

Abstract
Chromodomain helicase DNA binding proteins (CHDs) are characterized by N‐terminal tandem chromodomains and a central adenosine triphosphate‐dependent helicase domain. CHDs govern the cellular machinery's access to DNA, thereby playing critical roles in various cellular processes including transcription, proliferation, and DNA damage repair. Accumulating evidence demonstrates that mutation and dysregulation of CHDs are implicated in the pathogenesis of developmental disorders and cancer. However, we know little about genomic and transcriptomic alterations and the clinical significance of most CHDs in human cancer. We used TCGA and METABRIC datasets to perform integrated genomic and transcriptomic analyses of nine CHD genes in more than 10 000 primary cancer specimens from 32 tumor types, focusing on breast cancers. We identified associations among recurrent copy number alteration, gene expression, clinicopathological features, and patient survival. We found that CHD7 was the most commonly gained/amplified and mutated, whereas CHD3 was the most deleted across the majority of tumor types, including breast cancer. Overexpression of CHD7 was more prevalent in aggressive subtypes of breast cancer and was significantly correlated with high tumor grade and poor prognosis. CHD7 is required to maintain open, accessible chromatin, thus providing fine‐tuning of transcriptional regulation of certain classes of genes. We found that CHD7 expression was positively correlated with a small subset of classical oncogenes, notably NRAS, in breast cancer. Knockdown of CHD7 inhibits cell proliferation and decreases gene expression of several CHD7 targets, including NRAS, in breast cancer cell lines. Thus, our results demonstrate the oncogenic potential of CHD7 and its association with poor prognostic parameters in human cancer.
Keywords: breast cancer, chromatin remodeler, chromodomain helicase DNA binding protein, copy number alteration
Abbreviations
- AJCC
The American Joint Committee on Cancer
- ATP
adenosine triphosphate
- CAN
copy number alteration
- CDK8
cyclin‐dependent kinase 8
- CHD
chromodomain helicase DNA binding
- ChIP
chromatin immunoprecipitation
- CR
cysteine‐rich
- INO80
inositol requiring 80
- ISWI
imitation SWI
- METABRIC
Molecular Taxonomy of Breast Cancer International Consortium
- MYCN
v‐Myc avian myelocytomatosis viral oncogene neuroblastoma‐derived homolog
- NPI
Nottingham prognostic index
- NRAS
neuroblastoma RAS viral oncogene homolog
- NuRD
nucleosome remodeling histone deacetylase
- SANT
switching‐defective protein 3, adaptor 2, nuclear receptor corepressor, transcription factor IIIB
- SEC63
SEC63 homolog, protein translocation regulator
- SOX2
SRY‐box 2
- SRC
SRC proto‐oncogene, nonreceptor tyrosine
- SWI/SNF
switching‐defective/sucrose non‐fermentable
- TCGA
the cancer genome atlas
1. Introduction
Adenosine triphosphate (ATP)‐dependent chromatin remodelers govern the cellular machinery's access to DNA. Hence, they play critical roles in various cellular processes, including transcription, proliferation, and DNA damage repair (Clapier and Cairns, 2009; Murawska and Brehm, 2011). Eukaryotic chromatin remodelers are divided into the following four families according to their protein similarity and domain structure: switching‐defective/sucrose non‐fermentable (SWI/SNF), imitation SWI (ISWI), inositol requiring 80 (INO80), and chromodomain helicase DNA binding (CHD). The CHD family, which consists of nine members (CHD1–CHD9), is characterized by two consecutive chromodomains in the N‐terminal region and an ATPase domain in the central region (Barrett et al., 2007). Based on other significant structural motifs and functional complexes, CHD proteins are divided into three subfamilies (Alhazzazi et al., 2011). CHD1 and CHD2 are class I CHD proteins, which are distinguished by a C‐terminal DNA‐binding domain with a preference for binding AT‐rich DNA sequences (Delmas et al., 1993; Stokes and Perry, 1995). CHD3, CHD4, and CHD5 are class II CHD proteins, which are distinguished by a pair of N‐terminal plant homeodomain zinc finger domains and a lack of DNA‐binding domain (Schuster and Stoger, 2002). CHD6, CHD7, CHD8, and CHD9 are class III CHD proteins, which are distinguished by a C‐terminal duplicated Brahma and Kismet (BRK) domains, a switching‐defective protein 3, adaptor 2, nuclear receptor corepressor, transcription factor IIIB (SANT) domain, cysteine‐rich (CR) domain, and a DNA‐binding domain (Chiba et al., 1994; Schuster and Stoger, 2002; Shur and Benayahu, 2005). Regardless of overall protein domain structure, the function of CHD superfamily proteins is intimately tied to regulating gene expression by modulating chromatins.
Dysregulation of CHD chromatin remodelers is a pivotal event in various human diseases, notably cancer and developmental disorders (Li and Mills, 2014; Mills, 2017). For example, CHD1 is one of the most frequently deleted genes in prostate cancer. A recent study demonstrated that loss of CHD1 causes DNA repair defects and has the potential to enhance prostate cancer therapeutic responsiveness (Kari et al., 2016). Class II CHD proteins are the catalytic components of the nucleosome remodeling histone deacetylase (NuRD) complex (Stanley et al., 2013). CHD4 deficiency was shown to reduce the recruitment of homologous recombination repair factor BRCA1, and it impaired the efficiency of homologous recombination repair, which could affect the treatment of breast cancer characterized by BRCA1/BRCA2 mutations (Abdelmohsen et al., 2012). Using high‐throughput genomics, Geeleher et al. (2015) revealed that expression of CHD4 predicted the sensitivity of the histone deacetylase inhibitor vorinostat in a large panel of cancer cell lines. We previously reported that CHD3 and CHD4 are commonly mutated in a subset of breast cancers (Yu et al., 2017). Furthermore, a recent study revealed that CHD4 has an oncogenic role in maintaining epigenetic silencing of tumor suppressor genes in colorectal cancer (Xia et al., 2017). CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer (Colbert et al., 2014). Furthermore, loss‐of‐function mutations in several CHD genes are associated with developmental disorders and intellectual disability (Li and Mills, 2014). Notably, de novo mutations in CHD7 cause the CHARGE syndrome (coloboma, heart defects, atresia of the choanae, retarded growth and development, genitourinary hypoplasia, and ear abnormalities, including deafness and vestibular disorders), which is characterized by a unique combination of organ anomalies (Basson and van Ravenswaaij‐Arts, 2015).
Previous studies revealed the importance of several CHDs in cancer pathogenesis and therapeutic responsiveness (Geeleher et al., 2015; Kadoch and Crabtree, 2015; Li and Mills, 2014). However, our knowledge of the genomic and transcriptomic alterations of CHD genes and the clinical significance of those alterations in human cancer remains incomplete. In the present study, we performed a genotranscriptomic meta‐analysis of nine CHDs in more than 10 000 cancer samples across 32 tumor types. We identified the frequency of copy number alteration (CNA), mutations, and aberrant expression for each CHD in a broad spectrum of human cancers. We then focused on human breast cancer, one of the most common cancers, resulting in more than 450 000 deaths each year worldwide. We investigated the associations between recurrent CNA and gene expression level of each CHD, clinicopathological features, overall survival, and disease‐free survival of patients with breast cancer. Our studies demonstrate the oncogenic potential of CHD7 and prioritize a subset of CHDs for future research focused on understanding molecular mechanisms and therapeutic potentials.
