

Abstract
Multiple endocrine neoplasia (MEN) refers to a group of autosomal dominant disorders with generally high penetrance that lead to the development of a wide spectrum of endocrine and non-endocrine manifestations. The most frequent among these conditions is MEN type 1 (MEN1), which is caused by germline heterozygous loss-of-function mutations in the tumor suppressor gene MEN1. MEN1 is characterized by primary hyperparathyroidism (PHPT) and functional or nonfunctional pancreatic neuroendocrine tumors and pituitary adenomas. Approximately 10% of patients with familial or sporadic MEN1-like phenotype do not have MEN1 mutations or deletions. Recently, a novel MEN syndrome was discovered, initially in rats (MENX), and later in humans (MEN4), which is caused by germline mutations in the putative tumor suppressor CDKN1B. The most common phenotype of the 19 cases of MEN4 that have been described to date is PHPT followed by pituitary adenomas. Recently, somatic and germline mutations in CDKN1B were also identified in patients with sporadic PHPT, lymphoma, and breast cancer, demonstrating a novel role for CDKN1B as a tumor susceptibility gene for other neoplasms. In this review, we report on the genetic characterization and clinical features of MEN4.
Keywords: Multiple Endocrine Neoplasia, MEN4, MEN1, Neuroendocrine tumors, CDKN1B, p27
Introduction
Among the multiple endocrine neoplasia (MEN) syndromes, the most frequent is type 1 or MEN1 (OMIM #131100). MEN1 is characterized by primary hyperparathyroidism (PHPT) due to parathyroid gland hyperplasia, and functional or nonfunctional pancreatic neuroendocrine tumors (pNETs) and pituitary adenomas (Thakker, et al. 2012). MEN2, a less common entity, is characterized by medullary thyroid carcinoma (MTC), pheochromocytoma and PHPT. MEN2 is further divided into MEN2A (OMIM #171400) that typically manifests with MTC, pheochromocytoma, and PHPT, and MEN2B (OMIM #162300) that manifests with MEN2A features, although typically lacking PHPT, ganglioneuromas of the lips, tongue and colon, and a marfanoid habitus (Brandi, et al. 2001). The genetic cause of MEN1 was initially localized to 11q13 through positional cloning (Chandrasekharappa, et al. 1997; Larsson, et al. 1988), and later identified as germline heterozygous loss-of-function mutations in the tumor suppressor gene MEN1 (Agarwal, et al. 1997), which consists of 10 exons and codes for the protein menin. The genetic defect in the MEN2 syndromes is due to mutations in the RET (Rearranged in Transfection) proto-oncogene on chromosome 10q11 (Brandi et al. 2001). Mutations in MEN1 have been detected in ~90% of familial cases (including germline deletions) and fewer among patients with sporadic MEN1 (Brandi et al. 2001; Thakker et al. 2012). The numbers are higher for patients with one of the MEN2 syndromes: almost all have one or the other RET mutation (Brandi et al. 2001). Overall, approximately 10% of patients with familial MEN1-like phenotype and many more with sporadic MEN1 have negative MEN1 mutational analysis (Brandi et al. 2001; Namihira, et al. 1999); patients with a “mixed” MEN1/MEN2 phenotype have also been described that do not have mutations in MEN1 or RET (El-Maouche, et al. 2016).
A novel MEN syndrome was recently discovered, initially in rats where it was named “MENX” and then in humans, now known as MEN4 (OMIM #610755). MEN4 is caused by germline mutations in Cdkn1b in rats and CDKN1B in humans, coding for p27Kip1 (commonly referred to as p27 or KIP1, hereafter p27), a putative tumor suppressor gene regulating cell cycle progression. The most common phenotypic features of patients with MEN4 are parathyroid and pituitary neoplasias. Recently, somatic and germline mutations in CDKN1B were also identified in patients with sporadic PHPT, lymphoma, and breast cancer, demonstrating a novel role for CDKN1B as a tumor susceptibility gene for endocrine and other neoplasms. In this review, we present the clinical and genetic characterization of the MEN4 syndrome and talk about the role of p27 in other tumors.
