

Abstract
Our aim was to evaluate whether repetition of C‐ion (carbon ion beam) irradiation induces radioresistance as well as repeated X‐ray irradiation in cancer cell lines, and to find the key molecular pathway for radioresistance by comparing radioresistant cancer cells with their parental cells. A mouse squamous cell carcinoma cell line, NR‐S1, and radioresistant cancer cells, NR‐S1‐C30 (C30) and NR‐S1‐X60 (X60), established by repetition of C‐ion and X‐ray irradiation, respectively, were used. X‐ray and C‐ion sensitivity, changes in lysosome, mitochondria, intracellular ATP and reactive oxygen species (ROS) level, and mechanistic target of rapamycin (mTOR) signaling were evaluated. Moreover, the effect of rapamycin on radioresistance was also assessed. X‐ray and C‐ion resistance of C30 cells was moderate, and the resistance of X60 cells was the highest in this study. In X60 cells, the amount of lysosome, mitochondria, intracellular ATP and ROS level were significantly increased, and mTOR and p70S6K (ribosomal protein S6 kinase p70) phosphorylation were enhanced compared with C30 and NR‐S1 cells. The inhibition of mTOR signaling was effective for X‐ray and C‐ion radiosensitization in both cell lines, especially in X60 cells in which X‐ray and C‐ion resistance was decreased to the same level as that in NR‐S1 cells. Our results indicated that the contribution to generate X‐ray and C‐ion resistance was less for repeated C‐ion irradiations compared with repeated X‐ray irradiation. Moreover, we found that activated mTOR signaling contributes to X‐ray and C‐ion resistance in the X60 cancer cells.
Keywords: Acquired radioresistance, energy metabolism, mTOR signaling, rapamycin, repeated X‐ray and C‐ion irradiations
Although the development of radiotherapy involving stereotactic X‐ray irradiation1 and carbon ion beam (C‐ion) irradiation2 has improved therapeutic outcomes for early stage cancers,3, 4 poor tumor response has been observed in some cases for both X‐ray and C‐ion therapies,3, 4, 5, 6 possibly due to the existence of radioresistant cancer cells in the tumor. These radioresistant cancer cells possibly lead not only to local relapse but also to distant metastasis after radiotherapy. Moreover, once the tumor relapses after radiotherapy, it cannot be re‐treated by conventional radiotherapy because the surrounding normal tissue could not tolerate the additional irradiations. Therefore, it is a critical problem for patient survival.
Advanced techniques such as active scanning irradiation7 and a rotating gantry with a superconducting magnet8 have recently been developed. These techniques are able to further decrease the radiation exposure to the surrounding normal tissues by optimizing the dose distribution and escalating the radiation dose at the tumor.9 There are advantages in using these advanced techniques for re‐irradiation of local recurrent tumors after radiotherapy,10 because an accurate dose distribution is required for re‐irradiation to avoid additional damage to the surrounding normal tissues, the tolerant radiation dose of which is lower than that before radiotherapy. However, recurrent tumors after radiotherapy may contain radioresistant cancer cells. To obtain favorable outcomes for highly advanced radiotherapy involving C‐ion re‐irradiation, the nature of radioresistant cancer cells must be understood.
The characteristics of radioresistant cancer cells have been actively investigated in vitro. Such studies have indicated enhancement of DNA repair potential11, 12 and activation of survival pathways such as the DNA‐PK/AKT/GSK3β pathway in radioresistant cancer cells13. Previously, we initially demonstrated that repetition of X‐ray irradiation generates not only X‐ray resistance but also significant C‐ion resistance in the cancer cells, and DNA repair potential of the radioresistant cells was significantly enhanced.14 In contrast, the pivotal molecular mechanisms of the radioresistance to both X‐ray and C‐ion remain to be clarified. Moreover, there are no published reports evaluating whether repetition of C‐ion irradiation generates radiation resistance.
In this study, we aimed to evaluate whether repeated C‐ion irradiations generate radioresistant cancer cells as well as repeated X‐ray irradiations, and to find key molecules for radioresistance in the cancer cells through comparison of the characteristics among the repeated X‐ray irradiated cancer cells, the repeated C‐ion irradiated cancer cells and parent cells.
Materials and Methods
Cell lines and culture methods
Mouse squamous cell carcinoma cell line NR‐S1 was kindly provided by Dr Koichi Ando (Gunma University Heavy Ion Medical Center, Gunma, Japan). NR‐S1‐X60 (X60)14 and NR‐S1‐C30 (C30) cells were derived from NR‐S1 cells. These cells were maintained in DMEM (Wako, Osaka, Japan) containing 10% FBS (Sigma‐Aldrich, St. Louis, MO, USA) at 37°C, 5% CO2.
Establishment of C30 cells by repetitive C‐ion irradiations
Two days before C‐ion irradiation, 4.0 × 104 cells/mL of NR‐S1 cells were seeded onto cell culture flasks. NR‐S1 cells were irradiated with 5 Gy of C‐ion and cultured for 2 weeks. This procedure was repeated six times so that the cells were irradiated with a total of 30 Gy of C‐ion. After the final irradiation, the cells were cultured for 4 weeks (Fig. S1).
Assessment of cell survival by colony formation assay
After irradiation, an arbitrary number of harvested cells were seeded on cell culture dishes. Eights days after seeding, the cells were fixed with 10% buffered formalin and stained with 1% crystal violet solution (Wako). Colonies with more than 50 cells were counted. Plating efficiency was calculated by dividing colonies of non‐irradiated cells by seeded cells. Survival fractions were normalized by the plating efficiency.