2. Materials and methods
2.1. Genomic and clinical data on TCGA and METABRIC cancer samples
Genetic and expression alteration data from 11 313 tumor samples spanning 32 tumor types in provisional the cancer genome atlas (TCGA) studies were obtained from the cBio Cancer Genomics Portal (http://www.cbioportal.org) (Cerami et al., 2012; Gao et al., 2013; Jiang et al., 2016; Liu et al., 2016a). Integrative analysis of cancer genomics and clinical data has been described in detail earlier (Jiang et al., 2016; Liu et al., 2016a). In the cBio portal, the copy number for each CHD gene was generated by the genomic identification of significant targets in cancer (GISTIC) algorithm and categorized as copy number level per gene: ‘−2’ is a deep loss (possibly a homozygous deletion), ‘−1’ is a heterozygous deletion, ‘0’ is diploid, ‘1’ indicates a low‐level gain, and ‘2’ is a high‐level amplification. For mRNA expression data, the relative expression of an individual gene and the gene's expression distribution in a reference population were analyzed. The reference population consisted of tumors that are diploid for the gene in question. The returned value indicates the number of standard deviations from the mean of expression in the reference population (Z‐score). Somatic mutation data were obtained by exome sequencing (Cerami et al., 2012; Gao et al., 2013). Breast cancer subtype and clinicopathologic information were obtained from a previous publication and extracted via the UCSC Cancer Genomics Browser (genome‐cancer.ucsc.edu) and the cBio Cancer Genomics Portal (Cancer Genome Atlas Network, 2012; Cerami et al., 2012). Of the 960 breast cancer samples, 808 had subtype data available, namely 22 normal‐like, 405 luminal A, 185 luminal B, 66 HER2+, and 130 basal‐like breast cancers (Cerami et al., 2012; Liu et al., 2016a). The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) dataset contains ~ 2000 primary breast cancers with long‐term clinical follow‐up data. A detailed description of the dataset is presented in the original publication (Curtis et al., 2012). The CNAs and normalized expression data from the METABRIC database were downloaded with access permissions from the European Genome‐phenome Archive (https://www.ebi.ac.uk/ega) under accession number EGAC00000000005 as well as from the cBio Cancer Genomics Portal (Cerami et al., 2012). In the METABRIC dataset, 1974 samples had subtype data available, namely 199 normal‐like, 718 luminal A, 488 luminal B, 240 HER2+, and 329 basal‐like breast cancers (Curtis et al., 2012).
2.2. Semiquantitative PCRs
To assess gene expression at the mRNA level, RNA was prepared from human breast cancer cell lines and the MCF10A cell line by using an RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA) (Jiang et al., 2016). RNA was mixed with qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD, USA) and then converted to cDNA through a reverse transcription (RT) reaction for real‐time PCRs. Primer sets were obtained from Life Technologies (Carlsbad, CA, USA). A PUM1 primer set was used as a control. Semiquantitative RT‐PCR was performed using the FastStart Universal SYBR Green Master (Roche Diagnostics, Indianapolis, IN, USA) as described earlier (Jiang et al., 2016; Liu et al., 2016a).
2.3. Cell culture and growth assays
The SUM cell lines were obtained from Stephen P. Ethier, and all other cell lines in this study were obtained from American Type Culture Collection (Manassas, VA, USA). Our cultures for the SUM breast cancer cell lines and an immortalized, nontransformed human mammary epithelial MCF10A cell line have been described in detail (Liu et al., 2009; Yang et al., 2006). All cell lines were tested routinely and authenticated using cell morphology, proliferation rate, a panel of genetic markers, and contamination checks. To determine the contribution of endogenous CHD7 overexpression on the growth of human breast cancer in vitro, in the HCC1187 and SUM102 breast cancer cell lines, we knocked down CHD7 with small interfering RNA (siRNA). siRNA were purchased from Sigma Aldrich (St. Louis, MO, USA). As a negative control, we used a MISSION siRNA Universal Negative Control. For transfection, cells were seeded in appropriate cell culture plates and maintained overnight under standard conditions. Plate sizes, cell densities, and siRNA quantities depended on the cell line and the experimental setup; 10–30 nm siRNA was transfected using the MISSION siRNA transfection reagent according to the manufacturer's protocol (Sigma Aldrich). Five days after siRNA transfection, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assays were performed as described earlier (Jiang et al., 2016; Liu et al., 2016a).
2.4. Immunoblotting and antibodies
Immunoblot assays were performed as previously described (Jiang et al., 2016; Liu et al., 2016a). Briefly, whole‐cell lysates were prepared by scraping cells from the dishes into cold RIPA lysis buffer. After centrifugation, protein content was estimated by the Bradford method. A total of 20–50 μg of total cell lysate was resolved by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. Antibodies used in the study included anti‐CHD7 (1 : 1000; Bethyl Laboratories A300‐705A‐T, Montgomery, TX, USA) and anti‐β‐actin (1 : 5000; Sigma Aldrich A5441).
2.5. Statistical analysis
Statistical analyses were performed using r software (http://www.r-project.org) and graphpad Prism (version 6.03; GraphPad Software, Inc., La Jolla, CA, USA) (Jiang et al., 2016; Liu et al., 2016a). The significance of the difference in mRNA expression level for each CHD among different subtypes, stages, and grades of breast cancer samples was calculated using ANOVA and Welch's t‐test as described earlier (Jiang et al., 2016; Liu et al., 2016a). To analyze the relationships between CHD mRNA expression and overall patient survival in breast cancer, samples were divided into high and low expression groups for each CHD, based on mRNA expression Z‐scores in TCGA and METABRIC cancer samples.
3. Results
3.1. Copy number alternations of CHD genes in human cancers
As the first step in our systematic meta‐analysis to determine the spectrum of genetic alterations in CHD genes in human cancers, we queried CNAs of nine CHD genes compiled from 11 313 tumor samples spanning 32 tissue types in TCGA via cBioPortal (Table S1). The copy number for each CHD was generated by the copy number analysis algorithm GISTIC and categorized according to copy number level per gene as high‐level amplification, low‐level gain, diploid, heterozygous deletion, and homozygous deletion. We combined the copy number grouping into amplification/gain (high‐level amplification and low‐level gain), diploid, and deletion (heterozygous or homozygous deletions). Among 10 845 TCGA samples with CNA data available, we found that CHD6 and CHD7 were amplified/gained and CHD3 was deleted in more than 35% of patient samples. Among 32 tumor types, CHD7 was more frequently (> 50%) amplified/gained in 11 tumor types, including breast, lung, colorectal, and ovarian cancers, and CHD6 was more frequently (> 50%) amplified/gained in eight TCGA tumor types, whereas CHD3 was more frequently deleted (> 50%) in nine tumor types (Fig. 1A and Table S2). Furthermore, among 10 845 TCGA tumor samples, CHD7 had high‐level amplification in 362 cases (0.9%), in which five tumor types, namely breast, ovarian, uterine, and liver cancers and melanoma, exhibited CHD7 amplification in more than 5% of cases (Fig. 1A and Table S2). In contrast, CHD6 had high‐level amplification in 179 TCGA samples (0.47%), in which two types, colorectal and uterine cancers, exhibited CHD6 amplification in more than 5% of samples. We also found homozygous deletions of CHD1 and CHD3 in 119 (0.01%) and 103 (0.01%) TCGA cases, respectively. Strikingly, prostate cancer had a dramatically higher frequency of homozygous deletions of CHD1 and CHD3, in 10.2% and 6.7% of cases, respectively. In summary, among nine CHD genes, CHD6 and CHD7 had the highest frequency of genetic gain/amplification, whereas CHD1 and CHD3 were most commonly deleted in a spectrum of human cancers.
Figure 1.