Identification of a new syndrome predisposing to multiple endocrine neoplasias: MENX
In 2000, Franklin et al. (Franklin, et al. 2000) tested the theory that cyclin-dependent kinase inhibitor (CDKI) genes may function as tumor suppressors genes on mouse models. They showed that loss of both p18 and p27 function resulted in spontaneous development of a wide spectrum of neuroendocrine tumors (NET) affecting various organ systems, including the pituitary, adrenals, thyroid, parathyroid, and the gastroduedenal tract (Franklin et al. 2000). It was noted that somatic biallelic inactivation of CDKN1B, albeit a rare event, lead to several non-endocrine human tumors, suggesting that p27 is a haploinsufficient tumor suppressor and a potential candidate gene for tumorigenesis in humans (Philipp-Staheli, et al. 2001).
In 2002, an MEN-like syndrome in rats that did not involve mutations in the MEN or RET gene was described but the responsible genetic defect(s) remained unknown (Fritz, et al. 2002). The spontaneous development of multiple endocrine neoplasias (e.g.: bilateral pheochromocytoma, bilateral MTC, multigland parathyroid neoplasia, and pancreatic islet cells hyperplasia) within the first year of life with high penetrance characterized this syndrome as intermediate or combinatorial of both MEN1 and MEN2, termed “MENX” (Fritz et al. 2002).
In 2004, the MENX locus was mapped to the distal part of rat chromosome 4 by a genome-wide linkage analysis that excluded RET, which is present on the same chromosomal region (Piotrowska, et al. 2004). In 2006, Pellegata et al. fine-mapped the locus of interest to a ~3 Mb interval on the distal part of rat chromosome 4, and suitable candidate genes were identified and sequenced, including the Cdkn1b gene encoding the p27 protein (Pellegata, et al. 2006). By that time, it was known that the Cdkn1b−/− mice developed features of overgrowth with multiple tissue hyperplasias and pituitary adenomas of the intermediate lobe (Fero, et al. 1996; Kiyokawa, et al. 1996; Nakayama, et al. 1996). Indeed, Pellegata et al. identified in these rats an 8-bp tandem duplication on exon 2 of the Cdkn1b gene (p.G177fs) leading to a homozygous frameshift mutation encoding a protein predicted to code for an elongated mutant protein with a different C-terminus than the wild-type p27 (Pellegata et al. 2006). This protein was later found to be highly unstable and therefore absent, or present at low levels, in vivo in the mutant rat. The phenotype of this rat included increased body weight and a reduced life span of 10 ±2 months when compared with wild-type (healthy homozygous or heterozygous) littermates of approximately 24–30 months of age; this syndrome was described in rats as MENX (Pellegata et al. 2006).
CDKN1B Gene Function and Cyclins
In 1994, p27 was identified in molecular complexes of cyclin dependent kinases (CDK) as a member of the CDK inhibitors (CDKI) family that regulate cell cycle progression and arrest through their inhibitory function on several cyclin/CDKIs, particularly the transition from G1 to S phase (Hengst, et al. 1994; Polyak, et al. 1994). The gene encoding the protein p27, called CDKN1B (also referred to as p27), is located on chromosome 12p13 and has two coding exons resulting in a 2.5-kb-long coding region for a nuclear protein, and one noncoding exon. The human and mouse p27 genes share similar structures in their exon–intron, with >90% sequence homology in cDNA (Philipp-Staheli et al. 2001). The U-rich element located in the 5′UTR of p27 mRNA is necessary for efficient translation of p27 in proliferating and quiescent cells (Millard, et al. 2000; Philipp-Staheli et al. 2001).
In humans, two CDKI families were identified: the INK4a/ARF and Cip/Kip family. INK4 proteins strictly inhibits and binds to CDK monomers while Cip/Kip proteins bind to both cyclin and CDK, and can be inhibitory or activating. The Cip/Kip family proteins activate cyclin D and CDK4 or CDK6 complexes. CDK4 are inhibitors and include p15 (encoded by CDKN2B), p16/p14 (encoded by CDKN2A), p18 (encoded by CDKN2C) and p19 (encoded by CDKN2D). The kinase inhibitor proteins (CIP) includes p21 (encoded by CDKN1A), p27 and p57 (encoded by CDKN1C) (Sherr and Roberts 1999).
