

Abstract
Immunotherapeutics are gaining more traction in the armamentarium used to combat cancer. Specifically, in situ vaccination strategies have gained interest because of their ability to alter the tumor microenvironment to an antitumor state. Herein, we investigate whether flexuous plant virus-based nanoparticles formed by the potato virus X (PVX) can be used as an immunotherapeutic for in situ vaccine monotherapy. We further developed dual chemo-immunotherapeutics by incorporating doxorubicin (DOX) into PVX yielding a dual-functional nanoparticle (PVX−DOX) or by coadministration of the two therapeutic regimes, PVX immunotherapy and DOX chemotherapy (PVX+DOX). In the context of B16F10 melanoma, PVX was able to elicit delayed tumor progression when administered as an intratumoral in situ vaccine. Furthermore, the coadministration of DOX via PVX+DOX enhanced the response of the PVX monotherapy through increased survival, which was also represented in the enhanced antitumor cytokine/chemokine profile stimulated by PVX+DOX when compared to PVX or DOX alone. Importantly, coadministered PVX+DOX was better for in situ vaccination than PVX loaded with DOX (PVX−DOX). Whereas the nanomedicine field strives to design multifunctional nanoparticles that integrate several functions and therapeutic regimens into a single nanoparticle, our data suggest a paradigm shift; some therapeutics may need to be administered separately to synergize and achieve the most potent therapeutic outcome. Altogether, our studies show that development of plant viral nanoparticles for in situ vaccines for treatment is a possibility, and dual mechanistic therapeutics can increase efficacy. Nonetheless, combining immunotherapeutics with cytolytic chemotherapy requires detailed investigation to inform optimal integration of cytolytic and immunotherapies and maximize synergy and efficacy.
Keywords: Plant virus nanoparticle, cowpea mosaic virus, potato virus X, in situ vaccination, immunotherapy, chemotherapy, cancer, melanoma
Graphical abstract
Each year, over one million new cases of cancer are diagnosed in the United States alone.1 Current cancer therapy options include surgery, hormone therapy, radiation therapy, immunotherapy, and chemotherapy, either alone or in combination. However, available therapy options are often not effective especially for patients with advanced disease, and newer treatment options are urgently needed. Recent advances in cancer immunotherapy have demonstrated that modulation of the patient's immune system can result in dramatic antitumor activity. However, despite the enthusiasm surrounding clinical results using checkpoint inhibitor therapies,2 side effects can be limiting and many patients do not respond, so there continues to be a clear need to develop immune-based approaches that integrate with and support other therapy options. In particular workable strategies are needed to take advantage of unique neoantigens that are the result of mutations within each patient's tumor.
Vaccine strategies have considerable promise for cancer immunotherapy.3 In situ vaccination, the injection of an immunostimulatory reagent into a tumor to change the microenvironment from immunosuppressive to immunostimulatory, was the first form of cancer immunotherapy, practiced over 100 years ago.4–6 When effective it not only impacts the local tumor with an immune attack but also supports systemic antitumor immunity that attacks metastatic tumors not treated directly. In situ vaccination was studied by Coley when he injected live and killed bacteria into his patients' unresectable tumors. With this approach, the tumor itself is used as the antigen source and what is introduced is an adjuvant. One form of in situ vaccination with attenuated mycobacteria, BCG, has been used as the standard of care for over 40 years for superficial bladder cancer.7 Another form of in situ vaccination, recently FDA-approved, is talimogene laherparepvec (T-VEC) [Amgen]. T-VEC is an oncolytic viral therapy based on an attenuated herpes simplex virus and engineered to express granulocyte-macrophage colony-stimulating factor (GM-CSF). T-VEC is injected into identified tumors and the therapy is mediated through cytolytic activity of the virus, immune recognition of the virus, and immune cell recruitment and activation through secretion of GM-CSF.8–11
Using a plant virus-based nanotechnology, we recently demonstrated that virus-like particles (VLPs) from the icosahedral virus cowpea mosaic virus (CPMV, 30 nm in diameter) stimulate a potent antitumor immune response when applied as an in situ vaccine. Efficacy was demonstrated in mouse models of melanoma, breast cancer, ovarian cancer, and colon cancer. Data indicate that the effect is systemic and durable, resulting in immune-memory and protecting subjects from recurrence.12 While the underlying mechanism has not been elucidated in depth, initial studies in which VLPs were inhaled into the lungs of mice bearing B16F10 lung tumors revealed a subpopulation of lung antigen presenting cells (APC) that are MHC class II+ CD11b+ Ly6G+ neutrophils that ingest VLPs and are activated following VLP exposure.12 Further, the increase in this neutrophil population is accompanied by a decrease of myeloid-derived suppressor cells (MDSCs) that mediate immunosuppression in the tumor microenvironment.12 Similarly, others recently showed that filamentous nanoparticles from papaya mosaic virus (PapMV, 530 × 12 nm) also induce antitumor efficacy in the setting of melanoma,13 thus indicating that plant viruses offer a novel platform for development of next-generation in situ vaccination approaches. Here we set out to investigate the use of alternate plant viruses as in situ vaccines and their combination with chemotherapy regimes.
Recent clinical and preclinical research indicates that the combination of chemo- and immunotherapies can be beneficial because the therapy regimes can synergize to potentiate the therapy and improve patient outcomes.14,15 For chemo–immuno combination treatment, the use of the anthracycline doxorubicin (DOX) could be a particularly powerful approach, because DOX itself induces immunogenic cell death that elicits an antitumor immune response.16 The immune response is induced by calreticulin exposure on the surface of dying cells, which facilitates tumor cell phagocytosis by dendritic cells resulting in tumor antigen presentation.17 Furthermore, doxorubicin-killed tumor cells recruit intratumoral CD11c+ CD11b+ Ly6Chi myeloid cells, which present tumor antigens to T lymphocytes; therefore, the combination of doxorubicin with tumor vaccines or immunotherapies can synergize and potentiate the overall efficacy.18–23
In this study, we set out to address the following questions:
whether the flexuous particles formed by the potato virus X (PVX) would stimulate an antitumor response when used as in situ vaccine?
whether the combination of VLP-based in situ vaccine with DOX chemotherapy would potentiate therapeutic efficacy; specifically we asked whether the formulation as combinatorial nanoparticle where DOX is bound and delivered by PVX (PVX−DOX) or the coadministration of the therapeutic regimens (PVX+DOX) would be the most effective treatment strategy?
All studies were performed using a mouse model of melanoma.