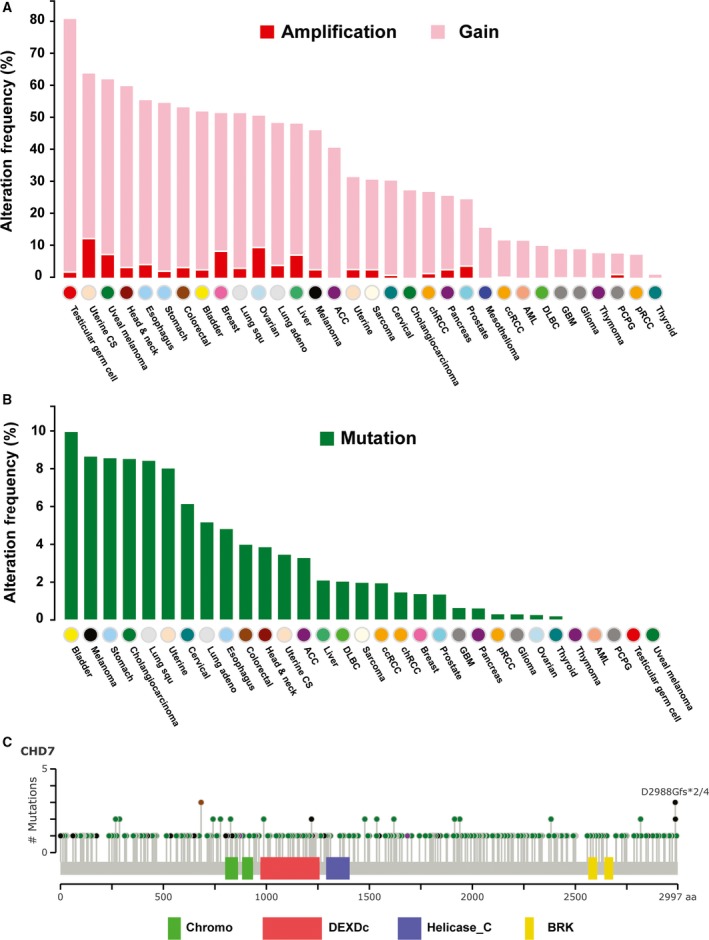
Genetic alterations of CHD7 in a spectrum of human cancers. (A) Frequencies of CHD7 gain and amplification across 32 TCGA tumor types. (B) Frequencies of CHD7 somatic mutation across 31 TCGA tumor types (excluding mesothelioma, as its mutation data were not available). (C) Mutational spectra of CHD7 gene in human tumors. The images show protein domains and the positions of CHD7 somatic mutations in 31 TCGA tumor types. A green dot indicates a missense mutation, a black dot indicates a truncated mutation, a brown dot indicates an in‐frame insertion or deletion, and a pink dot indicates other mutation. The data were obtained from TCGA database via cBioPortal.
3.2. Mutation frequencies and spectra of CHD genes in human cancers
To investigate the mutation frequencies and spectra of CHD genes in human cancers, we analyzed somatic mutation profiles of CHD genes in TCGA dataset. In 7978 sequenced TCGA tumor samples, four CHD genes—CHD4, CHD5, CHD6, and CHD7—exhibited mutations in more than 200 tumor samples. We previously reported the mutation spectra for CHD4 and CHD5 in TCGA breast cancers (Yu et al., 2017). Here, we analyzed the mutation spectra of CHD6 and CHD7 genes in human cancers.
In TCGA samples, 233 of 7978 sequenced tumor samples contained the following 274 CHD6 mutations: 234 missense, one in‐frame insertion and one in‐frame deletion, 35 truncating, and three other mutations (Table S3). In 31 tumor types (excluding mesothelioma, as its mutation data were not available), we found that CHD6 was mutated in more than 5% of five tumor types, namely uterine, stomach, bladder, colorectal cancers, lung adenocarcinoma, and melanoma. Melanoma had the most frequent mutations (41 of 368 melanoma samples, 11.1%; Table S3).
In TCGA samples, 236 sequenced tumor samples contained a total of 291 CHD7 mutations: 250 missense, two in‐frame insertions, 37 truncating, and two other mutations (Table S4). We found that CHD7 was mutated in more than 5% of eight tumor types, namely bladder, stomach, uterine, and cervical cancers, as well as cholangiocarcinoma, lung squamous cell carcinoma, lung adenocarcinoma, and melanoma (Fig. 1B). Among 31 tumor types, bladder cancer exhibited the most frequent CHD7 mutations (13 of 130 sequenced tumor samples, 10%; Fig. 1B). Similar to CHD7 mutations in the CHARGE syndrome, these mutations in human cancers were also distributed throughout the entire coding region of CHD7 genes (Fig. 1C) (Basson and van Ravenswaaij‐Arts, 2015). However, in contrast to the most prevalent loss‐of‐function mutation of CHD7, such as nonsense mutation and frameshift deletion or insertion in CHARGE syndrome, the most frequent (86%) CHD7 mutations in human cancers were missense mutations.
3.3. Molecular profiling of CHD7 genes in different subtypes of breast cancer
Breast cancer is the most common cancer and one of the leading causes of cancer death among women. Using gene expression profiling, breast cancer has been classified into five molecular subtypes: luminal A, luminal B, epidermal growth factor receptor 2‐enriched (HER2+), basal‐like, and normal‐like breast cancers (Cancer Genome Atlas Network, 2012; Perou et al., 2000; Riaz et al., 2013). Basal‐like (closely related to triple‐negative) breast cancer tends to occur in young women and presents with an aggressive course, recurrence, distant metastasis, and shorter survival (Bertucci et al., 2012; Burstein et al., 2015; Carey et al., 2006). In 960 TCGA breast cancers that have mRNA, CNA, and sequencing data, we found that the most commonly amplified/gained (> 50%) CHD gene was CHD7, and the most deleted (> 50%) CHD genes were CHD3 and CHD9. Notably, CHD7 was highly amplified in 8.85% but mutated in 1.25% and homozygously deleted in only 0.1% of TCGA breast cancers. In contrast, CHD6 exhibited high‐level amplification in only 1.98% of TCGA breast cancers. Furthermore, CHD7 was overexpressed (Z‐score ≥ 1) in 33.96% and CHD3 was underexpressed (Z‐score ≤ −1) in 35.1% of TCGA breast cancers (Table 1).
Table 1.
Frequency (%) of CHD genetic alterations and expression levels in 960 TCGA breast cancers
| Gene | Location | DNA alterations | mRNA expression levels | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Amp | Gain | Diploid | Hetloss | Homdel | Mutation | Z score ≥ 1 | 1 > Z score > −1 | Z score ≤ −1 | ||
| CHD1 | 5q15‐q21 | 0.21 | 18.13 | 55.83 | 25.00 | 0.83 | 0.63 | 8.44 | 89.79 | 1.77 |
| CHD2 | 15q26 | 3.54 | 13.75 | 57.60 | 25.00 | 0.10 | 0.94 | 11.35 | 69.58 | 19.06 |
| CHD3 | 17p13.1 | 0.10 | 5.42 | 33.44 | 60.31 | 0.73 | 1.46 | 6.25 | 58.65 | 35.10 |
| CHD4 | 12p13 | 3.33 | 21.88 | 60.42 | 14.17 | 0.21 | 2.08 | 19.27 | 64.58 | 16.15 |
| CHD5 | 1p36.31 | 0.73 | 6.56 | 53.23 | 38.85 | 0.63 | 1.46 | 1.67 | 98.33 | 0.00 |
| CHD6 | 20q12 | 1.98 | 40.10 | 51.35 | 6.46 | 0.10 | 1.98 | 21.04 | 64.79 | 14.17 |
| CHD7 | 8q12.2 | 8.85 | 44.06 | 41.25 | 5.73 | 0.10 | 1.25 | 33.96 | 55.31 | 10.73 |
| CHD8 | 14q11.2 | 0.63 | 17.40 | 59.79 | 22.08 | 0.10 | 1.46 | 17.92 | 60.00 | 22.08 |
| CHD9 | 16q12.2 | 1.88 | 12.60 | 29.38 | 55.10 | 1.04 | 1.46 | 9.58 | 71.88 | 18.54 |
Amp, high‐level amplification; Gain, low‐level gain; Hetloss, heterozygous deletion; Homdel, homozygous deletion. Numbers in bold indicate higher frequencies of DNA or mRNA alternations (Amp > 5%; Gain and Hetloss > 35%, and mRNA (Z score) up‐ or down‐regulation >30%).