p27 primarily inhibits cyclin E/CDK2 with high and low affinities, and undergoes inactivation at the posttranslational level by active cyclin/CDK2 complexes (Figure 1) (Sheaff, et al. 1997). p27 is regulated by ubiquitin-mediated proteasomal degradation via the mitogen-activated protein kinase (MAPK) and the phosphatidyl inositol-3 kinase (PI3K) pathways (Andreu, et al. 2005; Donovan, et al. 2001; Pagano, et al. 1995). Immunohistochemical staining of affected tissues in MEN4 did not detect expression of the p27 protein, suggesting that other mechanisms, likely posttranslational, such as phosphorylation and ubiquitination, may regulate p27 stability in these tumors. In sporadic tumors, point mutations of the CDKN1B coding region are not common (Kawamata, et al. 1995; Pietenpol, et al. 1995; Ponce-Castaneda, et al. 1995), despite LOH in some tumors (Pietenpol et al. 1995; Stegmaier, et al. 1995). In others, there is lower CDKN1B mRNA expression (Hengst and Reed 1996), and/or increased degradation (Chiappetta, et al. 2007; Pagano et al. 1995), or cytosolic mislocalization of the p27 protein (Min, et al. 2004).
Figure 1.
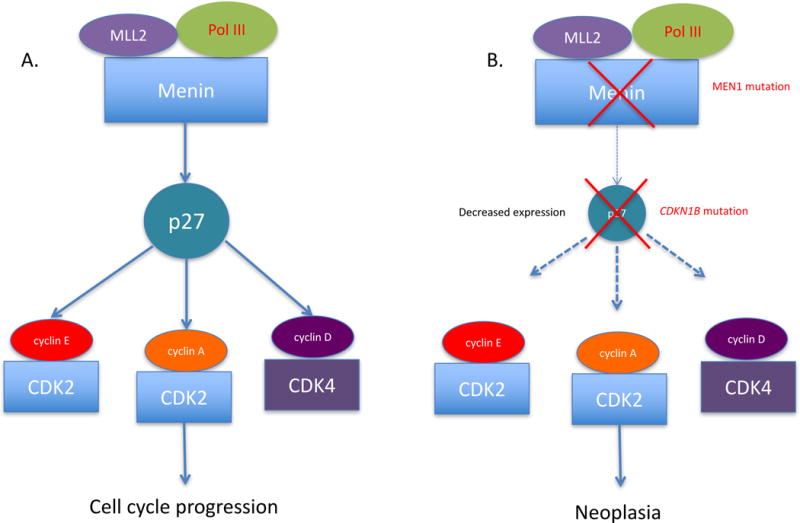
A pathway depicting the alterations in p27 expression in MEN1 and/or MEN4 that lead to tumorigenesis. Menin, encoded by MEN1, regulates the expression of p27 by forming a transcriptional activation complex with methyltransferase (MLL2) and RNA polymerase II (Pol III). Menin inactivation (MEN1 mutation) leads to decreased p27 expression. Mutations in CDKN1B, either solely or with MEN1 as a second germline hit, leads to a greater decrease in expression of p27 protein, triggering neoplasia.
The CDKN1B gene is not a classic tumor suppressor gene and does not always follow Knudson’s ‘two-mutation’ criterion for a tumor-suppressor gene (Fero, et al. 1998). Although the loss of one allele of p27 is a frequent event in many human cancers, the remaining allele is rarely mutated or lost by LOH in human cancers (Philipp-Staheli et al. 2001). In MEN1, p27 can act as a disease modifier associated with MEN1 germline mutations (Figure 1). The CDKN1B gene is transcriptionally regulated by menin through epigenetic mechanisms, such as promotion of histone modifications and maintenance of transcription at multiple loci encoding cell cycle regulators (Hughes, et al. 2004; Karnik, et al. 2005), suggesting a common pathway for tumorigenesis between MEN1 and MEN4 (Figure 1). It is known that menin regulates the expression of p27 by forming a transcriptional activation complex with methyltransferase (MLL2) and RNA polymerase II (POL III). Menin inactivation leads to decreased p27. Mutations in CDKN1B, either solely or in combination with a mutation in MEN1 lead to a greater decrease in expression of p27 protein, triggering neoplasia (Figure 1). In one study, Borsari et al. showed that MEN1 biallelic inactivation could be directly related to down-regulation of p27 expression through the inhibition of CDKN1B gene transcription (Borsari, et al. 2017).