For measuring radiosensitization, cells were cultured in medium containing 100 nM of rapamycin (Wako) for 24 h and concurrently irradiated with X‐ray or C‐ion. Immediately after irradiation, colony formation assays were performed, as described above.
Fluorescence staining of lysosome and mitochondria
Lysosome, mitochondria and nuclei were stained in 1 nM of Lysotracker‐Red DND‐99 (Life Technologies, Carlsbad, CA, USA), 1 nM of Mitotracker‐Green FM (Life Technologies) and 1 μM of Hoechst 33342 (Sigma‐Aldrich), respectively, by incubating for 20 min at 37°C in a CO2 incubator. The cells were observed by fluorescent microscope (BX‐51, Olympus, Tokyo, Japan) with 60× magnification objective lens.
Flow cytometric assay for lysosome, mitochondria and reactive oxygen species analysis
To stain lysosome, mitochondria, and reactive oxygen species (ROS), cells were cultured for 20 min with culture medium containing 1 nM of Lysotracker‐Red DND‐99, 1 nM of Mitotracker‐Green FM and 1 nM of CellRox‐Green. The fluorescent intensities were analyzed by Gallios (Beckman Coulter, Brea, CA, USA) and Kaluza software (version 1.3, Beckmann Coulter). Details are described in the (Doc. S1).
Measurement of ATP concentration per cell
Intracellular ATP was extracted as described by Yang et al.,15 and the concentration was measured using the ATP Bioluminescence Assay Kit CLS II (Roche Diagnostics, Basel, Switzerland). The luminescence was measured by ARVO, X3 (PerkinElmer, Waltham, MA, USA). Details are described in the (Doc. S2).
Western blotting analysis
A total of 20 μg of proteins was separated by SDS‐PAGE and then transferred to PDVF membrane. The membrane was blocked with 5% BSA or 5% non‐fat dry milk, then incubated with primary antibody and HRP‐conjugated secondary antibody (Table S1). Separated proteins were detected by Clarity Western ECL Substrate (Bio‐Rad laboratories, Hercules, CA, USA) and Las‐4000 (Bio‐Rad Laboratories), and were analyzed using Image‐J software (version 1.50i). Details are described in the (Doc. S3).
Irradiation
X‐ray irradiation was performed using TITAN‐320 (Shimadzu, Kyoto, Japan). The dose rate was approximately 1 Gy/min.
C‐ion irradiation was performed at the Heavy Ion Medical Accelerator in Chiba (HIMAC) at the National Institute of Radiological Sciences, Japan. The energy of the C‐ion beam and the dose rate were 290 MeV/nucleon and approximately 5 Gy/min, respectively. The cells were irradiated at the center of 6‐cm spread out Bragg peak.16 Additional information is provided in the (Doc. S4).
Statistics
The statistical difference in cell survival curve between each cell was assessed by two‐way analysis of variance (anova). The comparison of the results between more than three groups was performed by Dunnett's test. A P‐value <0.05 was regarded as a statistically significant difference.
Results
Repeated X‐ray irradiations, but not C‐ion irradiations generated the resistance to X‐ray and C‐ion
We previously established X60 cells by repetition of X‐ray irradiation, and reported that they were significantly resistant to both X‐ray and C‐ion irradiation.14 To evaluate whether repeated C‐ion irradiations conferred X‐ray and C‐ion resistance on NR‐S1 cells, we compared X‐ray and C‐ion sensitivities of C30 cells with those of NR‐S1 cells (Fig. 1). The D10 values, defined as the radiation dose that decreases survival fractions to 0.1, of X‐ray and C‐ion irradiation for C30 cells were 6.6 and 4.7 Gy, respectively, and for NR‐S1 cells were 5.9 and 4.0 Gy, respectively. Although the X‐ray and C‐ion resistance of C30 cells were modestly increased by the repeated C‐ion irradiations, statistical significance for both X‐ray and C‐ion sensitivity was not detected between C30 cells and the parental NR‐S1 cells (P = 0.26 and P = 0.23 for X‐ray and C‐ion sensitivity, respectively, Dunnett's test). In line with our previous report, the D10 values of X‐ray and C‐ion irradiation for X60 cells were 8.5 and 5.6 Gy, respectively, which were significantly higher than that for NR‐S1 cells and C30 cells (P < 0.01 for both X‐ray and C‐ion sensitivity, using Dunnett's test). These results indicated that X‐ray and C‐ion resistance was conferred by the repeated X‐ray irradiations, whereas the impact of the repeated C‐ion irradiations on the radiosensitivity of NR‐S1 cells was small.
Figure 1.
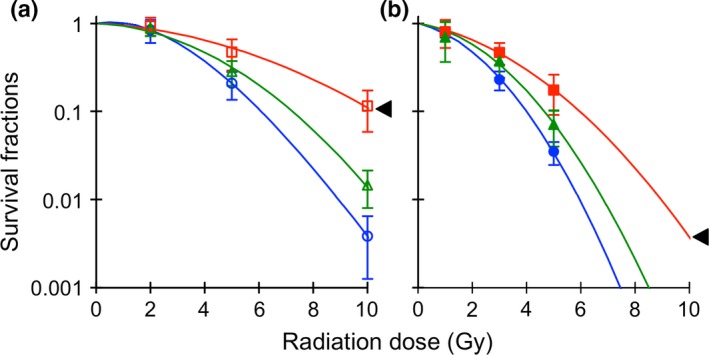
Difference in X‐ray (a) and C‐ion (b) sensitivity between NR‐S1, X60 and C30 cells. The blue, red and green lines show survival curves of NR‐S1, X60 and C30 cells, respectively. The triangles indicate statistical difference with respect to the survival curve of NR‐S1 cells (anova, P < 0.05).