To determine whether genetic alteration or mRNA expression of each CHD gene is specific to a breast cancer subtype, we analyzed CNA and mRNA expression across five breast cancer subtypes in TCGA cohort. The frequencies of copy number, somatic mutation, and expression level of CHD genes in five breast cancer subtypes are shown in Table S5. We found that CHD7 was more commonly (> 50%) amplified/gained in aggressive luminal B, HER2+, and basal‐like subtypes (Table S5). We also found that CHD7 was overexpressed (Z‐score ≥ 1) in more than 50% of luminal B and basal‐like breast cancers. Notably, among nine CHDs, CHD7 was overexpressed (Z‐score ≥ 1) in 71.54% of TCGA basal‐like breast cancers, compared to 20.49% of luminal A subtype (P < 0.01; Fig. 2A and Table S5).
Figure 2.
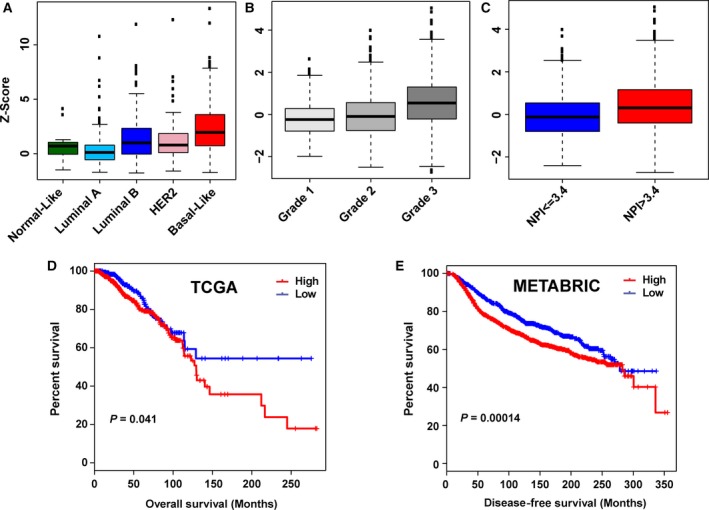
CHD7 expression is significantly associated with aggressiveness and poorer prognosis of breast cancer. (A) Expression levels of CHD7 across five subtypes of TCGA breast cancer samples. (B) CHD7 was significantly more highly expressed in Grade 3 compared with that in Grade 1 and Grade 2 METABRIC breast cancers (P < 0.001). (C) Patients with a poor prognosis (NPI > 3.4) have significantly increased levels of CHD7 expressed in their tumors compared with those with a good prognosis (P < 0.001). (D) Kaplan–Meier plots of overall survival associated with mRNA expression levels of CHD7 in TCGA breast cancers. (E) Kaplan–Meier plots of disease‐free survival associated with mRNA expression levels of CHD7 in METABRIC breast cancers.
To validate our findings from TCGA dataset regarding CHD genetic alterations in breast cancer, we conducted an independent analysis using the METABRIC breast cancer dataset, which contains ~ 2000 primary breast cancers with long‐term clinical follow‐up data. Here, too, CHD7 was the most commonly amplified/gained CHD gene (Table S6), although the frequency of gain/amplification in the METABRIC dataset was lower than that of the TCGA dataset, possibly due to the different CNA analysis platforms. Also, mRNA expression levels of CHD7 were significantly higher in basal‐like breast cancers (P < 0.01; Fig. S1 and Table S7).
3.4. CHD7 expression is significantly associated with poorer prognosis of breast cancer
To investigate the clinicopathological relevance of CHDs in breast cancer, we next examined expression levels of each CHD gene at different stages [The American Joint Committee on Cancer (AJCC)] and histologic grades of breast cancers. CHD7 was dramatically more highly expressed in advanced stages and in higher grades of breast cancer (Fig. 2B). The Nottingham prognostic index (NPI), a clinicopathological classification system based on tumor size, histologic grade, and lymph node status that is widely used in Europe for breast cancer prognostication, was also available in the METABRIC cohort (Galea et al., 1992). Thus, we compared expression levels of nine CHDs between patients with high NPI (> 3.4) versus those with low NPI (≤ 3.4). As shown in Fig. 2C and Table S8, we found that among the nine CHD genes, only CHD7 was significantly more highly expressed, while CHD1, CHD6, and CHD9 were underexpressed in samples with high NPI. Next, we analyzed the relationship between CHD mRNA expression and overall and disease‐free survival of patients with breast cancer. We found that higher mRNA levels of CHD7 were significantly associated with shorter overall survival of TCGA breast cancer patients (P < 0.05; Fig. 2D). We validated that higher mRNA levels of CHD7 were also significantly associated with shorter overall survival of METABRIC breast cancer patients (P < 0.001; Fig. S2). In the METABRIC dataset, disease‐free survival clinical information was available. We found that higher expression of CHD7 was significantly associated with shorter disease‐free survival (P < 0.001) in METABRIC breast cancer patients (Fig. 2E).
3.5. CHD7 regulates a subset of cancer‐associated genes
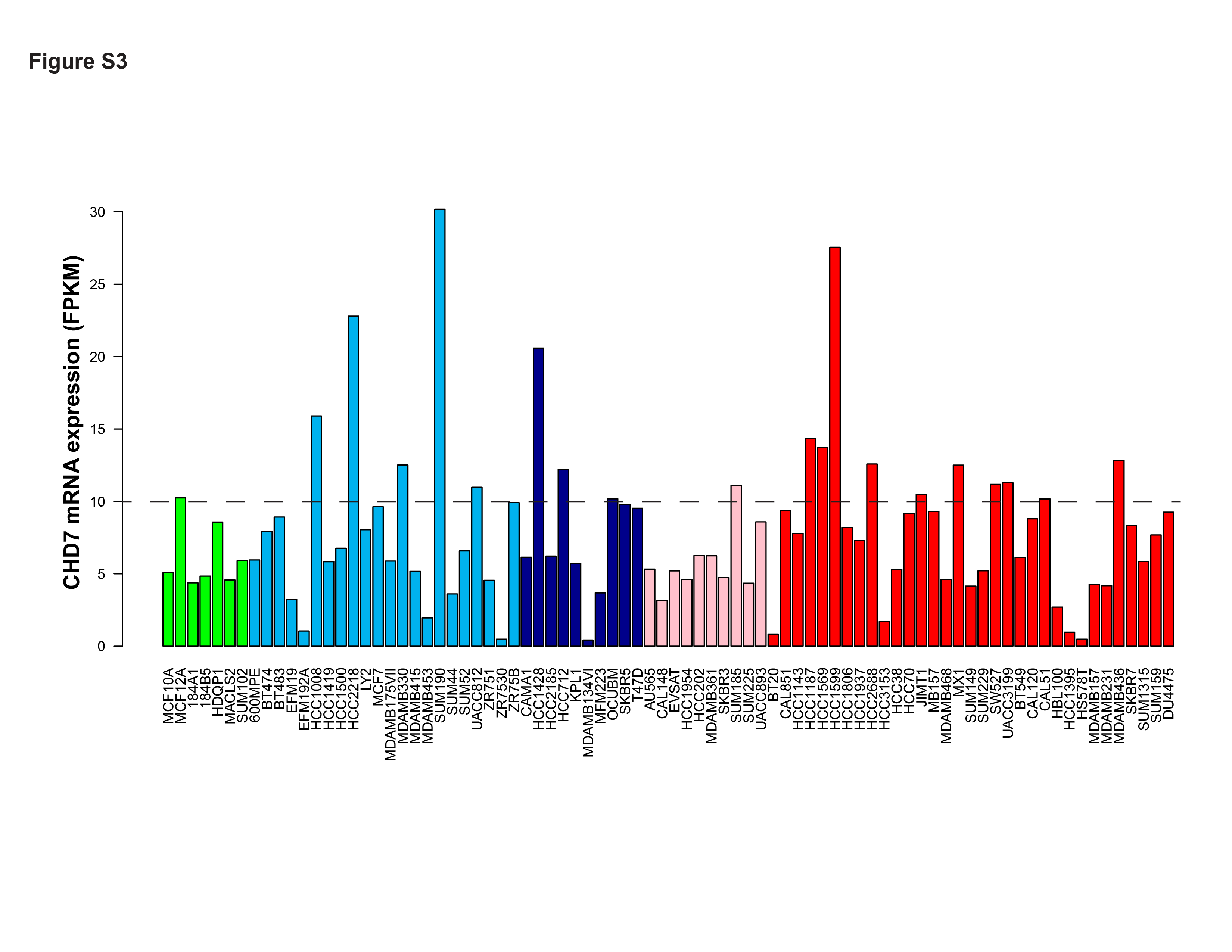