CDKN1B mutations that were initially identified in mice and humans behave as loss-of-function mutations and occur in heterozygosity. To date, most of the reported mutations in humans were missense and not found in controls. They were deemed pathogenic due to their in vivo or in vitro effects on the function of p27. Thus, given the small number of cases reported to date (described later in this review) and the lack of segregation with the disease phenotype in the reported families, assigning a possible pathogenic role for these mutations required functional studies for confirmation. These studies demonstrated that the MEN4-associated deletions led to a truncated p27 protein that is very unstable and rapidly degraded, in part, by the proteasome (Sherr and Roberts 1999). Missense mutations, on the other hand, led to a reduced binding to interacting partners or decreased nuclear localization (Table 1) (Sherr and Roberts 1999). Overall, CDKN1B mutations causing MEN4 affect p27’s cellular localization, stability or biding with Cdk2 or Grb2 (Agarwal, et al. 2009b).
Table 1.
Established cases of MEN4. Abbreviation: ACTH, Adrenocorticotropic hormone; FIPA, familial isolated pituitary adenoma; GH, growth hormone; NET, neuroendocrine tumor; PHPT, primary hyperparathyrodism; NA, not available
| CDKN1B Mutation | Age | Sex | Race | Clinical Manifestations | Family History | Ref. |
|---|---|---|---|---|---|---|
| p.W76X (c.692G>A) |
48 | F | Caucasian | Acromegaly PHPT |
3 family members with the same p27 variant. Father had acromegaly (not tested for the p27 variant). Sister had renal angiomyolipoma (positive for same p27 variant); her son has testicular cancer | (Pellegata et al. 2006) |
| p.K25fs (c.59_77dup) |
47 | F | Caucasian | Small-cell neuroendocrine cervical carcinoma at age 45 ACTH-dependent Cushing syndrome (Cushing disease) PHPT Multiple sclerosis at age 46 |
Negative | (Georgitsi et al. 2007) |
| ATG-7G>C in the 50 –UTR | 61 | F | NA | Bilateral nonfunctional adrenal masses Uterine fibroids PHPT |
Two asymptomatic daughters (ages 47 and 48), positive for same p27 variant | (Agarwal, et al. 2009a) |
| p.P95S (c.283C>T) |
50 | F | NA | Zollinger-Ellison Syndrome with masses in duodenum and tail of pancreas PHPT |
NA | (Agarwal et al. 2009a) |
| Stop>Q (c.595T>C) |
50 | F | NA | PHPT |
Monozygotic twin sisters positive for same variant with PHPT (1 parathyroid tumor) at age 66. Aunt and cousin have PHPT, not tested for p27 |
(Agarwal et al. 2009a) |
| p.P69L (c.678C>T) |
79 | F | Caucasian | Papillary thyroid carcinoma with neck lymph node metastases Bilateral multiple lung metastatic from bronchial carcinoid PHPT Non-functioning pituitary microadenoma Subcutaneous epigastric lipoma Type 2 diabetes mellitus |
Negative | (Molatore et al. 2010) |
| p.G9R (c.25G>A) |
68 | M | Caucasian | PHPT | NA | (Costa-Guda, et al. 2011) |
| p.P133T (c.397C>A) |
58 | F | Caucasian | PHPT (1 parathyroid tumor) | NA | (Costa-Guda, et al. 2011) |
| heterozygous GAGA deletion in the 5′-UTR (ATG-32–29del) (c.32_29delGAGA) |
69 | F | Hispanic | Gastric NET PHPT |
NA | ((Malanga et al. 2012) |
| p.A55T (c.163G>A) |
42 | F | Hispanic | Zollinger-Ellison Syndrome with gastrinoma and hepatic metastases PHPT |
Negative | (Belar, et al. 2012) |
| p.S125X (c.374_375delCT) |
53 | F | Caucasian | Gastrointestinal NET PHPT |
The patient’s 35-year-old son, who had no MEN4-associated clinical features, was the first reported male mutation carrier in p27 | (Tonelli, et al. 2014 |
| p.E126D (c.378G>C) |
15 | F | Caucasian | Early-onset PHPT Recurrent renal calculi and hypercalcemia |