Amount of lysosome, mitochondria, ROS level and ATP production were increased in the X‐ray and C‐ion‐resistant cancer cells
We found that X60 cells have numerous vesicles in the cytoplasm by microscopic observation (Fig. 2a–c and Fig. S2a–c). To evaluate what these vesicles were, we stained the cells with organelle‐specific fluorescent dyes and confirmed that they were lysosomes.17 The lysosome digests intra‐cellular and extra‐cellular components including damaged mitochondria to maintain cellular homeostasis.18 Therefore,we speculated that the contents of cellular organelles including lysosomes and mitochondria should be significantly different between each cell line.
Figure 2.
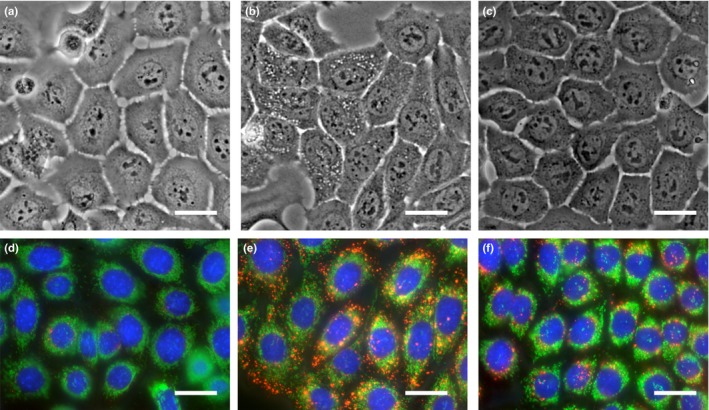
Morphological difference between NR‐S1 (a, d), X60 (b, e) and C30 (c, f). Upper panels (a–c) show images under bright light in normal culture conditions. Lower panels (d–f) show fluorescence image of lysosome, mitochondria and nucleus, which are stained in red, green and blue, respectively. The scale bars indicate 25 μm.
The size and number of lysosome in X60 cells were significantly larger than C30 cells and NR‐S1 cells (Fig. 2d–f, Fig. S2d–f). Flow cytometry was used to quantitatively compare lysosomes and mitochondria in each cell. The fluorescent intensity indicator of lysosomes, Lysotracker‐Red, was 4.8‐fold higher in X60 cells than that in NR‐S1 cells, but there was no significant difference observed between C30 cells and NR‐S1 cells (Fig. 3a).
Figure 3.
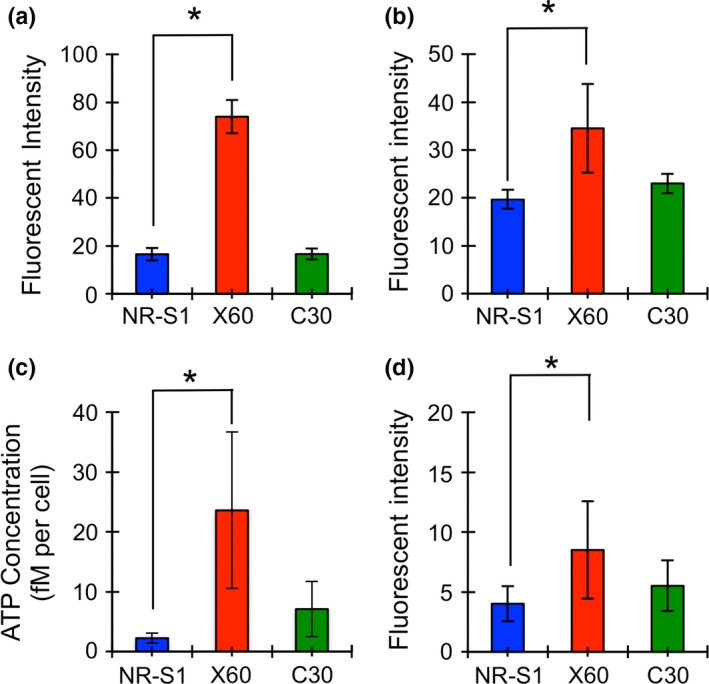
Amount of lysosome (a), mitochondria (b), intracellular ATP concentration (c) and reactive oxygen species (ROS) level (d). Blue, red and green boxes represent the values of NR‐S1, X60 and C30 cells. The boxes and error bars show mean value and standard deviation, respectively, of at least three independent experiments. The asterisk shows statistical difference (P < 0.05, Dunnett's test).
An obvious difference in the number of mitochondria between each cell could not be detected by microscopy (Fig. 2d–f, Fig. S2d–f). However, by flow cytometry, we found that the intensity of mitochondria in X60 cells was 1.8‐fold higher than that in NR‐S1 cells. Although the value in C30 cells was 1.2‐fold higher compared with that in NR‐S1 cells, it was not statistically significant (Fig. 3b).
It is known that lysosomes store large amounts of ATP.19, 20, 21, 22, 23 In addition, mitochondria produce ATP, and simultaneously release ROS into the cytoplasm.24 Therefore, we further assessed the intracellular concentration of ATP (Fig. 3c) and ROS (Fig. 3d) using a luminescence based assay and flow cytometory, respectively. ATP concentration in X60 cells was 10.5‐fold higher than that in NR‐S1 cells. Although there was a 3.2‐fold increase in C30 cells compared to NR‐S1 cells, statistical difference could not be detected. The intracellular ROS level in X60 cells was 2.1‐fold higher than that in NR‐S1 cells. The value in C30 cells was 1.4‐fold higher than that in NR‐S1 cells, but again the difference was not statistically significant. These results indicated that the radioresistant property may be correlated with the promotion of lysosome and mitochondria metabolism, such as through energy production.