Results and Discussion
PVX is a filamentous plant virus, measuring 515 × 13 nm, and is comprised of 1270 identical coat proteins. While different in its physical nature compared to the 30 nm-sized icosahedrons formed by CPMV, PVX and PapMV share similar organization of the nucleoproteins arranged as flexuous soft matter filaments. To test whether PVX would stimulate an antitumor response when employed as an in situ vaccine, we used the B16F10 melanoma model. B16F10 is a highly aggressive and poorly immunogenic tumor model used extensively for immunotherapy studies; it also has served as a model for the evaluation of the immunotherapeutic potential of virus-based therapies.24,25 Its low immunogenicity makes it an attractive platform to investigate new immunostimulatory therapies. B16F10 isografts were induced intradermally on the right flank of C57BL/6J mice. Eight days post induction (tumor starting volume <100 mm3), mice were randomized (n = 3) and treated weekly intratumorally with PBS or 100 μg of PVX or CPMV. Tumor volumes were measured daily and mice were sacrificed when tumors reached >1000 mm3. Treatment with CPMV or PVX alone significantly slowed tumor growth rate and extended survival time compared to PBS (Figure 1) but there was no significant difference between CPMV and PVX treatment. These data indicate that PVX, like CPMV, can stimulate an antitumor response when used as an in situ vaccine.
Figure 1.
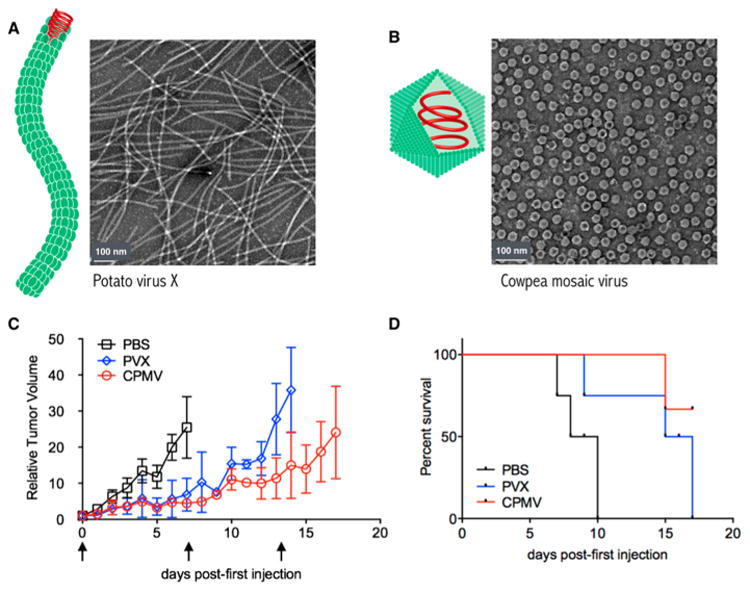
(A,B) Schematic and transmission electron micrographs of (A) PVX and (B) CPMV. (C,D) Tumor treatment study. Tumors were induced with an intradermal injection of 125 000 cells/mouse. Mice (n = 3) were treated with 100 μg of PVX or CPMV (or PBS control) once weekly, starting ∼8 days post induction when tumors measured <100 mm3. Arrows indicate injection days; mice were sacrificed when tumor volumes reached 1000 mm3. (C) Tumor growth curves shown as relative tumor volume. (D) Survival rates of treated mice.
Although the immunostimulatory potential of some plant viruses, bacteriophages, and mammalian vectors has long been recognized, leading to their development as epitope display platforms and vaccines for infectious disease, chronic diseases, addiction, and cancer (reviewed in ref 26), the application of plant viruses and their VLPs as in situ vaccines is a novel and intriguing direction that enables a new field of clinical development and scientific inquiry. We and others demonstrated potent efficacy of VLPs from CPMV12 and virus-nanoparticles formed by PapMV13 and here we also demonstrate efficacy of PVX particles. While in our previous study RNA-free “empty” CPMV (eCPMV) was utilized, in the present study RNA-containing CPMV and PVX particles were studied. The use of RNA-containing particles is expected to increase immunostimulation due to inclusion of single-stranded RNA, a natural ligand for Toll-like receptors 7 and 8.16–19 However, it should be noted that the observed antitumor immune-stimulation is not simply a result of TLR7/8 signaling based on RNA delivery, because we previously reported potent efficacy of RNA-free eCPMV.12 The proteinaceous, highly ordered, and multivalent structures formed by the capsids whether arranged helically or icosahedrally also appear to function as pathogen-associated molecular patterns (PAMPs), which are recognized by the innate immune system and elicit robust cellular and humoral immune responses.12 While more research is required to detail the mechanism of immune-activation, the previous studies with eCPMV12 and PapMV13 indicate the following principles: intratumoral administration of plant viruses or their VLPs leads to a localized, innate immune response (with neutrophils being a primary responder); localized cytokine and chemokine secretion then leads to changes in immune cell populations within the tumor microenvironment with immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), being down-regulated and CD8+T-cells infiltrating. The interplay between innate and adaptive immune-activation then results in systemic antitumor efficacy including the mounting of immune memory. It is important to remember that the virus-based nanoparticles here do not codeliver any tumor-specific epitopes; the tumor itself contains the antigen and the working hypothesis is that through initial cell lysis, triggered by infiltrating neutrophils, activated myeloid cells and T cells, a “soup” of tumor-associated and neoantigens is generated and functional antigen presentation develops. Future research dissecting the role and epitope specificity of the T cells will aid in understanding the mechanism of plant virus-nanoparticle in situ vaccination. In this paper, however, we took a different direction and analyzed how best to combine the plant virus nanoparticle in situ vaccine with chemotherapy.
Having established that PVX in situ vaccination slows tumor growth, we went ahead with a chemo–immuno combination therapy approach. We hypothesized that the combination of chemotherapy delivery, either coadministered (as physical mixture, PVX+DOX) or codelivered (as complexed version, PVX−DOX), would enhance the antitumor effect. The underlying idea was that the chemotherapy would debulk the tumor to provide a burst of tumor antigens in the context of immunogenic cell death. This fosters specific immune recognition and response to those antigens; in turn, VLP-mediated immunostimulation would further augment antitumor immunity.