Mother (aged 46) and maternal grandfather (aged 74 years) carried the same missense mutation but both were normocalcemic with normal PTH levels | (Elston, et al. 2015) |
| 4-bp deletion within the 5′-UTR (c.-456_-453delCCTT) |
62 | F | Caucasian | Acromegaly (somatotropinoma) Well-differentiated nonfunctional pancreatic NET |
Negative | (Occhi et al. 2013) |
| heterozygous mutation in the 5′-UTR region (c.-29_-26delAGAG) |
6 | F | Caucasian | Gigantism (somatotropinoma) | Negative | (Sambugaro, et al. 2015) |
| p.I119T (c.356T>C) |
NA | NA | NA | AIP mutation-negative FIPA, presenting with a GH-secreting adenoma | The p.I119T change was found in one member of a two person homogeneous FIPA family with somatotropinomas | (Tichomirowa, et al. 2012). |
| p.K96Q (c.286A>C) |
NA | NA | NA |
AIP mutation-negative FIPA, presenting with hyperprolactinemia, due to a suspected prolactinoma that was treated chronically with cabergoline when referred Breast cancer at the age of 41 |
The unaffected sister of this patient was also a carrier of this variant. | (Tichomirowa, et al. 2012). |
| c.-80C>T | NA | NA | NA | PHPT | Negative | (Borsari et al. 2017). |
| c.-29_-26delAGAG | NA | NA | NA | PHPT | Negative | (Borsari et al. 2017). |
| c.397C>A | NA | NA | NA | PHPT | Negative | (Borsari et al. 2017). |
Translation of CDKN1B involves regulatory elements within its 5′UTR, such as the upstream ORF (uORF) (Gopfert, et al. 2003). Mutations in CDKN1B 5′UTR have been studied: the GAGA deletion encompassing nucleotides −32/−29 of 5′-UTR of CDKN1B significantly impairs transcription and, possibly, translation of p27 mRNA and its expression in tumor cells (Malanga, et al. 2012), while the 4-bp deletion that modifies the regulatory uORF in the 5′UTR of the CDKN1B leads to an in vivo and in vitro reduction of p27 expression (Occhi, et al. 2013). The common CDKN1B rs2066827 polymorphism (described later) may influence the clinical outcome (i.e.: tumor formation) of patients with MEN1 (Longuini, et al. 2014), a finding that should be ascertained in further studies.
MEN4 in humans
In 2008, MENX was renamed to MEN4 during the 11th International Workshop on MENs in Delphi, Greece (Alevizaki and Stratakis 2009). It was there that MEN4 was accepted as the latest member of the MEN syndromes affecting humans (Lee and Pellegata 2013). To date, only 19 cases having CDKN1B germline mutations have been reported in the medical literature (Table 1). The incidence of CDKN1B mutations in patients with a MEN1-related phenotype is difficult to estimate, but it is likely to be in the range of 1.5–3.7% (Agarwal et al. 2009b; Georgitsi, et al. 2007; Molatore, et al. 2010).
The first case of MEN4 in humans was reported in 2006 by Pellegata et al (Pellegata et al. 2006) in a 3-generation family with a negative mutation in MEN1. The family history consisted of acromegaly in the father, severe hypertension (possibly due to endocrine hypertension) in the brother who died at 39 years old, and MEN1-like features in the proband, who was a 48-year-old Caucasian female with a 3-cm somatotropinoma causing acromegaly. The pathology revealed an invasive pituitary adenoma that stained for growth hormone with a high mitotic activity and cell atypia. Later, the same patient developed PHPT, likely due to parathyroid hyperplasia. Sequencing of the CDKN1B gene showed a germline heterozygous nonsense mutation at codon 76 (c.692G>A, p.W76X), causing premature truncation of the p27 protein (Pellegata et al. 2006). Family screening showed that the proband’s older sister presented with a renal angiomyolipoma at age 55 years, with no p27 staining, and was also a carrier of the mutation (Molatore et al. 2010). Her son had testicular cancer.