Breast cancer cell lines and related animal models are essential tools with which to study cancer biology and to test novel therapeutic strategies. Thus, we next examined CHD7 expression in a panel of breast cancer cells. Figure S3 shows the expression level of CHD7 based on RNA sequencing data from 78 breast cancer cell lines compared with four normal mammary epithelial cell lines (Marcotte et al., 2016). Compared with MCF10A, an immortalized but nontumorigenic breast epithelial cell line, mRNA levels of CHD7 were more than twofold higher in 19 breast cancer cell lines, nine of them belonging to the basal subtype. We next performed qRT‐PCR assays and demonstrated that mRNA expression levels of CHD7 in HCC1187 and SUM102 breast cancer cell lines were more than twofold higher than that in MCF10A cells (data not shown). To assess the contribution of endogenous CHD7 overexpression on the transformation of human breast cancer, we examined the effects of knocking down CHD7 in HCC1187 and SUM102 cells. We obtained three siRNA targeting different regions of CHD7 genes. qRT‐PCR and western blot assays revealed that two siRNA decreased the expression of CHD7 at mRNA and protein levels (Fig. 3A). As shown in Fig. 3B, CHD7 knockdown slowed HCC1187 and SUM102 cell growth to ~ 70% of the growth of the nonsilenced control.
Figure 3.
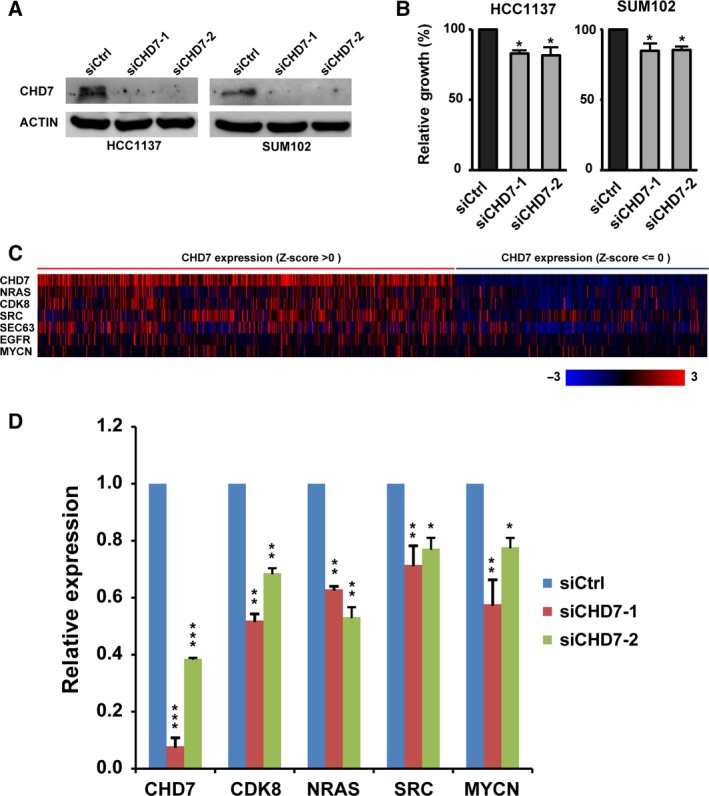
Knockdown of CHD7 inhibits cell proliferation and decreases gene expression of several CHD7 targets in breast cancer cell lines. (A) Knockdowns of CHD7 in HCC1187 and SUM102 cells with two different shRNA were confirmed by western blot assays. (B) Bar graph shows relative cell growth after knocking down CHD7 in HCC1187 and SUM102 breast cancer cells (*P < 0.05). Data are expressed as mean ± SD. (C) mRNA expression heatmap of CHD7 and six CHD7 candidate target genes in 960 TCGA breast cancers. (D) qRT‐PCR assays show that CHD7 knockdown inhibited expression of four genes (NRAS,CDK8,SRC, and MYCN) in HCC1187 cells (*P < 0.05, **P < 0.01, and ***P < 0.001, Student's t‐test).
A previous study that used the approach of chromatin immunoprecipitation (ChIP) on tiled microarrays revealed that CHD7 targets active gene enhancer elements to modulate expression of specific sets of genes (Schnetz et al., 2009). Notably, CHD7 physically interacted with SRY‐box 2 (SOX2) transcriptional factor and regulated a set of common target genes associated with cancer and developmental disorders (Engelen et al., 2011). Using approaches of ChIP followed by massively parallel DNA sequencing and mRNA microarray, Engelen et al. identified 46 genes that are bound and activated by CHD7 and SOX2 in a neural stem cell model. To determine which of the 46 genes had higher positive or negative correlation with CHD7 expression in breast cancer, we analyzed the Spearman and Pearson correlations between expression levels of CHD7 and each CHD7‐SOX2‐targeted gene in TCGA breast cancer specimens from the cBioPortal database (Table S9). Higher weight was assigned to the Spearman correlation coefficient. We found that expression levels of four genes—cyclin‐dependent kinase 8 (CDK8), neuroblastoma RAS viral oncogene homolog (NRAS), SRC proto‐oncogene, nonreceptor tyrosine (SRC), and SEC63 (SEC63 homolog, protein translocation regulator)—were positively correlated (Spearman's r > 0.3) with CHD7 expression (Fig. 3C). Furthermore, expression of the classical breast cancer oncogenes EGFR (epidermal growth factor receptor) and v‐Myc avian myelocytomatosis viral oncogene neuroblastoma‐derived homolog (MYCN) was also positively correlated with CHD7 expression, with Spearman's r = 0.24 and 0.12, respectively (Fig. 3C and Table S9). Next, we measured mRNA expression levels of six CHD7 candidate targets in HCC1187 cells after knocking down CHD7. We found that expression levels of four genes (NRAS, CDK8, SRC, and MYCN) decreased in CHD7‐knockdown HCC1187 cells (Fig. 3D). We also found that expression levels of NRAS and MYCN, but not others, decreased in CHD7‐knockdown SUM102 cells (Fig. S4). Thus, CHD7 likely modulates expression of a set of genes that are critical for cancer pathogenesis.
4. Discussion
In this study, we used a systematic genomics approach to assess the oncogenic properties of nine CHD genes across human tumor types. We found that CHD6 and CHD7 were most commonly gained/amplified or mutated, whereas CHD1 and CHD3 were most deleted in a spectrum of human cancers. Integrated genomic, transcriptomic, clinicopathological data, and in vitro siRNA‐mediated knockdown assays revealed the oncogenic potential of CHD7 in multiple cancer types, notably those arising from breast.