Mechanistic target of rapamycin phosphorylation was enhanced in X‐ray and C‐ion‐resistant cancer cells
It is known that the central control of energy production is regulated by the mechanistic target of rapamycin (mTOR) signaling. mTOR is localized on the lysosomal surface in nutrition‐abundant culture condition.25 In addition, mTOR is phosphorylated at serine 2448, which is the most typical phosphorylation site of mTOR, by ribosomal protein S6 kinase B1 (p70S6K) in response to intracellular ATP level and nutrition condition (e.g. amount of amino acid in the cells).25, 26 P70S6K is a downstream protein of the mTOR signaling and its phosphorylation at threonine 386 is generally measured to assess mTOR activity.27, 28
The increase in energy production and in the amount of lysosome in X60 cells may suggest that the mTOR pathway is promoted in X60 cells. Therefore, we evaluated whether mTOR and p70S6K expression and phosphorylation was different in each cell (Fig. 4). Western blotting analysis showed that the mTOR phosphorylation in X60 cells was significantly promoted compared with that in NR‐S1 cells, although mTOR and p70S6K expression levels were similar (Fig. 4a–c, Fig. S3). Interestingly, the p70S6K band for X60 slightly shifted to a larger size (Fig. 4b). Phosphorylated p70S6K on Threonine 386 was detected as double bands in NR‐S1 and C30 cells, but the lower band of phosphorylated p70S6K was reduced in X60 (Fig. 4b,c).
Figure 4.
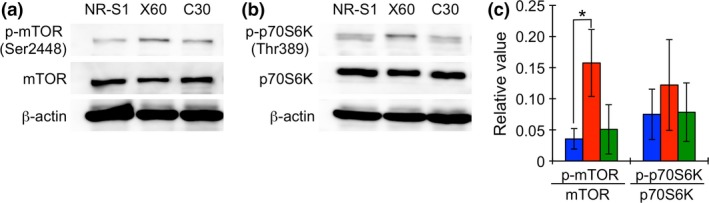
Protein expression and phosphorylation of mTOR (a) and p70S6K (b) in each cell, and the phosphorylation ratio of each protein (c). Blue, red and green boxes in Figure 4(c) represent the values of NR‐S1, X60 and C30 cells. Boxes and error bars show mean value and standard deviation of at least three independent experiments. The asterisk shows statistical difference (P < 0.05, Dunnett's test).
These results indicated that p70S6K was additionally modified posttranslationaly in X60 cells, and suggested that the mTOR signaling closely relates to X‐ray and C‐ion resistance in X60 cells.
Rapamycin treatment decreased the X‐ray and C‐ion resistance
To evaluate the impact of the mTOR signal pathway on X‐ray and C‐ion resistance, radiosensitization in each cell was evaluated by rapamycin, a mTOR pathway inhibitor. We first measured plating efficiency in non‐irradiated conditions to assess the effect of rapamycin treatment for each cell. The plating efficiency of X60 cells cultured under control conditions was significantly higher than that of NR‐S1 cells (Fig. 5a). Adding 100 nM of rapamycin into the culture medium decreased the plating efficiency of X60 cells compared with that of NR‐S1 and C30 cells (Fig. 5a), while the plating efficacy of NR‐S1 and C30 cells was not changed by the rapamycin treatment. These results indicated that the rapamycin treatment might suppress some properties in X60 cells, which were promoted by the repetitive X‐irradiation.
Figure 5.
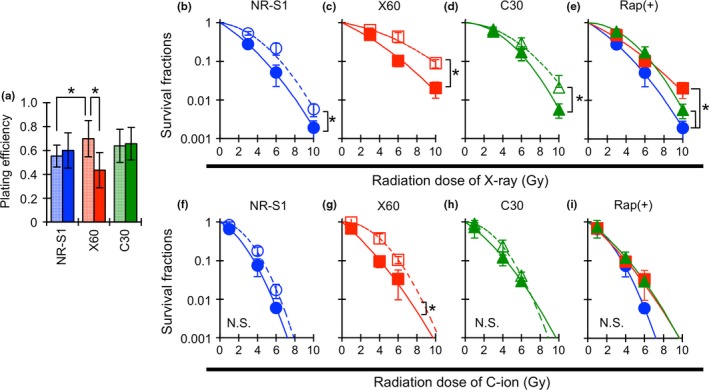
Effect of rapamycin on plating efficiency (a), X‐ray (b–e) and C‐ion (f–i) sensitivity. Cells were treated with 100 nM of rapamycin or 0.1% methanol for 24 h, and then irradiated with the indicated doses of X‐ray or C‐ion. Blue, red and green boxes in (a) represent the plating efficiency of NR‐S1, X60 and C30 cells. The meshed and filled boxes, respectively, show 0.1% methanol and rapamycin treated cells. The values and error bars show mean value and standard deviation of at least three independent experiments. The asterisk shows statistical difference (P < 0.05, Dunnett's test and t‐test). The blue circle, red square and green triangle in (b–i) show survival fractions for NR‐S1, X60 and C30 cells, respectively, and the close symbols with the solid curve and the open symbols with dashed curve show the survival fractions with and without rapamycin, respectively. Asterisks adjacent to the curves indicate that the curves were statistically difference (P < 0.05, anova). N. S. means no significant difference between the curves.