To obtain the PVX−DOX complex (Figure 2A), purified PVX was loaded with DOX by incubating a 5000 molar excess of DOX with PVX for 5 days; excess DOX was removed by ultracentrifugation. Incubation criteria were optimized: increasing molar excess of DOX resulted in extensive aggregation and further increasing incubation time did not increase loading capacity (data not shown). The PVX−DOX complex was characterized by agarose gel electrophoresis, UV/visible spectroscopy, and transmission electron microscopy (TEM) (Figures 2B–D). TEM imaging confirmed particle integrity following DOX loading. The PVX−DOX formulation stability was confirmed after 1 month of storage at 4 °C; DOX release was not apparent and the particles remained intact (data not shown). UV/visible spectroscopy was used to determine the number of DOX attached per PVX. In conjunction with PVX- and DOX specific extinction coefficients, the Beer–Lambert law was used to determine the concentrations of both PVX and DOX in solution. The ratio of DOX to PVX concentration was then used to determine DOX loading. Each PVX was loaded with ∼850 DOX per PVX. Agarose gel electrophoresis analysis indicated that DOX was indeed associated with PVX and not free in solution, as free DOX was not detectable in the PVX−DOX sample. The association of DOX with PVX may be explained based on hydrophobic interactions and π−π stacking of the planar drug molecules and polar amino acids.
Figure 2.
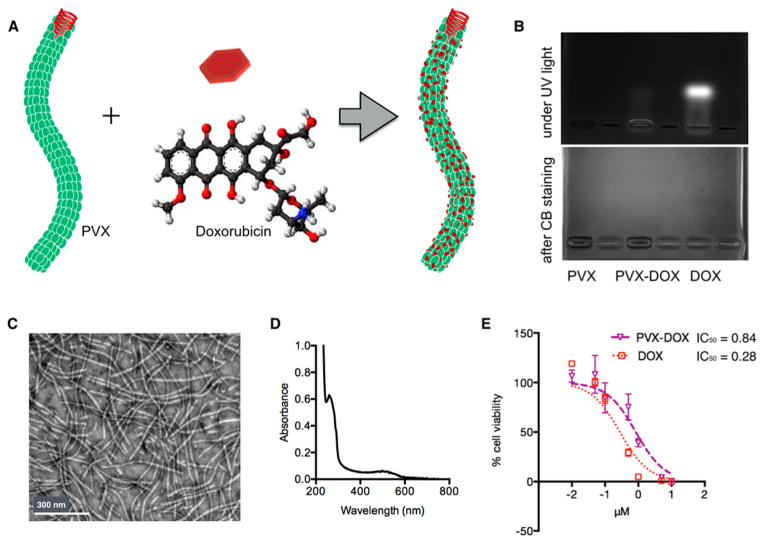
Synthesis and characterization of PVX−DOX. (A) Scheme of DOX loading onto PVX. (B) Agarose gel electrophoresis of PVX, PVX−DOX, and free DOX under UV light (top) and after Coomassie Blue staining (bottom). PVX preparations do not have electrophoretic mobility in these gels and thus are detectable in the wells of the gel; the fluorescent signal from PVX-DOX in the well indicates formation of the PVX-DOX complex was formed. Free DOX migrates toward the cathode. (C) TEM images of negatively stained PVX−DOX. (D) UV/visible spectrum of PVX−DOX. (E) Efficacy of PVX−DOX versus DOX in B16F10 cells after 24 h exposure (MTT assay); IC50 values in μM.
Efficacy of the PVX−DOX complex was confirmed using B16F10 melanoma cells (Figure 2E). DOX conjugated to PVX maintained cell killing ability, although with decreased efficacy resulting in an IC50 value of 0.84 μM versus 0.28 μM for free DOX. Similar trends have been reported with synthetic27 and virus-based nanoparticles for DOX delivery.28,29 The reduced efficacy may be explained by reduced cell uptake and required endolysosomal processing when DOX is delivered by nanoparticles.
To test the hypothesis that a combination of chemo-immunotherapy would potentiate the efficacy of PVX alone, DOX-loaded PVX (PVX−DOX) and PVX+DOX combinations were tested in the B16F10 murine melanoma model. The combination of PVX+DOX served to test whether merely the combination of the therapies or the codelivery (PVX−DOX) would enhance the overall efficacy. PVX+DOX was combined less than 30 min before injection to ensure that the two therapies did not have time to interact. PBS, PVX alone, and free DOX were used as controls. When tumors were <100 mm3, mice were treated every other day intratumorally with PVX−DOX, PVX+DOX, or corresponding controls (n = 6). Tumor volumes were measured daily and mice were sacrificed when tumors reached >1000 mm3. PVX was administered at 5 mg kg−1 (corresponding to a dose of 0.065 mg kg−1 DOX). Clinically, doxorubicin is administered at doses of 1–10 mg kg−1, intravenously. If 1–10% of the injected dose reaches the tumor site, the resulting intratumoral dose would equate to 0.01–1 mg kg−1; thus our intratumoral dose is within a clinically relevant range of DOX.
Although there was no statistical difference in tumor growth rate or survival time between PVX−DOX complex versus PVX or DOX alone, PVX+DOX did significantly slow tumor growth rate versus PVX and DOX alone (Figures 3A,B). Thus, the data indicate that the combination of DOX chemotherapy and PVX immunotherapy indeed potentiates efficacy, however the formulation as a combined nanoparticle, PVX−DOX, did not improve the treatment. The lack of statistically significant enhancement of efficacy of the PVX− DOX complex versus immuno- or chemo-monotherapy may be explained by the fact that the therapies synergize best when they act on their own. DOX targets replicating cancer cells to induce cell death, and PVX likely associated with immune cells to stimulate an antitumor effect, most likely through activation of signaling cascades through PAMP receptors or other danger signal recognition molecules. Indeed, we found that PVX was colocalized with F4/80+ macrophages within the tumor tissue (Figure 3C), which may cause killing of immune cells rather than cancer cells. Even if the nanoparticles do not exhibit cytotoxic effect on the immune cell population, the sequestration of PVX−DOX in the immune cells would lower the antitumor efficacy of the complex. (In in vitro cell uptake assays, we confirmed that there are no differences in phagocyte cell uptake comparing PVX versus PVX−DOX or PVX+DOX; data not shown).
Figure 3.