All CHD proteins are characterized by two consecutive chromodomains in the N‐terminal region. The chromodomain belongs to a larger, structurally related family of protein domains called Royal family domains, which include Tudor, malignant brain tumor (MBT), pro‐trp‐trp‐pro (PWWP), and Agenet domains. Based on the ChromoHub database, the human genome encodes 29 proteins that contain a chromodomain (Fig. S5), including nine CHDs and eight chromobox proteins (CBX 1–8). Notably, all nine CHD proteins have double chromodomains, but all other chromodomain‐containing proteins have only a single chromodomain. In general, the chromodomain binds methyl marks on histones with high specificity; such binding is coordinated by a hydrophobic cage formed by two, three, or four well‐conserved aromatic residues (Yap and Zhou, 2011). For example, the chromodomain is responsible for the direct interaction between heterochromatin protein 1 and trimethylated H3K9. Human CHD1 recognizes the di‐ and trimethylation of H3K4 through its two chromodomains and induces gene transcription. The chromodomains of CHD7 have a unique specificity for the monomethylated H3K4 mark, and recent studies have shown that CHD7 tracks H3K4 monomethylation patterns at enhancer motifs (Bajpai et al., 2010; Schnetz et al., 2009). Furthermore, CHD proteins contain additional epigenetic effector domains, including the plant homeodomain in class II family and SANT domain in class III family domains. Thus, chromodomains, together with other epigenetic recognition domains, likely play regulatory roles in recruiting CHD proteins to specific chromatin regions.
Chromodomain helicase DNA binding proteins are highly conserved from yeast to humans and have essential roles in controlling fundamental cellular processes of development (Hota and Bruneau, 2016). For example, CHD1 is essential during preimplantation embryonic development. The CHD3/4‐containing NuRD complex is involved in synapse formation and heart development (Garnatz et al., 2014; Yamada et al., 2014). CHD5 has critical roles during spermatogenesis (Govin et al., 2004). CHD7 is involved in a variety of stem cell differentiation and cell‐fate decisions. De novo mutations of CHD genes can lead to severe developmental disorders, including CHARGE syndrome, autism, and intellectual disability. Notably, de novo heterozygous mutations of the CHD7 gene are the primary cause of the CHARGE syndrome (Schnetz et al., 2009). Most CHD7 mutations in patients with CHARGE syndrome are nonsense mutations or frameshift deletions; missense mutations of CHD7 are associated with milder CHARGE symptoms. The conditional deletion of chd7 in mice recapitulates most symptoms of CHARGE syndrome (Sperry et al., 2014). Most CHD7 mutations found in CHARGE syndrome affect ATP‐dependent chromatin remodeling (Basson and van Ravenswaaij‐Arts, 2015). A mutation in the first chromodomain (S834F) associated with CHARGE syndrome completely suppressed CHD7's remodeling activity (Bouazoune and Kingston, 2012). In this study, we revealed that in human cancer most CHD7 mutations are missense, and those mutations were distributed throughout the entire coding region of the CHD7 gene. We found that, of 291 CHD7 mutations in TCGA tumors, 11 mutations were located at chromodomains and 25 mutations were at the ATP‐dependent helicase domain. Much work is needed to decipher the biological impacts and underlying mechanisms of these CHD7 mutations on cancer pathogenesis.
Among nine CHDs, CHD1 and CHD3 were most commonly deleted in a spectrum of human cancers. Strikingly, homozygous deletions of CHD1 and CHD3 were found in 10.2% and 6.7% of prostate cancers, respectively. These findings agree with and consolidate prior reports on the genetic alterations and tumor‐suppressive functions of CHD1 and CHD3 in human cancer. Previous studies revealed that homozygous deletion of CHD1 is the second most common genetic event in prostate cancer after PTEN deletion (Liu et al., 2012). Inactivation of CHD1 is correlated with anchorage‐independent growth (Yu et al., 2015) and enhances the invasiveness of prostate cancer (Liu et al., 2012; Rodrigues et al., 2015). In breast cancer, the most commonly deleted/underexpressed CHD gene was CHD3. CHD3 is localized to 17p13.1, the TP53 region. A recent study demonstrated that deletions linked to TP53 loss drive cancer through p53‐independent mechanisms (Liu et al., 2016b). In that study, a shRNA library targeting the ~ 100 protein‐coding genes (excluding TP53) in mouse chromosome 11B3 (syntenic to human 17p13.1) was screened for tumor‐suppressive activity in mouse models. Among 17 identified genes, CHD3 was considered a potential tumor suppressor (Liu et al., 2016b).
A notable finding from our study is the dysregulation of CHD7 in a subset of human cancers. CHD7 was highly amplified in more than 5% of samples among 11 tumor types, including breast, lung, colorectal, and ovarian cancers. A previous study revealed that CHD7 is genetically altered in response to tobacco smoke in small‐cell lung cancer; either it has an in‐frame duplication of exons 3–7, or it is expressed as a fusion with Pvt1 oncogene (PVT1) (Pleasance et al., 2010). Another study found that CHD7 is highly expressed in human gliomas (Ohta et al., 2016). Recently, Colbert et al. reported that CHD7 was dysregulated in over 90% of their pancreatic ductal adenocarcinoma samples. Low CHD7 expression was associated with higher recurrence‐free survival and overall survival in patients receiving adjuvant gemcitabine (Colbert et al., 2014).
Studies of CHD7 in the CHARGE syndrome not only highlight the critical role of CHD7 in development but also indicate that CHD7 modulates central pathways in tumorigenesis. For example, CHD7 could be recruited to the p53 promoter and repress the expression of p53. Thus, loss of CHD7 contributed to the inappropriate activation of p53 and promoted the CHARGE phenotypes (Van Nostrand and Attardi, 2014). Another interesting study showed that CHD7 cooperates with SOX2 to regulate a small set of genes, such as NOTCH and Sonic Hedgehog pathway genes and classical oncogenes NRAS and SRC; these genes play critical roles in stem cell development and tumorigenesis (Engelen et al., 2011; Puc and Rosenfeld, 2011). SOX2 also plays a key role in the stem‐like cancer phenotype, particularly in squamous cell carcinoma (Ferone et al., 2016). Notably, a multiplatform analysis of 12 cancer types revealed that basal‐like breast cancer shares similar molecular features with squamous cell carcinoma. We also found that CHD7 and SOX2 (3q26) were likely cogained/amplified (P < 0.0001) in breast cancer. In this study, we revealed that CHD7 likely regulates a small set of oncogenes, such as NRAS and MYCN, in breast cancer. We speculate that CHD7 allows transcription factors and the general transcription machinery access to DNA, thus promoting activation of certain classes of oncogenes in a cell type‐specific manner, which subsequently contributes to tumorigenesis.
5. Conclusion
We conducted a large‐scale genomic analysis of nine CHDs in human cancer, focusing on breast cancer. We found that CHD6 and CHD7 were the most commonly gained/amplified or mutated, whereas CHD1 and CHD3 were the most deleted CHDs in a spectrum of human cancers. Integrated genomic, transcriptomic, and clinicopathological data in ~ 3000 primary breast cancers revealed that different subtypes of breast cancer had distinctive copy number and expression patterns for each CHD. CHD7 was the most upregulated CHD gene in breast cancer and was significantly associated with aggressiveness and poor prognosis of patients. Knockdown of CHD7 inhibited cell proliferation in breast cancer cell lines. We found that CHD7 expression was positively correlated with a small subset of classical oncogenes, notably NRAS and MYCN, and validated that CHD7 knockdown downregulated expression of them. Our findings provide a strong foundation for further mechanistic research and for developing therapies that target CHD7 or other CHDs in human cancer.