The combination of rapamycin significantly decreased the X‐ray and C‐ion resistance in X60 cells (Fig. 5b−i). The sensitivity enhancement ratio (SER), which was calculated by dividing the survival fractions at D10 value of control medium‐treated cells by that of rapamycin treated cells, was 1.3, 1.5 and 1.2 for both X‐ray and C‐ion irradiation in NR‐S1, X60 and C30 cells, respectively. In particular, rapamycin treatment was significantly effective for both X‐ray and C‐ion sensitization in X60 cells (Fig. 5c,g). The survival fractions at D10 dose of X60 cells after X‐ray and C‐ion irradiation were decreased to the same level as that of C30 cells (Fig. 5e) and that of both NR‐S1 and C30 cells (Fig. 5i), respectively.
These results showed that mTOR signaling commonly contributed to radioresistance in each cell, although the contribution in X60 cells was relatively higher compared with that in NR‐S1 and C30 cells. Moreover, these results indicated that acquired radioresistance might be regulated by several independent pathways, and that mTOR signaling, one of the key pathways, contributes to the acquisition of radioresistance by one or more mechanisms.
Discussion
Difference in radioresistance induction between repeated X‐ray and C‐ion irradiations
Our results clearly demonstrated that only repeated X‐ray irradiations, but not repeated C‐ion irradiation, to the NR‐S1 cells generated X‐ray and C‐ion resistance (Fig. 1). Although further study using other cell lines is necessary to explain the reason for radiation‐type specific radioresistance induction and its molecular mechanisms, it may be possible to interpret the potential underlying mechanisms for the differences in mechanisms between the effect of repeated X‐ray and that of C‐ion irradiations from previous reports. Several reports indicated that the generation of radioresistant cells might be associated with selection of the radioresistant cells.29, 30, 31, 32 Other studies showed that the relatively low dose of X‐ray and g‐ray irradiation, values of which ranged from 2 to 4 Gy, enriched cancer stem cells (CSC) in the human cancer cell lines.29, 30, 31 In contrast, several groups reported that C‐ion irradiation did not enrich the CSC fraction, while the X‐ray irradiation increased CSC fraction under both in vitro and in vivo conditions.33, 34 These reports indicate that C‐ion irradiation is able to effectively kill both CSC and non‐CSC, while X‐ray irradiation has weaker cytotoxic effect on CSC than non‐CSC. Likewise, the enrichment of CSC fractions by repeated X‐ray irradiation, but not C‐ion irradiation, might be a possible mechanism for the difference observed between the X‐ray repetitive and the C‐ion repetitive irradiation in our study. In contrast, other mechanisms are also assumed to be responsible for the radioresistance. If the selection of CSC by repeated X‐ray irradiations is, indeed, the dominant mechanism, the radiosensitivity of X60 cells should gradually diminish over time to levels observed in NR‐S1 cells because CSC is a minority in the cell population and the growth rate of CSC is lower than that of non‐CSC.35 However, X60 cells retained the radioresistant phenotype for more than a month.14 This suggests that enrichment of radioresistant population, such as CSC, is not the only reason for the radioresistance of X60 cells, and means that X60 cells might be composed of a large number of radioresistant non‐CSC. Furthermore, we previously showed that the DNA contents of X60 cells were significantly increased compared with those of NR‐S1 cells,14 which indicated that genomic alterations or rearrangements were induced in X60 cells. Numerous genetic amplifications or mutations in important genes, such as Tp53, may result from the repeated X‐ray irradiation in these radioresistant cells. Although further investigations are essential, the mechanisms of radioresistance induction may be different between repeated X‐ray and C‐ion irradiations.
Promotion of mTOR phosphorylation in X60 cells and its association with radioresistance
Generally, mTOR is known as a central mediator of many signaling pathways, including phosphatidylinositol 3‐kinase (PI3K)/AKT pathway.28 In normal culture condition, the binding of growth factors such as insulin like growth factor (IGF) and WNT ligand activates AKT. This is followed by phosphorylation of tuberous sclerosis complex 2 (TSC2) and Ras homolog enriched in brain (Rheb), and then mTOR is phosphorylated. Downstream targets of mTOR, including p70S6K, are consequently phosphorylated to promote cell proliferation and survival. Activation of the mTOR signal pathway is enhanced in response to intracellular ATP concentration.36
In our study, intracellular ATP concentration (Fig. 3), and phosphorylation of mTOR and p70S6K (Fig. 4) were increased in X60 cells. These results indicated that mTOR signaling might be promoted in X60 cells. Notably, the phosphorylation level of mTOR did not decrease during this study, although there were no additional X‐ray irradiations, meaning that mTOR was constitutively activated over the long period in the X60 cells. It is reported that the mTOR and p70S6K phosphorylation in cancer cells was increased by cytotoxic stimuli, such as X‐ray irradiation.37, 38, 39, 40 However, to our knowledge, its duration has not been elucidated yet. The constitutive enhancement of mTOR signaling in X60 cells was likely to be induced by many other mechanisms, such as the downregulation of TSC2,41 missense mutation of the MTOR gene,42 and the promotion of GSK3β and Akt signaling.28 To clarify the underlying mechanisms of mTOR‐mediated radioresistance, it is important to elucidate the difference in impact of the repeated X‐ray and C‐ion irradiations.