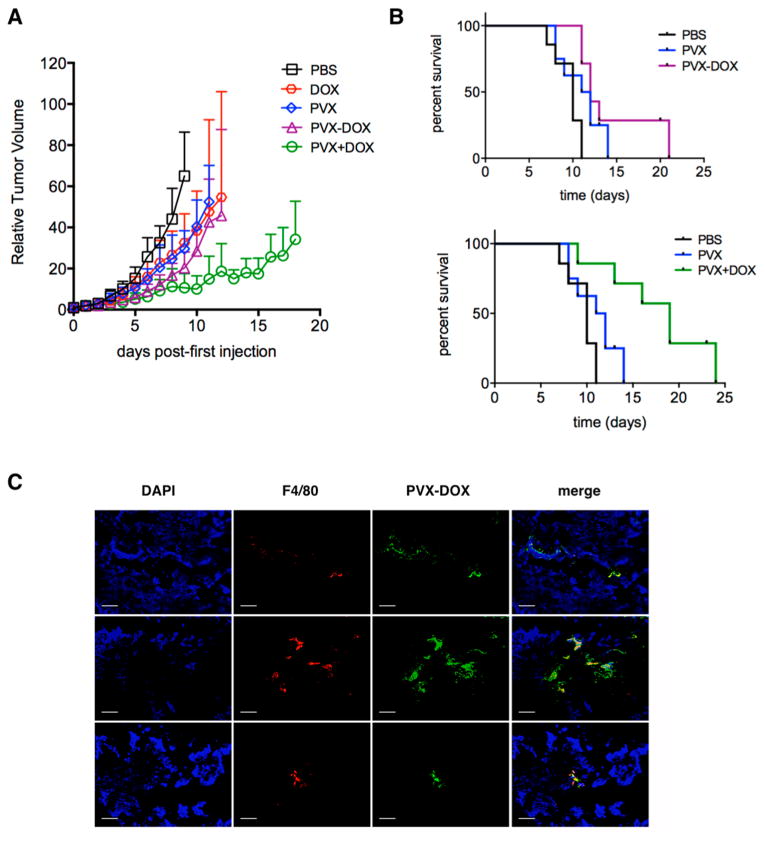
Chemo-immunotherapy treatment of B16F10 tumors. Groups (n = 6) were treated with PBS, PVX, DOX, PVX−DOX, or PVX+DOX. PVX was administered at a dose of 5 mg kg−1, DOX was administered at a dose of 0.065 mg kg−1. Treatment started ∼8 days post induction when tumors measured <100 mm3, and injections were repeated every other day until tumors reached >1000 mm3. (A) Tumor growth curves shown as relative tumor volume. Statistical significance was detected comparing PVX versus PVX+DOX. (B) Survival rates of treated mice. (C) Immunofluorescence imaging of three representative PVX−DOX tumor sections after weekly dosing of PVX−DOX (animals received two doses of PVX and were collected when tumors reached >1000 mm3). Tumors treated with PVX−DOX (rows 1–3) were sectioned and stained with DAPI (blue), F4/80 (red), and PVX (green). Scale bar = 100 μm.
To gain insight into the underlying immunology, we performed cytokine/chemokine profiling using a 32-plex MILLIPLEX Luminex assay. Tumors were treated with PBS, PVX, DOX, PVX−DOX, or PVX+DOX and harvested 24 h after the first injection. Profiles were obtained using tumor homogenates and normalized to total protein levels by the bicinchoninic acid (BCA) assay. The PVX+DOX group repeatedly showed significantly higher particular cytokine and chemokine levels compared to any other group (Figure 4). Specifically, interferon gamma (IFNγ)and IFNγ-stimulated or synergistic cytokines were elevated. These included but may not be limited to Regulated on Activation, Normal T Cell Expressed and Secreted (RANTES/CCL5), Macrophage Inflammatory Protein 1a (MIP-1a/CCL3), Monocyte Chemo-attractant Protein (MCP-1/CCL2), Monokine Induced by Gamma interferon (MIG/CXCL9), and IFNγ-induced protein 10 (IP-10). IFNγ is a multifunctional type II interferon critical for inducing a pro-inflammatory environment and antiviral responses and is often associated with effective tumor immunotherapy responses. Under the influence of IFNγ, these chemokines mediate the influx of monocytes, macrophages, and other immune cells.30 Interestingly, the induction of IFNγ was not associated with the increased expression of its master positive regulator, IL-12, thus in this context the increased expression of IFNγ is IL-12-independent (data not shown). In the tumor microenvironment, activation of the IFNγ pathway is in accordance with other work, where viruses were applied as an in situ vaccine.12,13,31 Stimulation of the IFNγ pathway alleviates the immuno-suppressive tumor microenvironment promoting an antitumor immune response. The molecular receptors and signaling cascades are yet to be elucidated, but the body of data indicates IFNγ to be a key player for viral-based in situ vaccination approaches.
Figure 4.
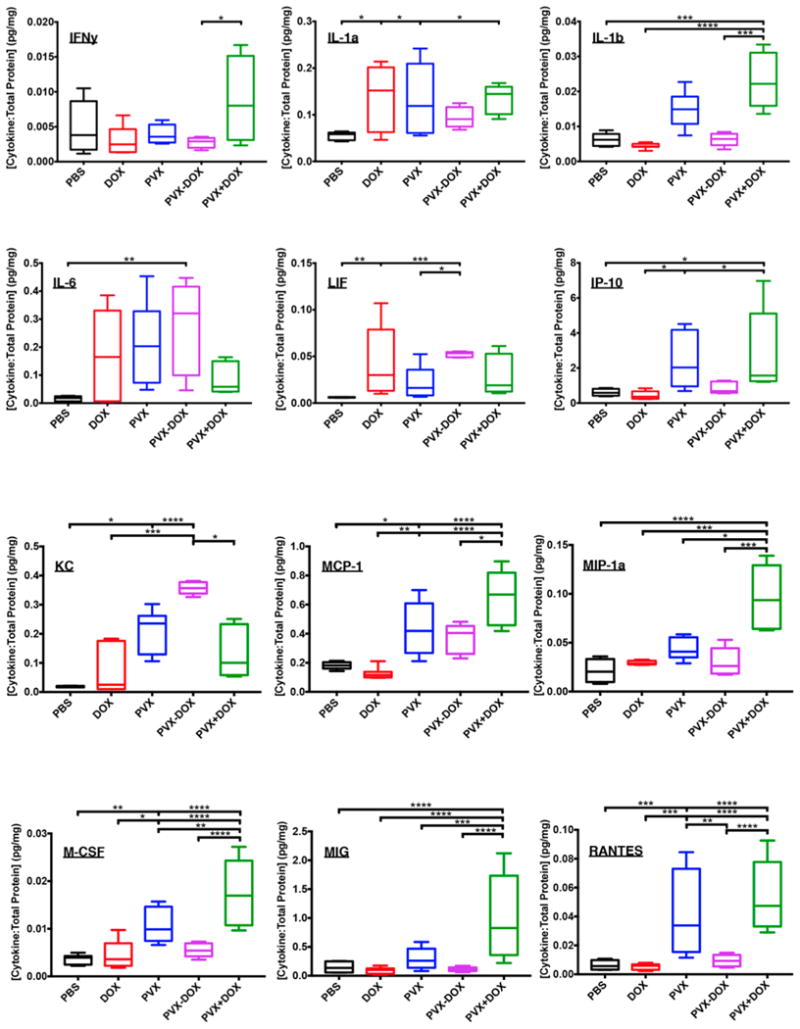
Luminex Multiplex Cytokine/Chemokine profiles. Groups (n = 4) were treated with PBS, PVX, DOX, PVX−DOX, or PVX+DOX. PVX was administered at a dose of 5 mg kg−1, DOX was administered at a dose of 0.065 mg kg−1. After one injection, tumors were harvested at 24 h post injection for analysis. Data shown as ratio of cytokine concentration: total protein (or cytokine per total protein in pg/mg). * = <0.05, ** = <0.01, *** = <0.001, **** = <0.0001).