Subsequently, Georgitsi et al. studied 37 patients (36 Dutch, 1 German) with a MEN1-like phenotype who did not have MEN1 gene mutations or deletions. They identified a 19-bp duplication in exon 1 (c.59_77dup, p.K25fs; heterozygous frameshift mutation at codon 25) of CDKN1B in a 47-year-old Dutch woman with a small-cell neuroendocrine cervical carcinoma that was first diagnosed at the age of 45 years and in which LOH of wild-type CDKN1B was observed. The patient also had a corticotropinoma causing Cushing disease at 46 years of age, and PHPT that was diagnosed a year later (Georgitsi et al. 2007). This second report further expanded the clinical spectrum of MEN4.
In 2009, Agarwal et al. (Agarwal et al. 2009b) identified three potentially pathogenic changes in CDKN1B (c.-7G>C; c.283C>T, p.P95S; c.595T>C, p.X199QextX*60 or stop>Q) after screening a total of 196 consecutive index cases of clear or suspected MEN1 and no identifiable germline mutations in MEN1. The c.-7G>C and c.595T>C variants showed decreased expressivity of p27 when compared to wild-type p27, while the c.283C>T variant did not affect the protein expression, but rather its ability to bind Grb2 (Agarwal et al. 2009b; Moeller, et al. 2003). The patient with the c.-7G>C variant (mutation at the −7 position in the Kozak sequence ATG-7G>C) had a parathyroid tumor, bilateral adrenocortical masses (first and only report so far of adrenal tumors in MEN4) and uterine fibroids, while the patient with the c.283C>T variant had PHPT and masses in both the duodenum and pancreas. Later in 2010, Molatore et al. extended their preliminary observation and reported a novel germline missense variant in CDKN1B (c.678C>T, p.P69L) in a 79-year-old Caucasian woman of Italian ancestry with multiple typical and metastatic bronchial NET tumors, subcutaneous epigastric lipoma, nonfunctioning pituitary microadenoma, parathyroid adenoma, and papillary thyroid carcinoma (pT1bN1M0) (Molatore et al. 2010). Subsequently, the same group studied 90 patients with presumably sporadic PHPT as the sole presentation of MEN4, and reported two novel mutations in CDKN1B. The first patient was a 68-year-old man with PHPT that had a heterozygous germline single nucleotide change at base 25 in CDKN1B exon 1 (c.25G>A, p.G9R). The second patient was a 53-year-old woman with mild PHPT and a heterozygous single nucleotide substitution (c.397C>A, pP133T) in CDKN1B (Costa-Guda, et al. 2011). A year later, Malang et al. reported on a 69-year-old female with a gastric NET and PHPT who was positive for a heterozygous GAGA deletion in the 5′-UTR of CDKN1B. This patient was found after screening 15 Spanish index cases with menin mutation-negative MEN1 (Malanga et al. 2012).
Belar et al. studied 79 different cases of sporadic and familial cases with MEN1 phenotype and identified a novel missense mutation (c.163G>A, p.A55T) in CDKN1B in a Spanish woman with a corticotropinoma, Zollinger-Ellison syndrome (ZES) with hepatic metastasis that was diagnosed at 42 years of age and PHPT at 51 years (Belar, et al. 2012). In a different report, Tonelli et al. described a 53-year-old Italian woman that had presented with PHPT and gastrointestinal NET due to a germline frameshift mutation in CDKN1B (c.371delCT) (Tonelli, et al. 2014). The patient’s 35-year-old son, who had no MEN4-associated clinical features, was the first reported male mutation carrier in CDKN1B. In 2015, Pardi et al. characterized this germline mutation in CDKN1B c.374_375delCT (p.S125X) confirming the pathogenic role of this mutation in MEN4 (Pardi, et al. 2015). Early-onset PHPT was identified in a 15-year-old girl with a germline CDKN1B variant (p.E126D) that was predicted to be damaging. There was no family history to suggest a syndromic association (Elston, et al. 2015). This report is believed to describe the youngest published case of MEN4 to date.