Author contributions
XC, XG, YJ, and ZY designed research studies, analyzed data, and wrote manuscript. XC, XG, YJ, HY, LL, and WS conducted experiments and analyzed data. LL and ZY edited the manuscript.
Supporting information
Fig. S1. Expression levels of CHD7 across five subtypes of METABRIC breast cancer samples.
{kind=link}
Fig. S2. Kaplan‐Meier plots of overall survival associated with mRNA expression levels of CHD7 in METABRIC breast cancers.
{kind=link}
Fig. S3. Expression levels of CHD7 based on RNA sequencing data from 78 breast cancer cell lines compared with four normal mammary epithelial cell lines.
{kind=link}
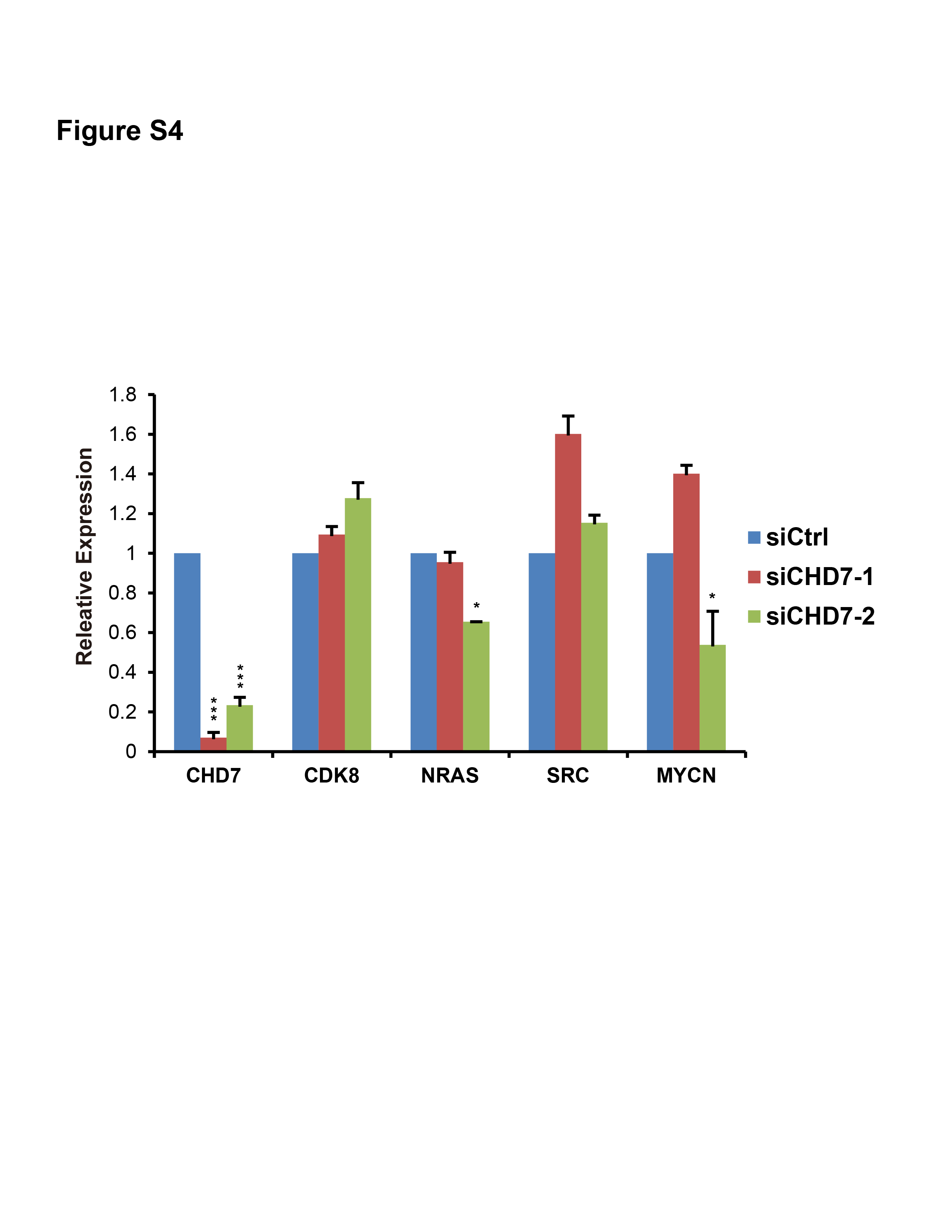
Fig. S4. Expression levels of NRAS and MYCN, but not others, decreased in CHD7‐knockdown SUM102 cells (*P < 0.05 and ***P < 0.001, Student's t‐test).
{kind=link}
Fig. S5. Phylogenetic analysis of chromodomain‐containing proteins. The image was obtained from the ChromoHub database (http://www.thesgc.org).
{kind=link}
Table S1. The number of samples and data type in 32 TCGA databases.
Table S2. Frequency (%) of CHD genetic alterations in 32 tumor types from TCGA database.
Table S3. Mutations of CHD6 in TCGA tumors.
Table S4. Mutations of CHD7 in TCGATumors.
Table S5. Frequency (%) of CHD genetic alterations and expression levels in five subtypes of TCGA breast cancers.
Table S6. Frequency (%) of genetic and transcriptional alterations of CHDs in 1980 METABRIC breast cancers.
Table S7. Frequency (%) of CHD genetic alterations and expression levels in five subtypes of Metabric breast cancers.
Table S8. Expression levels of CHD genes associated with NPI score in METABRIC breast cancer.
Table S9. Correlation between mRNA expression of CHD7 and 46 candidate target genes in TCGA breast cancers.
Acknowledgements
This work was partially supported by grants from the NIH/NCI R21CA175244‐01A1 and the Office of Research on Women's Health (OWRH) of NIH, the Department of Defense (DoD) Prostate Cancer Program PC130259, to Dr. Z‐QY, by funding from the China International Postdoctoral Exchange Fellowship Program to HY. We thank Hui Liu and Qianhui Huang for technical contributions. We thank Dr. Stephen P. Ethier for providing the SUM breast cancer cell lines.