Other noteworthy cytokines/chemokines that were up-regulated include interleukin-1β (IL-1β) and Macrophage Colony-Stimulating Factor (M-CSF). IL-1β is known to be an early pro-inflammatory cytokine activated by many PAMPs and danger associated molecular patterns (DAMPs).32 IL-1β signaling was also observed in our earlier work with CPMV,12 and data suggest that initial recognition of the viral in situ vaccine by innate surveillance cells is promoting immune activation. IL-1β and M-CSF are both major recruiters and activators of monocytes and macrophages to the site of challenge.30 In particular, M-CSF enhances monocyte functions including phagocytic activity and cytotoxicity for tumor cells, while inducing synthesis of inflammatory cytokines such as IL-1, TNFα, and IFNγ in monocytes.33,34 PVX monotherapy appears to follow a similar trend of increased expression of cytokines/chemokines with further enhanced response through combination with DOX when coadministered (PVX+DOX) but reduced response when directly coupled together as PVX−DOX.
Conclusion
In this study, we demonstrate that PVX stimulates an antitumor immune response when used as an in situ vaccine. To date, the only other plant virus-based platforms reported to show efficacy as in situ vaccines are CPMV12 and PapMV.13 While further research is required to comprehend the underlying mechanism, data indicate that the plant virus-based nanoparticles activate the innate immune system locally; this innate immune activation is thought to overcome the immunosuppressive tumor microenvironment restarting the cancer immunity cycle leading to systemic elimination of cancer cells through the adaptive immune system. The potency of the approach is intriguing and questions remain regarding the underlying immunology as well as the engineering design space, that is, whether the antitumor effect is a property of any (plant) virus-based nanomaterial or whether shape, size, and molecular composition of the virus relate to its efficacy. While the antitumor immunostimulation of PapMV was attributed to its packaged RNA and stimulation of toll-like receptors (TLRs),13 our previous studies with RNA-free empty CPMV (eCPMV) indicated that the RNA cargo is not required.12 It is likely that innate receptors such as pattern recognition receptors (PRR) play a key role recognizing the multivalent nature of the plant virus nanoparticles.
Bacteriophages, mammalian viral gene-delivery vectors, as well as oncolytic viruses have been explored in the context of cancer immunotherapy. Yet the underlying principles are distinct: some bacteriophages interact with cancer cells via displayed KGD (Lys-Gly-Asp) motifs, and observations of reduced tumor growth were attributed to interactions of the bacteriophage proteins with integrins expressed in tumor vasculature and tumor cells.35–37 Research also highlighted the immune-modulatory nature of bacteriophages, their interaction and stimulation of dendritic cells, as well as their potential role in oncolytic therapy, in particular in colon cancer.38 To the best of our knowledge, successful in situ vaccination with bacteriophages has not been demonstrated yet.
Mammalian viruses have been studied and developed in the context of oncolytic viriotherapy and they can function as in situ vaccines.5 In general, oncolytic viriotherapy builds on viruses that target and infect cancer cells, either through natural or engineered cell tropism. The infection activates the immune system and cells expressing viral proteins will be attacked and eliminated by the host immune system; cross-priming of T cells will also result in cell killing of noninfected cells resulting in reduced tumor burden. In addition to the immunostimulatory properties, through viral RNAs and expression of viral proteins, mammalian virus-based vectors have also been engineered to express immunostimulatory molecules to activate and prime antitumor responses through dual-pronged mechanisms. T-VEC, for example, is an oncolytic virus engineered to express granulocyte-macrophage colony-stimulating factor (GM-CSF) to recruit and activate immune cells, in addition to tumor cell killing based on its cytolylic activity.8–11
Together, these data highlight the potential of virus-based technologies as in situ vaccines for cancer immunotherapy. There are many novel virus-based materials in the pipeline; plant viruses can be considered safer for use in humans compared to mammalian viruses39 because they do not replicate in or infect mammals. They can be administered at doses of up to 100 mg (1016 particles) per kg body weight without clinical toxicity.40,41 While used here as a topical treatment, the plant virus-based nanoparticles show excellent blood and tissue compatibility,42,43 thus also opening the door for possible intravenous, systemic administration. Compared to bacteriophages, plant viruses also offer advantages: their manufacture does not involve fermentation in bacteria, which may contain LPS or other toxic contaminants.44–46 Furthermore, bacteriophages, but not plant viruses, play a role in the human microbiome, and careful considerations must be made when applying phages as therapies as the interplay and/or balance of the microbiome could be shifted.47 Although each system, synthetic or viral, has its advantages and disadvantages, each system also has its unique attributes. Future studies are needed to systemically dissect whether some viruses are more potent as in situ vaccine than others and which design criteria are the governing factors; that is, it needs to be addressed how size, shape, and molecular composition impact the efficacy.
The combination of the DOX chemotherapeutic with PVX was more efficacious than the monotherapies when coadministered as the PVX+DOX formulation but not when physically linked in the PVX−DOX formulation. It is clear that further research is needed to scrutinize the underlying immunology, however, it is fair to conclude that the Luminex panel is in good agreement with the efficacy read-out. Data show that PVX+DOX induced a higher immune mediator profile within the tumor microenvironment, in turn resulting in increased efficacy against B16F10 melanoma. A key conclusion to draw from these studies is that the combination of chemo- and immunotherapy indeed is a powerful tool–yet the formulation of the two regimes into a single, multifunctional nanoparticle may not always be the optimal approach.