In addition, the concurrent treatment of rapamycin significantly decreased the X‐ray and C‐ion resistance in X60 cells (Fig. 5c,e,g,i). Rapamycin suppresses the mTORC1 function by inhibiting interaction of mTOR with FK506‐binding protein 12kD (FKBP12).43 Therefore, our result indicated that the mTOR signaling, especially mTORC1 functions, largely contributes to the X‐ray and C‐ion resistance in X60 cells. Influence of rapamycin and its derivate Everolimus on radiosensitivity has been evaluated by some groups.37, 38, 39, 40 Their results showed that these drugs induced cell cycle arrest at G1 phase, apoptotic, and antophagic cell death after concurrent treatment with X‐ray irradiation, resulting in a significant enhancement of X‐ray sensitivity. Furthermore, inhibition of mTOR signaling suppressed DNA repair potential.44, 45 Previously, we reported that DNA repair potential, which is represented by the disappearance of phosphorylated H2A histone family member X (γ‐H2AX) foci after X‐ray and C‐ion irradiation, was enhanced in the X60 cells compared with NR‐S1 cells.14 Therefore, suppression of the enhanced DNA repair by rapamycin might also contribute to decrease the radioresistance in X60 cells. The precise mechanisms of radiosensitization by mTOR signaling inhibition must be elucidated in further studies.
With regard to mTOR signaling, serine 2448 and threonine 386 are typical phosphorylation residues of mTOR and p70S6K, respectively. Serine 2448 is phosphorylated with the activation of mTOR signaling. Moreover, Threonine 386 of p70S6K is phosphorylated by mTORC1, and it further phosphorylates the serine 2448 of mTOR.46 In our study, western blot analysis showed that 100 nM of rapamycin did not alter the phosphorylation of p70S6K on threonine 386 and mTOR on serine 2448, although the band position corresponding to the p70S6K of X60 cells was clearly shifted into a lower position (Fig. S4), indicating the pan‐phosphorylation of p70S6K might be decreased by rapamycin treatment. These results mean that mTOR signaling remained even after treatment with 100 nM rapamycin, the concentration of which showed a slight cytotoxic effect on X60 cells. A number of reports have indicated that the suppression of mTOR phosphorylation by rapamycin is incomplete,38, 40, 46, 47 and our result is consistent with these reports. Furthermore, mTOR and p70S6K are phosphorylated by other signaling pathways, including MAPK, Hippo, Notch and WNT pathway.28 In addition, rapamycin treatment itself activates the stress signaling though Akt, and this results in the phosphorylation of mTOR and p70S6K.48, 49, 50 This feedback loop is known to be the cause of the rapamycin resistance in cancer cells. These signaling pathways could underlie the reason why rapamycin treatment did not lead to a significant decrease in the phosphorylation of mTOR on serine 2448 and p70S6K on threonine 386 in our study. Although evaluation of other signaling pathways in X60 cells is necessary to reveal the pivotal mechanisms, our results indicate that mTOR serves a key role in the X‐ray and C‐ion resistance in X60 cells.
In conclusion, we revealed for the first time that repeated C‐ion irradiation to cancer cells did not significantly change X‐ray and C‐ion sensitivity, whereas repeated X‐ray irradiations generate significant X‐ray and C‐ion resistance. This finding supports the theory that C‐ion irradiation is superior to X‐ray irradiation with regard to the acquisition of radioresistance in radiation therapy. Moreover, we found that mTOR signaling contributes to X‐ray and C‐ion resistance in the X60 cells, and rapamycin could be an attractive candidate for an effective radiation sensitizer.
Disclosure Statement
The authors have no conflicts of interest to declare.
Supporting information
Fig. S1. Irradiation protocol for establishment of C30 cells.
Fig. S2. Magnified images of the cells in Figure 2. Individual images (a–f) show the magnified images of Figure 2a–f, respectively. The scale bar represents 10 μm.
Fig. S3. Protein expression and phosphorylation of p70S6K (a) on threonine 421/serine 424 in each cell, and the phosphorylation ratio of each protein (b). Blue, red and green boxes in (b) represent the values of NR‐S1, X60 and C30 cells, respectively. Boxes and error bars show mean value and standard deviation of at least three independent experiments.
Fig. S4. Effect of rapamycin on mechanistic target of rapamycin (mTOR) signaling. (a) and (b) show the results of western blotting and band shift length of p70S6K, respectively. The band shift values were calculated by subtracting the peak position of the rapamycin treated group (+) from that of the 0.1% methanol treated group (−). Vertical profile of p70S6K expression in NR‐S1 (c), X60 (d) and C30 cells (e) are shown. The solid and dashed lines show protein distribution on the western blot of the rapamycin‐treated lane and the 0.1% methanol‐treated lane, respectively. Horizontal solid and dashed lines show the peak position of the band for the rapamycin‐treated group and the 0.1% methanol‐treated group, respectively. These values were obtained from more than three independent experiments. The results showed that there was no significant difference in mTOR phosphorylation between the three cell lines. However, p70S6K phosphorylation was markedly changed by rapamycin treatment (a). Although the band intensity of p70S6K phosphorylation on threonine 389 was approximately the same in each cell, there was a clear shift of the band to a lower molecular size, especially in NR‐S1 and X60 cells after rapamycin treatment (b–d). However, the band shift in the C30 cells was not observed regardless of rapamycin treatment (b,e). These results indicated that the p70S6K phosphorylation in the X60 and the NR‐S1 cells was suppressed by 100 nM of rapamycin although there was no change observed for phosphorylation of mTOR itself as well as for the typical phosphorylation site of p70S6K at threonine 386.
Table S1. Antibodies used in this study.
Doc. S1. Flow cytometric assay for lysosome, mitochondria and reactive oxygen species (ROS) analysis
Doc. S2. Measurement of ATP concentration per cell.
Doc. S3. Western blotting analysis.
Doc. S4. Irradiation.
Acknowledgments
This work was supported by JPSP KAKENHI grant numbers 15K15467, and was performed as a research project with heavy ions at NIRS‐HIMAC (No. 15J183).