More recently, a number of MEN4 cases with pituitary involvement have been described. Occhi et al. identified a 4-bp deletion (c.-456_-453delCCTT) within the 5′-UTR of CDKN1B in a 62-year-old female with acromegaly and a well-differentiated non-functioning pNET (Occhi et al. 2013). Subsequently, Sambugaro et al. identified a patient with gigantism, first diagnosed at 6 years of age, and later confirmed to be due to a novel heterozygous mutation in the CDKN1B 5′-UTR region (c.-29_-26delAGAG) that led to reduction in CDKN1B mRNA levels (Sambugaro, et al. 2015). In a total of 124 affected subjects with a pituitary adenoma, Tichomirowa et al. identified two point mutations (~2% of the cases studied) in CDKN1B; p.I119T (c.356T>C) and p.K96Q (c.286A>C) in two patients from an AIP-negative FIPA family (Tichomirowa, et al. 2012). These variants altered p27 function or structure in vitro, but it should be noted that the p.K96Q variant did not segregate with pituitary adenomas in one kindred.
A recent study further expanded on the clinical and genetic spectrum of a MEN1-like syndrome. Borsari et al. studied 147 patients with typical parathyroid adenomas causing PHPT and found three germline CDKN1B variants (c.-80C>T, c.-29_-26delAGAG, c.397C>A) with reduction of CDKN1B gene transcription rate, and loss of p27 expression in the tumor carrying the c.-29_-26delAGAG variant (Borsari et al. 2017). Co-existence of MEN4 with other MEN syndromes have not been reported to date, although possible, as described in a case report of a patient with clinical findings of MEN1 (harboring a germline mutation in MEN1) and a MEN2-like phenotype (with RET polymorphisms p.G691S and p.A982C) (El-Maouche et al. 2016). Since only 19 cases of MEN4 have been reported in the medical literature, the clinical penetrance and precise tumor spectrum of MEN4 are still to be defined.
Clinical manifestations of MEN4
Although considerable overlap in the clinical manifestations of the MEN syndromes exists (Figure 2, Table 2), the relatively small number of cases reported so far does not allow conclusion to be drawn on the possible clinical differences between MEN4 and the other MEN syndromes. A recent study of 293 MEN1 mutation-positive and 30 MEN1 mutation-negative cases, all with the MEN1 phenotype showed that the mutation-negative cohort developed disease manifestations later in life with improved life expectancy (de Laat, et al. 2016). Although the mutation-negative cohort might have had MEN4, it is difficult to draw conclusion on life expectancy or penetrance of disease from this cohort. It appears that menin mutation-negative MEN1 cases should undergo a careful assessment for possible MEN4; still, confirmation of an MEN4 diagnosis should only be made with genetic testing for CDKN1B mutations.
Figure 2.
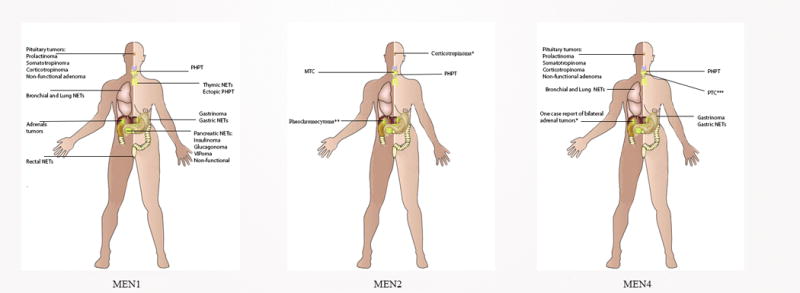
The variable clinical phenotypes of MENs.
*Recent case of Cushing Disease reported in MEN2B
**Pheochromocytomas have been described in mouse models of MEN4
***On case of metastatic papillary thyroid carcinoma
Table 2.
