1‐ALK fusion identified by deep RNA sequencing of anaplastic large cell lymphoma. Genes Chromosom Cancer 2013; 52: 1097–102. [DOI] [PubMed] [Google Scholar]
- 29. van de Krogt JA, Vanden Bempt M, Finalet Ferreiro J et al ALK‐positive anaplastic large cell lymphoma with the variant RNF213‐, ATIC‐ and TPM3‐ALK fusions is characterized by copy number gain of the rearranged ALK gene. Haematologica 2017; ; pii: haematol.2016.146571. https://doi.org/10.3324/haematol.2016.146571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawrence B, Perez‐Atayde A, Hibbard MK et al TPM3‐ALK and TPM4‐ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000; 157: 377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma Z, Hill DA, Collins MH et al Fusion of ALK to the Ran‐binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosom Cancer 2003; 37: 98–105. [DOI] [PubMed] [Google Scholar]
- 32. Debiec‐Rychter M, Marynen P, Hagemeijer A, Pauwels P. ALK‐ATIC fusion in urinary bladder inflammatory myofibroblastic tumor. Genes Chromosom Cancer 2003; 38: 187–90. [DOI] [PubMed] [Google Scholar]
- 33. Panagopoulos I, Nilsson T, Domanski HA et al Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int J Cancer 2006; 118: 1181–6. [DOI] [PubMed] [Google Scholar]
- 34. Takeuchi K, Soda M, Togashi Y et al Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1‐ALK: reappraisal of anti‐ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res 2011; 17: 3341–8. [DOI] [PubMed] [Google Scholar]
- 35. Chen YB, Xu J, Skanderup AJ et al Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun 2016; 7: 13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cajaiba MM, Jennings LJ, Rohan SM et al ALK‐rearranged renal cell carcinomas in children. Genes Chromosom Cancer 2016; 55: 442–51. [DOI] [PubMed] [Google Scholar]
- 37. Thorner PS, Shago M, Marrano P, Shaikh F, Somers GR. TFE3‐positive renal cell carcinomas are not always Xp11 translocation carcinomas: report of a case with a TPM3‐ALK translocation. Pathol Res Pract 2016; 212: 937–42. [DOI] [PubMed] [Google Scholar]
- 38. Kelly LM, Barila G, Liu P et al Identification of the transforming STRN‐ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A 2014; 111: 4233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vanden Borre P, Schrock AB, Anderson PM et al Pediatric, adolescent, and young adult thyroid carcinoma harbors frequent and diverse targetable genomic alterations, including Kinase fusions. Oncologist 2017; 22: 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Armstrong F, Duplantier MM, Trempat P et al Differential effects of X‐ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene 2004; 23: 6071–82. [DOI] [PubMed] [Google Scholar]
- 41. Chihara D, Fanale MA. Management of anaplastic large cell lymphoma. Hematol Oncol Clin North Am 2017; 31: 209–22. [DOI] [PubMed] [Google Scholar]
- 42. Medeiros LJ, Elenitoba‐Johnson KS. Anaplastic large cell lymphoma. Am J Clin Pathol 2007; 127: 707–22. [DOI] [PubMed] [Google Scholar]
- 43. Cools J, Wlodarska I, Somers R et al Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large‐cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosom Cancer 2002; 34: 354–62. [DOI] [PubMed] [Google Scholar]
- 44. Box JK, Paquet N, Adams MN et al Nucleophosmin: from structure and function to disease development. BMC Mol Biol 2016; 17: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hitchcock‐DeGregori SE, Barua B. Tropomyosin structure, function, and interactions: a dynamic regulator. Subcell Biochem 2017; 82: 253–84. [DOI] [PubMed] [Google Scholar]
- 46. Armstrong F, Lamant L, Hieblot C, Delsol G, Touriol C. TPM3‐ALK expression induces changes in cytoskeleton organisation and confers higher metastatic capacities than other ALK fusion proteins. Eur J Cancer 2007; 43: 640–6. [DOI] [PubMed] [Google Scholar]
- 47. Palaskar S, Koshti S, Maralingannavar M, Bartake A. Inflammatory myofibroblastic tumor. Contemp Clin Dent 2011; 2: 274–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jeanneau M, Gregoire V, Desplechain C et al ALK rearrangements‐associated renal cell carcinoma (RCC) with unique pathological features in an adult. Pathol Res Pract 2016; 212: 1064–6. [DOI] [PubMed] [Google Scholar]
- 49. Debelenko LV, Raimondi SC, Daw N et al Renal cell carcinoma with novel VCL‐ALK fusion: new representative of ALK‐associated tumor spectrum. Mod Pathol 2011; 24: 430–42. [DOI] [PubMed] [Google Scholar]
- 50. Kusano H, Togashi Y, Akiba J et al Two cases of renal cell carcinoma harboring a novel STRN‐ALK fusion gene. Am J Surg Pathol 2016; 40: 761–9. [DOI] [PubMed] [Google Scholar]