Cancer Sci 108 (2017) 2004–2010
Funding Information
Japan Society for the Promotion of Science., (15K15467), NIRS‐HIMAC (No. 15J183).
References
- 1. Nagata Y, Hiraoka M, Shibata T et al Prospective trial of stereotactic body radiation therapy for both operable and inoperable T1N0M0 non‐small cell lung cancer: Japan Clinical Oncology Group Study JCOG0403. Int J Radiat Oncol Biol Phys 2015; 93: 989–96. [DOI] [PubMed] [Google Scholar]
- 2. Miyamoto T, Baba M, Sugane T et al Carbon ion radiotherapy for stage I non‐small cell lung cancer using a regimen of four fractions during 1 week. J Thorac Oncol 2007; 2: 916–26. [DOI] [PubMed] [Google Scholar]
- 3. Onishi H, Shirato H, Nagata Y et al Hypofractionated stereotactic radiotherapy (HypoFXSRT) for stage I non‐ small cell lung cancer: updated results of 257 patients in a Japanese multi‐ institutional study. J Thorac Oncol 2007; 2(Suppl 3): S94–100. [DOI] [PubMed] [Google Scholar]
- 4. Koto M, Miyamoto T, Yamamoto N, Nishimura H, Yamada S, Tsujii H. Local control and recurrence of stage I non‐small cell lung cancer after carbon ion radiotherapy. Radiother Oncol 2004; 71: 147–56. [DOI] [PubMed] [Google Scholar]
- 5. Kamada T, Tsujii H, Tsuji H et al Efficacy and safety of carbon ion radiotherapy in bone and soft tissue sarcomas. J Clin Oncol 2002; 20: 4466–71. [DOI] [PubMed] [Google Scholar]
- 6. Onishi H, Araki T, Shirato H et al Stereotactic hypofractionated high‐dose irradiation for stage I nonsmall cell lung carcinoma: clinical outcomes in 245 subjects in a Japanese multiinstitutional study. Cancer 2004; 101: 1623–31. [DOI] [PubMed] [Google Scholar]
- 7. Mori S, Shibayama K, Tanimoto K et al First clinical experience in carbon ion scanning beam therapy: retrospectiveanalysis of patient positional accuracy. J Radiat Res 2012; 53: 760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iwata Y, Noda K, Murakami T et al Development of a compact superconducting rotating‐gantry for heavy‐ion therapy. J Radiat Res 2014; 55(Suppl 1): i24–5. [Google Scholar]
- 9. Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol 2010; 7: 37–43. [DOI] [PubMed] [Google Scholar]
- 10. Jensen AD, Poulakis M, Nikoghosyan AV et al Re‐irradiation of adenoid cystic carcinoma: analysis and evaluation of outcome in 52 consecutive patients treated with raster‐scanned carbon ion therapy. Radiother Oncol 2015; 114: 182–8. [DOI] [PubMed] [Google Scholar]
- 11. Lynam‐Lennon N, Reynolds JV, Pidgeon GP, Lysaght J, Marignol L, Maher SG. Alterations in DNA repair efficiency are involved in the radioresistance of esophageal adenocarcinoma. Radiat Res 2010; 174: 703–11. [DOI] [PubMed] [Google Scholar]
- 12. Pearce AG, Segura TM, Rintala AC, Rintala‐Maki ND, Lee H. The generation and characterization of a radiation‐resistant model system to study radioresistance in human breast cancer cells. Radiat Res 2001; 156: 739–50. [DOI] [PubMed] [Google Scholar]
- 13. Shimura T, Kakuda S, Ochiai Y et al Acquired radioresistance of human tumor cells by DNA‐ PK/AKT/GSK3β‐mediated cyclin D1 overexpression. Oncogene 2010; 29: 4826–37. [DOI] [PubMed] [Google Scholar]
- 14. Sato K, Imai T, Okayasu R, Shimokawa T. Heterochromatin domain number correlates with X‐ray and carbon‐ion radiation resistance in cancer cells. Radiat Res 2014; 182: 408–19. [DOI] [PubMed] [Google Scholar]
- 15. Yang NC, Ho WM, Chen YH, Hu ML. A convenient one‐step extraction of cellular ATP using boiling water for the luciferin‐luciferase assay of ATP. Anal Biochem 2002; 306: 323–7. [DOI] [PubMed] [Google Scholar]
- 16. Belli M, Bettega D, Calzolari P et al Effectiveness of monoenergetic and spread‐out Bragg peak carbon‐ions for inactivation of various normal and tumour human cell lines. J Radiat Res 2008; 49: 597–607. [DOI] [PubMed] [Google Scholar]
- 17. Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007; 8: 622–32. [DOI] [PubMed] [Google Scholar]
- 18. Kitamura N, Nakamura Y, Miyamoto Y et al Mieap, a p53‐inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS ONE 2011; 6: e16060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Appelqvist H1, Wäster P, Kågedal K, Öllinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013; 5: 214–26. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Z, Chen G, Zhou W et al Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 2007; 9: 945–53. [DOI] [PubMed] [Google Scholar]
- 21. Dou Y, Wu HJ, Li HQ et al Microglial migration mediated by ATP‐induced ATP release from lysosomes. Cell Res 2012; 22: 1022–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J, Liu W, Yang J. ATP‐containing vesicles in stria vascular marginal cell cytoplasms in neonatal rat cochlea are lysosomes. Sci Rep 2016; 6: 20903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao Q, Zhao K, Zhong XZ et al SLC17A9 protein functions as a lysosomal ATP transporter and regulates cell viability. J Biol Chem 2014; 289: 23189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 2014; 14: 709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sancak Y, Bar‐Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator‐Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 2013; 14: 133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bar‐Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol 2014; 24: 400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 2014; 15: 155–62. [DOI] [PubMed] [Google Scholar]