It has long been recognized that nanoparticles are preferentially ingested by phagocytic cells. Figure 3C confirms this and shows that PVX−DOX is concentrated in F4/80+ macrophages within the tumor. The basis for reduced efficacy of PVX−DOX as compared to PVX+DOX could simply be because when phagocytes ingest PVX−DOX, it reduces the concentration of DOX available to react with tumor cell DNA. If as seems likely, the cells that respond to PVX at least initially are phagocytes, then a nonexclusive alternative could be that ingestion of PVX−DOX by phagocytes leads to a different response than ingestion of PVX by itself by those cells, thus blunting the immune response stimulated by PVX.
Chemo- and immunotherapy regimes have been recognized to synergize and several reports have highlighted the potential of combinatorial nanoparticles to deliver chemotherapies while stimulating the immune system. Although the nanomedicine field strives to design multifunctional nanoparticles that integrate several functions and therapeutic regimens into a single nanoparticle (reviewed in ref 48), our data indicate minimal improvement in efficacy using the combinatorial PVX−DOX nanoparticles. Significant therapeutic efficacy with prolonged survival is only achieved when the therapeutic regimes, PVX immunotherapy and DOX chemotherapy, are coadministered separately allowing each drug to act on their own, leading to potent antitumor effects.
Methods
PVX and CPMV Production
PVX was propagated in Nicotiana benthamiana plants and purified as previously reported.49 CPMV was propagated in Vigna unguiculata plants and purified as previously reported.50
Synthesis of PVX−DOX
PVX (2 mg mL−1 in 0.1 M potassium phosphate buffer (KP), pH 7.0) was incubated with a 5000 molar excess of doxorubicin (DOX) at a 10% (v/v) final concentration of DMSO for 5 days at room temperature with agitation. PVX−DOX was purified twice over a 30% (w/v) sucrose cushion using ultracentrifugation (212 000g for 3 h at 4 °C) and resuspended overnight in 0.1 M KP, pH 7.0. PVX−DOX filaments were analyzed using UV/visible spectroscopy, transmission electron microscopy, and agarose gel electrophoresis.
UV/Visible Spectroscopy
The number of DOX per PVX filament was determined by UV/visible spectroscopy, using the NanoDrop 2000 spectrophotometer. DOX loading was determined using the Beer–Lambert law and DOX (11 500 M−1 cm−1 at 495 nm) and PVX (2.97 mL mg−1 cm−1 at 260 nm) extinction coefficients.
Transmission Electron Microscopy
TEM imaging was performed after DOX loading to confirm integrity of PVX− DOX filaments. PVX−DOX samples (0.1 mg mL−1, in dH2O) were placed on carbon-coated copper grids and negatively stained with 0.2% (w/v) uranyl acetate. Grids were imaged using a Zeiss Libra 200FE transmission electron microscope, operated at 200 kV.
Agarose Gel Electrophoresis
To confirm DOX attachment, PVX−DOX filaments were run in a 0.8% (w/v) agarose gel (in TBE). PVX−DOX and corresponding amounts of free DOX or PVX alone were loaded with 6× agarose loading dye. Samples were run at 100 V for 30 min in TBE. Gels were visualized under UV light and after staining with 0.25% (w/v) Coomassie blue.
Cell Culture and Cell Viability Assay
B16F10 cells (ATCC) were cultured in Dulbecco's modified Eagle's media (DMEM, Life Technologies), supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals) and 1% (v/v) penicillin– streptomycin (penstrep, Life Technologies). Cells were maintained at 37 °C, 5% CO2. Confluent cells were removed with 0.05% (w/v) trypsin-EDTA (Life Technologies), seeded at 2 × 103 cells/100 μL/well in 96-well plates, and grown overnight at 37 °C, 5% CO2. The next day, cells were washed two times with PBS and incubated with free DOX or PVX−DOX corresponding to 0, 0.01, 0.05, 0.1, 0.5, 1, 5, or 10 μM DOX for 24 h, in triplicate. A PVX only control corresponded to the amount of PVX in the highest PVX−DOX sample. Following incubation, cells were washed two times to remove unbound DOX or particles. Fresh medium (100 μL) was added and cells were returned to the incubator for 48 h. Cell viability was assessed using an MTT proliferation assay (ATCC); the procedure was as the manufacturer suggested.
Animal Studies
All experiments were conducted in accordance with Case Western Reserve University's Institutional Animal Care and Use Committee. C57BL/6J male mice (Jackson Laboratory) were used. B16F10 tumors were induced intradermally into the right flank of C57BL/6J mice (1.25 × 105 cells/50 μL media). Animals were monitored and tumor volumes were calculated as V = 0.5 (a × b2); where a is length and b is width of the tumor. Animals were sacrificed when tumor volume reached >1000 mm3. Treatment schedule: Eight days post-tumor induction (day 0), mice were randomly assigned to the following groups (n = 3): PBS, PVX, or CPMV. Mice were treated intratumorally (20 μL), every 7 days, with 5 mg kg−1 PVX or CPMV. Mice were sacrificed when tumors reached a volume >1000 mm3. For chemo-immunotherapy combination therapy, mice were randomly assigned to the following groups (n = 6): PBS, PVX, DOX, conjugated PVX−DOX, or PVX+DOX mixtures. PVX+DOX samples were prepared less than 30 min before injections and are considered not bound to each other. Mice were treated intratumorally (20 μL) every other day with 5mgkg−1 PVX or PVX−DOX or the corresponding dose of DOX (∼0.065 mg kg−1). Mice were sacrificed when tumors reached a volume >1000 mm3.
Immunostaining
When tumor reached volumes <100 mm3, mice were randomly assigned to the following groups (n = 3): PBS or PVX−DOX. Mice were treated intratumorally (20 μL), every 7 days, with 5 mg kg−1 PVX−DOX. Mice were sacrificed when tumors reached a volume >1000 mm3 and tumors were collected for analysis. Tumors were frozen in optimal cutting temperature compound (Fisher). Frozen tumors were cut into 12 μm sections. Sections were fixed in 95% (v/v) ethanol for 20 min on ice. Following fixation, tumor sections were permeabilized with 0.2% (v/v) Triton X-100 in PBS for 2 min at room temperature for visualization of intracellular markers. Then, tumor sections were blocked in 10% (v/v) goat serum (GS) in PBS for 60 min at room temperature. PVX and F4/80 were stained using rabbit anti-PVX antibody (1:250 in 1% (v/v) GS/PBS) and rat anti-mouse F4/80 (1:250 in 1% (v/v) GS/PBS) for 1–2 h at room temperature. Primary antibodies were detected using secondary antibody staining: Alexa-Fluor488-labeled goat anti-rabbit antibody (1:500 in 1% (v/v) GS/PBS) and AlexaFluor555-labeled goat-anti-rat antibody (1:500 in 1% (v/v) GS/PBS) for 60 min at room temperature. Tumor sections were washed three times with PBS in between each step. Following the final wash, coverslips were mounted using Fluoroshield with DAPI. Slides were imaged on a Zeiss Axio Observer Z1 motorized FL inverted microscope. Fluorescence intensity was analyzed using ImageJ 1.47d (http://imagej.nih.gov/ij).