- 51. Ji JH, Oh YL, Hong M et al Identification of driving ALK fusion genes and genomic landscape of medullary thyroid cancer. PLoS Genet 2015; 11: e1005467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Amin SM, Haugh AM, Lee CY et al A comparison of morphologic and molecular features of BRAF, ALK, and NTRK1 fusion spitzoid neoplasms. Am J Surg Pathol 2017; 41: 491–8. [DOI] [PubMed] [Google Scholar]
- 53. Guan J, Wolfstetter G, Siaw J et al Anaplastic lymphoma kinase L1198F and G1201E mutations identified in anaplastic thyroid cancer patients are not ligand‐independent. Oncotarget 2017; 8: 11566–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Choi YL, Soda M, Yamashita Y et al EML4‐ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010; 363: 1734–9. [DOI] [PubMed] [Google Scholar]
- 55. Sakamoto H, Tsukaguchi T, Hiroshima S et al CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011; 19: 679–90. [DOI] [PubMed] [Google Scholar]
- 56. Bown N. Neuroblastoma tumour genetics: clinical and biological aspects. J Clin Pathol 2001; 54: 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010; 362: 2202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Attiyeh EF, London WB, Mossé YP et al Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 2005; 353: 2243–53. [DOI] [PubMed] [Google Scholar]
- 59. Carpenter EL, Mossé YP. Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol 2012; 9: 391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee CC, Jia Y, Li N et al Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J 2010; 430: 425–37. [DOI] [PubMed] [Google Scholar]
- 61. De Brouwer S, De Preter K, Kumps C et al Meta‐analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res 2010; 16: 4353–62. [DOI] [PubMed] [Google Scholar]
- 62. Berry T, Luther W, Bhatnagar N et al The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012; 22: 117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. El Demellawy D, McGowan‐Jordan J, de Nanassy J, Chernetsova E, Nasr A. Update on molecular findings in rhabdomyosarcoma. Pathology 2017; 49: 238–46. [DOI] [PubMed] [Google Scholar]
- 64. Nishimura R, Takita J, Sato‐Otsubo A et al Characterization of genetic lesions in rhabdomyosarcoma using a high‐density single nucleotide polymorphism array. Cancer Sci 2013; 104: 856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yoshida A, Shibata T, Wakai S et al Anaplastic lymphoma kinase status in rhabdomyosarcomas. Mod Pathol 2013; 26: 772–81. [DOI] [PubMed] [Google Scholar]
- 66. Okubo J, Takita J, Chen Y et al Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene 2012; 31: 4667–76. [DOI] [PubMed] [Google Scholar]
- 67. Cazes A, Louis‐Brennetot C, Mazot P et al Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res 2013; 73: 195–204. [DOI] [PubMed] [Google Scholar]
- 68. Fransson S, Hansson M, Ruuth K et al Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosom Cancer 2015; 54: 99–109. [DOI] [PubMed] [Google Scholar]
- 69. Gustafson S, Medeiros LJ, Kalhor N, Bueso‐Ramos CE. Anaplastic large cell lymphoma: another entity in the differential diagnosis of small round blue cell tumors. Ann Diagn Pathol 2009; 13: 413–27. [DOI] [PubMed] [Google Scholar]
- 70. Le Rhun E, Chamberlain MC, Zairi F et al Patterns of response to crizotinib in recurrent glioblastoma according to ALK and MET molecular profile in two patients. CNS Oncol 2015; 4: 381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cao L, Yu Y, Bilke S et al Genome‐wide identification of PAX3‐FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 2010; 70: 6497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Gaal JC, Flucke UE, Roeffen MH et al Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J Clin Oncol 2012; 30: 308–15. [DOI] [PubMed] [Google Scholar]
- 73. Ardini E, Magnaghi P, Orsini P, Galvani A, Menichincheri M. Anaplastic Lymphoma Kinase: role in specific tumours, and development of small molecule inhibitors for cancer therapy. Cancer Lett 2010; 299: 81–94. [DOI] [PubMed] [Google Scholar]
- 74. Amin HM, Medeiros LJ, Ma Y et al Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene 2003; 22: 5399–407. [DOI] [PubMed] [Google Scholar]
- 75. Sequist LV, Gettinger S, Senzer NN et al Activity of IPI‐504, a novel heat‐shock protein 90 inhibitor, in patients with molecularly defined non‐small‐cell lung cancer. J Clin Oncol 2010; 28: 4953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Roskoski R. Anaplastic lymphoma kinase (ALK) inhibitors in the treatment of ALK‐driven lung cancers. Pharmacol Res 2017; 117: 343–56. [DOI] [PubMed] [Google Scholar]