- 29. Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012; 7: e43628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lagadec C, Vlashi E, Della Donna L et al Survival and self‐renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res 2010; 12: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun L, Moritake T, Zheng YW et al In vitro stemness characterization of radio‐resistant clones isolated from a medulloblastoma cell line ONS‐76. J Radiat Res 2013; 54: 61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shimura T, Noma N, Oikawa T et al Activation of the AKT/cyclin D1/Cdk4 survival signaling pathway in radioresistant cancer stem cells. Oncogenesis 2012; 1: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cui X, Oonishi K, Tsujii H et al Effects of carbon ion beam on putative colon cancer stem cells and its comparison with X‐rays. Cancer Res 2011; 71: 3676–87. [DOI] [PubMed] [Google Scholar]
- 34. Takahashi A, Ma H, Nakagawa A et al Carbon‐ion beams efficiently induce cell killing in X‐ray resistant human squamous tongue cancer cells. Int J Med Phys Clin Engn Oncol 2014; 3: 133–42. [Google Scholar]
- 35. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8: 755–68. [DOI] [PubMed] [Google Scholar]
- 36. Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science 2001; 294: 1102–5. [DOI] [PubMed] [Google Scholar]
- 37. Albert JM, Kim KW, Cao C, Lu B. Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol Cancer Ther 2006; 5: 1183–9. [DOI] [PubMed] [Google Scholar]
- 38. Nagata Y, Takahashi A, Ohnishi K et al Effect of rapamycin, an mTOR inhibitor, on radiation sensitivity of lung cancer cells having different p53 gene status. Int J Oncol 2010; 37: 1001–10. [DOI] [PubMed] [Google Scholar]
- 39. Wei F, Liu Y, Guo Y et al miR‐99b‐targeted mTOR induction contributes to irradiation resistance in pancreatic cancer. Mol Cancer 2013; 12: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang D, Xiang J, Gu Y et al Inhibition of mammalian target of rapamycin by rapamycin increases the radiosensitivity of esophageal carcinoma Eca109 cells. Oncol Lett 2014; 8: 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee CH, Inoki K, Karbowniczek M et al Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J 2007; 26: 4812–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino‐acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010; 29: 2746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 2013; 497: 217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen H, Ma Z, Vanderwaal RP et al The mTOR inhibitor rapamycin suppresses DNA double‐strand break repair. Radiat Res 2011; 175: 214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Udayakumar D, Pandita RK, Horikoshi N et al Torin2 suppresses ionizing radiation‐induced DNA damage repair. Radiat Res 2016; 185: 527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser‐2448 is mediated by p70S6 kinase. J Biol Chem 2005; 280: 25485–90. [DOI] [PubMed] [Google Scholar]
- 47. Cao C, Subhawong T, Albert JM et al Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res 2006; 66: 10040–7. [DOI] [PubMed] [Google Scholar]
- 48. Sun SY, Rosenberg LM, Wang X et al Activation of Akt and eIF4E survival pathways by rapamycin‐mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65: 7052–8. [DOI] [PubMed] [Google Scholar]
- 49. O'Reilly KE, Rojo F, She QB et al mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66: 1500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF‐1R‐dependent mechanism. Oncogene 2007; 26: 1932–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Irradiation protocol for establishment of C30 cells.
Fig. S2. Magnified images of the cells in Figure 2. Individual images (a–f) show the magnified images of Figure 2a–f, respectively. The scale bar represents 10 μm.
Fig. S3. Protein expression and phosphorylation of p70S6K (a) on threonine 421/serine 424 in each cell, and the phosphorylation ratio of each protein (b). Blue, red and green boxes in (b) represent the values of NR‐S1, X60 and C30 cells, respectively. Boxes and error bars show mean value and standard deviation of at least three independent experiments.
Fig. S4. Effect of rapamycin on mechanistic target of rapamycin (mTOR) signaling. (a) and (b) show the results of western blotting and band shift length of p70S6K, respectively. The band shift values were calculated by subtracting the peak position of the rapamycin treated group (+) from that of the 0.1% methanol treated group (−). Vertical profile of p70S6K expression in NR‐S1 (c), X60 (d) and C30 cells (e) are shown. The solid and dashed lines show protein distribution on the western blot of the rapamycin‐treated lane and the 0.1% methanol‐treated lane, respectively. Horizontal solid and dashed lines show the peak position of the band for the rapamycin‐treated group and the 0.1% methanol‐treated group, respectively. These values were obtained from more than three independent experiments. The results showed that there was no significant difference in mTOR phosphorylation between the three cell lines. However, p70S6K phosphorylation was markedly changed by rapamycin treatment (a). Although the band intensity of p70S6K phosphorylation on threonine 389 was approximately the same in each cell, there was a clear shift of the band to a lower molecular size, especially in NR‐S1 and X60 cells after rapamycin treatment (b–d). However, the band shift in the C30 cells was not observed regardless of rapamycin treatment (b,e). These results indicated that the p70S6K phosphorylation in the X60 and the NR‐S1 cells was suppressed by 100 nM of rapamycin although there was no change observed for phosphorylation of mTOR itself as well as for the typical phosphorylation site of p70S6K at threonine 386.
Table S1. Antibodies used in this study.
Doc. S1. Flow cytometric assay for lysosome, mitochondria and reactive oxygen species (ROS) analysis
Doc. S2. Measurement of ATP concentration per cell.
Doc. S3. Western blotting analysis.
Doc. S4. Irradiation.