Luminex Assay
Intradermal melanomas were induced in C57BL/6J male mice (Jackson) as described above. Eight days post-tumor induction (day 0), mice were randomly assigned to the following groups (n = 4): PBS, PVX, PVX−DOX, or PVX+DOX. Mice were treated intratumorally (20 μL) once and tumors where harvested at 24 h.p.i. Tumors were weighed and homogenized in T-PERBuffer (ThermoFisher) at 1 mL of buffer/100 mg of tissue. T-PERBuffer was supplemented with cOmplete Protease Inhibitor Cocktail tablets (Roche) at one tablet per 8 mL of buffer. After homogenization, homogenizer was rinsed with 0.5 mL of HBSS (ThermoFisher) and added to the homogenate. Homogenate was centrifuged at 9000g for 10 min at 2–8 °C. Supernatants were frozen and kept at −80 °C until analyses. Millipore Milliplex MAP mouse 32-plex was run at the CRWU Bioanalyte Core.
Acknowledgments
This work was funded in part by a Research Scholar Award from the American Cancer Society (128319-RSG-15-144-01-CDD to N.F.S.), training grants from the National Institutes of Health (R25 CA148052 to K.L.L., T32 EB007509 to K.L.L., T32 GM007250 to A.A.M., TL1 TR000441 to A.A.M.), and a Case Western Reserve University (CWRU) Council to Advance Human Health (CAHH) Award (to N.F.S.). Amy M. Wen (CWRU) is thanked for assistance with TEM imaging.
Footnotes
Notes: The authors declare no competing financial interest.
ORCID: Nicole F. Steinmetz: 0000-0002-0130-0481
References
- 1.American Cancer Society. Global Cancer Facts & Figures. 3rd. American Cancer Society; Atlanta: 2015. [Google Scholar]
- 2.Spain L, Larkin J. Immunotherapy. 2016;8(6):677–9. doi: 10.2217/imt.16.6. [DOI] [PubMed] [Google Scholar]
- 3.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Nat Rev Cancer. 2016;16(4):219–33. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 4.Hammerich L, Bhardwaj N, Kohrt HE, Brody JD. Immunotherapy. 2016;8(3):315–30. doi: 10.2217/imt.15.120. [DOI] [PubMed] [Google Scholar]
- 5.Hammerich L, Binder A, Brody JD. Mol Oncol. 2015;9(10):1966–81. doi: 10.1016/j.molonc.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pierce RH, Campbell JS, Pai SI, Brody JD, Kohrt HE. Hum Vaccines Immunother. 2015;11(8):1901–9. doi: 10.1080/21645515.2015.1049779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamat AM, Flaig TW, Grossman HB, Konety B, Lamm D, O'Donnell MA, Uchio E, Efstathiou JA, Taylor JA., 3rd Nat Rev Urol. 2015;12(4):225–35. doi: 10.1038/nrurol.2015.58. [DOI] [PubMed] [Google Scholar]
- 8.Harrington KJ, Puzanov I, Hecht JR, Hodi FS, Szabo Z, Murugappan S, Kaufman HL. Expert Rev Anticancer Ther. 2015;15(12):1389–403. doi: 10.1586/14737140.2015.1115725. [DOI] [PubMed] [Google Scholar]
- 9.Kohlhapp FJ, Zloza A, Kaufman HL. Drugs Today (Barc) 2015;51(9):549–58. doi: 10.1358/dot.2015.51.9.2383044. [DOI] [PubMed] [Google Scholar]
- 10.Johnson DB, Puzanov I, Kelley MC. Immunotherapy. 2015;7(6):611–9. doi: 10.2217/imt.15.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Killock D. Nat Rev Clin Oncol. 2015;12(8):438. doi: 10.1038/nrclinonc.2015.106. [DOI] [PubMed] [Google Scholar]
- 12.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S. Nat Nanotechnol. 2015;11(3):295–303. doi: 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lebel ME, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A. Nano Lett. 2016;16(3):1826–32. doi: 10.1021/acs.nanolett.5b04877. [DOI] [PubMed] [Google Scholar]
- 14.Bagalkot V, Lee IH, Yu MK, Lee E, Park S, Lee JH, Jon S. Mol Pharmaceutics. 2009;6(3):1019–28. doi: 10.1021/mp800177f. [DOI] [PubMed] [Google Scholar]
- 15.Roy A, Singh MS, Upadhyay P, Bhaskar S. Int J Pharm. 2013;445(1–2):171–80. doi: 10.1016/j.ijpharm.2013.01.045. [DOI] [PubMed] [Google Scholar]
- 16.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G. J Exp Med. 2005;202(12):1691–701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 18.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, Martins I, Schlemmer F, Michaud M, Kepp O, Sukkurwala AQ, Menger L, Vacchelli E, Droin N, Galluzzi L, Krzysiek R, Gordon S, Taylor PR, Van Endert P, Solary E, Smyth MJ, Zitvogel L, Kroemer G. Immunity. 2013;38(4):729–41. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Machiels JP, Reilly RT, Emens LA, Ercolini AM, Lei RY, Weintraub D, Okoye FI, Jaffee EM. Cancer Res. 2001;61(9):3689–97. [PubMed] [Google Scholar]
- 20.Eralp Y, Wang X, Wang JP, Maughan MF, Polo JM, Lachman LB. Breast Cancer Res. 2004;6(4):R275–83. doi: 10.1186/bcr787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehrke MJ, Verstovsek S, Maccubbin DL, Ujhazy P, Zaleskis G, Berleth E, Mihich E. Int J Cancer. 2000;87(1):101–9. doi: 10.1002/1097-0215(20000701)87:1<101::aid-ijc15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 22.Ewens A, Luo L, Berleth E, Alderfer J, Wollman R, Hafeez BB, Kanter P, Mihich E, Ehrke MJ. Cancer Res. 2006;66(10):5419–5426. doi: 10.1158/0008-5472.CAN-05-3963. [DOI] [PubMed] [Google Scholar]