References
- Abdelmohsen K, Srikantan S, Kang MJ and Gorospe M (2012) Regulation of senescence by microRNA biogenesis factors. Ageing Res Rev 11, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhazzazi TY, Kamarajan P, Joo N, Huang JY, Verdin E, D'Silva NJ and Kapila YL (2011) Sirtuin‐3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 117, 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada‐Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T and Wysocka J (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett A, Santangelo S, Tan K, Catchpole S, Roberts K, Spencer‐Dene B, Hall D, Scibetta A, Burchell J, Verdin E et al (2007) Breast cancer associated transcriptional repressor PLU‐1/JARID1B interacts directly with histone deacetylases. Int J Cancer 121, 265–275. [DOI] [PubMed] [Google Scholar]
- Basson MA and van Ravenswaaij‐Arts C (2015) Functional insights into chromatin remodelling from studies on CHARGE Syndrome. Trends Genet 31, 600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Finetti P and Birnbaum D (2012) Basal breast cancer: a complex and deadly molecular subtype. Curr Mol Med 12, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouazoune K and Kingston RE (2012) Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc Natl Acad Sci USA 109, 19238–19243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC et al (2015) Comprehensive genomic analysis identifies novel subtypes and targets of triple‐negative breast cancer. Clin Cancer Res 21, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S et al (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295, 2492–2502. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba H, Muramatsu M, Nomoto A and Kato H (1994) Two human homologues of Saccharomyces cerevisiae SWI2/SNF2 and Drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res 22, 1815–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR and Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78, 273–304. [DOI] [PubMed] [Google Scholar]
- Colbert LE, Petrova AV, Fisher SB, Pantazides BG, Madden MZ, Hardy CW, Warren MD, Pan Y, Nagaraju GP, Liu EA et al (2014) CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res 74, 2677–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas V, Stokes DG and Perry RP (1993) A mammalian DNA‐binding protein that contains a chromodomain and an SNF2/SWI2‐like helicase domain. Proc Natl Acad Sci USA 90, 2414–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, Szumska D, Kockx C, van Ijcken W, Dekkers DH et al (2011) Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43, 607–611. [DOI] [PubMed] [Google Scholar]
- Ferone G, Song JY, Sutherland KD, Bhaskaran R, Monkhorst K, Lambooij JP, Proost N, Gargiulo G and Berns A (2016) SOX2 is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell 30, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea MH, Blamey RW, Elston CE and Ellis IO (1992) The Nottingham Prognostic Index in primary breast cancer. Breast Cancer Res Treat 22, 207–219. [DOI] [PubMed] [Google Scholar]
- Gao JJ, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun YC, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnatz AS, Gao Z, Broman M, Martens S, Earley JU and Svensson EC (2014) FOG‐2 mediated recruitment of the NuRD complex regulates cardiomyocyte proliferation during heart development. Dev Biol 395, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Lenkala D, Wang F, LaCroix B, Wang J, Karovic S, Maitland ML, Huang R, Loboda A, Nebozhyn M et al (2015) Predicting response to histone deacetylase inhibitors using high‐throughput genomics. J Invest Med 63, 683–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govin J, Caron C, Lestrat C, Rousseaux S and Khochbin S (2004) The role of histones in chromatin remodelling during mammalian spermiogenesis. Eur J Biochem 271, 3459–3469. [DOI] [PubMed] [Google Scholar]
- Hota SK and Bruneau BG (2016) ATP‐dependent chromatin remodeling during mammalian development. Development 143, 2882–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liu L, Shan W and Yang ZQ (2016) An integrated genomic analysis of Tudor domain‐containing proteins identifies PHD finger protein 20‐like 1 (PHF20L1) as a candidate oncogene in breast cancer. Mol Oncol 10, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C and Crabtree GR (2015) Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, Sirma H, Simon R, Will H, Dobbelstein M et al (2016) Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep 17, 1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W and Mills AA (2014) Architects of the genome: CHD dysfunction in cancer, developmental disorders and neurological syndromes. Epigenomics 6, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Bollig‐Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP and Yang ZQ (2009) Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28, 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, Spitzer B, Bosbach B, Kastenhuber ER, Baslan T et al (2016b) Deletions linked to TP53 loss drive cancer through p53‐independent mechanisms. Nature 531, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Lindberg J, Sui G, Luo J, Egevad L, Li T, Xie C, Wan M, Kim ST, Wang Z et al (2012) Identification of novel CHD1‐associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene 31, 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu L, Holowatyj A, Jiang Y and Yang ZQ (2016a) Integrated genomic and functional analyses of histone demethylases identify oncogenic KDM2A isoform in breast cancer. Mol Carcinog 55, 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Sayad A, Brown KR, Sanchez‐Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB et al (2016) Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 164, 293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills AA (2017) The chromodomain helicase DNA‐binding chromatin remodelers: family traits that protect from and promote cancer. Cold Spring Harb Perspect Med 7, pii: a026450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murawska M and Brehm A (2011) CHD chromatin remodelers and the transcription cycle. Transcription 2, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S, Yaguchi T, Okuno H, Chneiweiss H, Kawakami Y and Okano H (2016) CHD7 promotes proliferation of neural stem cells mediated by MIF. Mol Brain 9, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA et al (2000) Molecular portraits of human breast tumours. Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C et al (2010) A small‐cell lung cancer genome with complex signatures of tobacco exposure. Nature 463, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puc J and Rosenfeld MG (2011) SOX2 and CHD7 cooperatively regulate human disease genes. Nat Genet 43, 505–506. [DOI] [PubMed] [Google Scholar]
- Riaz M, van Jaarsveld MT, Hollestelle A, Prager‐van der Smissen WJ, Heine AA, Boersma AW, Liu J, Helmijr J, Ozturk B, Smid M et al (2013) miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation‐specific miRNAs. Breast Cancer Res 15, R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour‐Fard A, Loda M, Lucia MS, Wu M, Shi L, Cimic A et al (2015) Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res 75, 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Zhang X, Song L, Wang Z, Laframboise T et al (2009) Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res 19, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster EF and Stoger R (2002) CHD5 defines a new subfamily of chromodomain‐SWI2/SNF2‐like helicases. Mamm Genome 13, 117–119. [DOI] [PubMed] [Google Scholar]
- Shur I and Benayahu D (2005) Characterization and functional analysis of CReMM, a novel chromodomain helicase DNA‐binding protein. J Mol Biol 352, 646–655. [DOI] [PubMed] [Google Scholar]
- Sperry ED, Hurd EA, Durham MA, Reamer EN, Stein AB and Martin DM (2014) The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Dev Dyn 243, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley FK, Moore S and Goodarzi AA (2013) CHD chromatin remodelling enzymes and the DNA damage response. Mutat Res 750, 31–44. [DOI] [PubMed] [Google Scholar]
- Stokes DG and Perry RP (1995) DNA‐binding and chromatin localization properties of CHD1. Mol Cell Biol 15, 2745–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL and Attardi LD (2014) Guilty as CHARGED: p53's expanding role in disease. Cell Cycle 13, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, Nie Y, Cai Y, Zhang YW, Yu LR et al (2017) CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 31, 653–668.e657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, Fioravante D, Regehr WG, Gygi SP, Georgopoulos K et al (2014) Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron 83, 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZQ, Streicher KL, Ray ME, Abrams J and Ethier SP (2006) Multiple interacting oncogenes on the 8p11‐p12 amplicon in human breast cancer. Cancer Res 66, 11632–11643. [DOI] [PubMed] [Google Scholar]
- Yap KL and Zhou MM (2011) Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry 50, 1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Jiang Y, Liu L, Shan W, Chu X, Yang Z and Yang ZQ (2017) Integrative genomic and transcriptomic analysis for pinpointing recurrent alterations of plant homeodomain genes and their clinical significance in breast cancer. Oncotarget 8, 13099–13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Swatkoski S, Holly A, Lee LC, Giroux V, Lee CS, Hsu D, Smith JL, Yuen G, Yue J et al (2015) Oncogenesis driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9. Proc Natl Acad Sci USA 112, E1724–E1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression levels of CHD7 across five subtypes of METABRIC breast cancer samples.
Fig. S2. Kaplan‐Meier plots of overall survival associated with mRNA expression levels of CHD7 in METABRIC breast cancers.
Fig. S3. Expression levels of CHD7 based on RNA sequencing data from 78 breast cancer cell lines compared with four normal mammary epithelial cell lines.
Fig. S4. Expression levels of NRAS and MYCN, but not others, decreased in CHD7‐knockdown SUM102 cells (*P < 0.05 and ***P < 0.001, Student's t‐test).
Fig. S5. Phylogenetic analysis of chromodomain‐containing proteins. The image was obtained from the ChromoHub database (http://www.thesgc.org).
Table S1. The number of samples and data type in 32 TCGA databases.
Table S2. Frequency (%) of CHD genetic alterations in 32 tumor types from TCGA database.
Table S3. Mutations of CHD6 in TCGA tumors.
Table S4. Mutations of CHD7 in TCGATumors.
Table S5. Frequency (%) of CHD genetic alterations and expression levels in five subtypes of TCGA breast cancers.
Table S6. Frequency (%) of genetic and transcriptional alterations of CHDs in 1980 METABRIC breast cancers.
Table S7. Frequency (%) of CHD genetic alterations and expression levels in five subtypes of Metabric breast cancers.
Table S8. Expression levels of CHD genes associated with NPI score in METABRIC breast cancer.
Table S9. Correlation between mRNA expression of CHD7 and 46 candidate target genes in TCGA breast cancers.