- 23.Zhu S, Waguespack M, Barker SA, Li S. Clin Cancer Res. 2007;13(14):4252–60. doi: 10.1158/1078-0432.CCR-06-2894. [DOI] [PubMed] [Google Scholar]
- 24.Baird JR, Byrne KT, Lizotte PH, Toraya-Brown S, Scarlett UK, Alexander MP, Sheen MR, Fox BA, Bzik DJ, Bosenberg M, Mullins DW, Turk MJ, Fiering S. J Immunol. 2013;190(1):469–78. doi: 10.4049/jimmunol.1201209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Saffold S, Cao X, Krauss J, Chen W. J Immunol. 1998;161(10):5516–24. [PubMed] [Google Scholar]
- 26.Lee KL, Twyman RM, Fiering S, Steinmetz NF. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;4:554–78. doi: 10.1002/wnan.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoo HS, Lee KH, Oh JE, Park TG. J Controlled Release. 2000;68(3):419–31. doi: 10.1016/s0168-3659(00)00280-7. [DOI] [PubMed] [Google Scholar]
- 28.Lockney DM, Guenther RN, Loo L, Overton W, Antonelli R, Clark J, Hu M, Luft C, Lommel SA, Franzen S. Bioconjugate Chem. 2011;22(1):67–73. doi: 10.1021/bc100361z. [DOI] [PubMed] [Google Scholar]
- 29.Ren Y, Wong SM, Lim LY. Bioconjugate Chem. 2007;18(3):836–43. doi: 10.1021/bc060361p. [DOI] [PubMed] [Google Scholar]
- 30.Arango Duque G, Descoteaux A. Front Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tahtinen S, Gronberg-Vaha-Koskela S, Lumen D, Merisalo-Soikkeli M, Siurala M, Airaksinen AJ, Vaha-Koskela M, Hemminki A. Cancer Immunol Res. 2015;3(8):915–25. doi: 10.1158/2326-6066.CIR-14-0220-T. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Castejon G, Brough D. Cytokine Growth Factor Rev. 2011;22(4):189–95. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, Brys L, Abels C, Lahmar Q, Ergen C, Vereecke L, Tacke F, De Baetselier P, Van Ginderachter JA, Laoui D. Cancer Res. 2016;76(1):35–42. doi: 10.1158/0008-5472.CAN-15-0869. [DOI] [PubMed] [Google Scholar]
- 34.Nakoinz I, Ralph P. Cell Immunol. 1988;116(2):331–40. doi: 10.1016/0008-8749(88)90235-3. [DOI] [PubMed] [Google Scholar]
- 35.Dabrowska K, Zembala M, Boratynski J, Switala-Jelen K, Wietrzyk J, Opolski A, Szczaurska K, Kujawa M, Godlewska J, Gorski A. Arch Microbiol. 2007;187(6):489–98. doi: 10.1007/s00203-007-0216-y. [DOI] [PubMed] [Google Scholar]
- 36.Dabrowska K, Opolski A, Wietrzyk J, Switala-Jelen K, Godlewska J, Boratynski J, Syper D, Weber-Dabrowska B, Gorski A. Anticancer Res. 2004;24(6):3991–5. [PubMed] [Google Scholar]
- 37.Dabrowska K, Opolski A, Wietrzyk J, Switala-Jelen K, Boratynski J, Nasulewicz A, Lipinska L, Chybicka A, Kujawa M, Zabel M, Dolinska-Krajewska B, Piasecki E, Weber-Dabrowska B, Rybka J, Salwa J, Wojdat E, Nowaczyk M, Gorski A. Acta Virol. 2004;48(4):241–8. [PubMed] [Google Scholar]
- 38.Budynek P, Dabrowska K, Skaradzinski G, Gorski A. Arch Microbiol. 2010;192(5):315–20. doi: 10.1007/s00203-010-0559-7. [DOI] [PubMed] [Google Scholar]
- 39.Manchester M, Singh P. Adv Drug Delivery Rev. 2006;58(14):1505–22. doi: 10.1016/j.addr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Kaiser CR, Flenniken ML, Gillitzer E, Harmsen AL, Harmsen AG, Jutila MA, Douglas T, Young MJ. Int J Nanomedicine. 2007;2(4):715–33. [PMC free article] [PubMed] [Google Scholar]
- 41.Singh P, Prasuhn D, Yeh RM, Destito G, Rae CS, Osborn K, Finn MG, Manchester M. J Controlled Release. 2007;120(1–2):41–50. doi: 10.1016/j.jconrel.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruckman MA, Randolph LN, Vanmeter A, Hern S, Shoffstall AJ, Taurog RE, Steinmetz NF. Virology. 2014;449:163–73. doi: 10.1016/j.virol.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee KL, Shukla S, Wu M, Ayat NR, El Sanadi CE, Wen AM, Edelbrock JF, Pokorski JK, Commandeur U, Dubyak GR, Steinmetz NF. Acta Biomater. 2015:166–179. doi: 10.1016/j.actbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma JK, Barros E, Bock R, Christou P, Dale PJ, Dix PJ, Fischer R, Irwin J, Mahoney R, Pezzotti M, Schillberg S, Sparrow P, Stoger E, Twyman RM, European Union Framework 6 Pharma-Planta, C EMBO Rep. 2005;6(7):593–9. doi: 10.1038/sj.embor.7400470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer R, Emans N. Transgenic Res. 2000;9(4–5):279–99. doi: 10.1023/a:1008975123362. discussion 277. [DOI] [PubMed] [Google Scholar]
- 46.Fischer R, Emans N, Schuster F, Hellwig S, Drossard J. Biotechnol Appl Biochem. 1999;30(Pt 2):109–12. [PubMed] [Google Scholar]
- 47.De Paepe M, Leclerc M, Tinsley CR, Petit MA. Front Cell Infect Microbiol. 2014;4:39. doi: 10.3389/fcimb.2014.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fontana F, Liu D, Hirvonen J, Santos HA. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017 doi: 10.1002/wnan.1421. [DOI] [PubMed] [Google Scholar]
- 49.Lee KL, Uhde-Holzem K, Fischer R, Commandeur U, Steinmetz NF. Methods Mol Biol. 2014;1108:3–21. doi: 10.1007/978-1-62703-751-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leong HS, Steinmetz NF, Ablack A, Destito G, Zijlstra A, Stuhlmann H, Manchester M, Lewis JD. Nat Protoc. 2010;5(8):1406–17. doi: 10.1038/nprot.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]