Phenotypic spectrum across the MEN syndromes. Abbreviation: CD, Cushing disease; MTC, Medullary Thyroid Carcinoma; NET, Neuroendocrine tumors; PHPT, primary hyperparathyroidism;PGL, paraganglioma; PTC, Papillary Thyroid Carcinoma.
| MEN Type |
Pituitary Tumors |
Acromegaly | CD | PHPT | MTC | PTC | Pheochromo cytoma |
PGL | Adrenal tumors |
Pancreatic NET |
Lung/Bronchial NET |
Gastric NET |
Thymic NET |
Gastrinoma | Skin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MEN1 | + | + | + | + | + | + | + | + | + | + | + | ||||
| MEN2A | + | + | + | + | + | ||||||||||
| MEN2B | + | + | + | + | + | ||||||||||
| MEN4 | + | + | + | + | * | ** | + | + | + | + | + |
A single case report of Cushing disease in a patient with MEN2B
A single case report of metastatic PTC in a patient with MEN4
Not yet found in humans
1. Primary Hyperparathyroidism (PHPT)
PHPT due to parathyroid neoplasia affects approximately 80% (15/19 cases, Table 1) of the reported cases of MEN4 to date. PHPT occurs at a later age in MEN4 than in MEN1 (mean age ~56 years vs. ~25 years, respectively) with a female predominance (Lee and Pellegata 2013). Interestingly, none of the reported cases of MEN4 to date had PHPT recurrence after surgical resection, which might indicate that PHPT in MEN4 might represent an overall milder disease spectrum than MEN1. The indications for parathyroid surgery in MEN4 are the same as for MEN1, although there are no specific guidelines to date on management of PHPT in MEN4. The surgical approach in MEN4-related PPHT should be individualized; some patients may be treated as in MEN1, and undergo three-and-a-half gland resection with close follow up for disease recurrence.
2. Pituitary Adenomas
Pituitary involvement in MEN4 is the second most common manifestation of the disease, affecting approximately 37% of the reported cases to date (7/19 cases, Table 1). The types of pituitary adenomas in MEN4 vary, including non-functional, somatotropinoma, prolactinoma or corticotropinoma. The age of diagnosis for these lesions also varies widely, from 30–79 years (see Table 2). In general, pituitary tumors in MEN4 are present with reduced aggressiveness, but may exert a variable degree of morbidity depending on the hormonal functional status, size, and presence of invasion or mitotic index. Conversely, pituitary adenomas are characterized by a larger size and a more aggressive presentation in MEN1.
Gigantism or acromegaly is reported in MEN4 (Crona, et al. 2015; Occhi et al. 2013; Sambugaro et al. 2015). Mutations in CDKN1B in sporadic gigantism or acromegaly and among pediatric patients with pituitary adenomas appear to be very rare (Schernthaner-Reiter, et al. 2016; Stratakis, et al. 2010). Genetic alterations of CDKN1B in somatotropinoma, corticotropinoma, non-functioning pituitary adenomas and sporadic pNET are also very infrequent (Dahia, et al. 1998; Ikeda, et al. 1997; Lee and Pellegata 2013; Lindberg, et al. 2007; Stratakis et al. 2010; Takeuchi, et al. 1998). Cushing disease has been reported in MEN4 due to a heterozygous 19-bp duplication (c.59_77dup19) in CDKN1B, leading to a truncated protein (Georgitsi et al. 2007). It should be noted that corticotropinomas are also observed in MEN1 (Verges, et al. 2002; Stratakis et al. 2010;), and very rarely in MEN2B (Kasturi, et al. 2017). Interestingly, one study found the common CDKN1B rs2066827 polymorphism to play a role in corticotropinoma susceptibility and tumorigenesis through a yet unidentified mechanism or, maybe, epigenetic factors (Sekiya, et al. 2014). The management of pituitary tumors in MEN4 is like other sporadic or familial cases. Routine surveillance for the development of pituitary tumors in patients with MEN4 should be performed on a case-by-case basis and following existing guidelines for other MENs.
3. Neuroendocrine duodeno-pancreatic tumors
Only a few cases of NETs in the context of MEN4 have been reported to date (7/19 cases, Table 1). These include duodeno- or gastric-pNETs, that could be nonfunctioning or hormonally active and may secrete several substances, including gastrin, insulin, ACTH, or vasoactive intestinal polypeptide (VIP). NETs in MEN may be associated with various clinical syndromes. Gastrin-secreting tumors (gastrinomas) lead to peptic and gastric ulcerations due to excess release of gastrin levels and are the leading cause of NET in MEN1. The clinical syndrome associated with this is ZES, which has been reported in two cases of MEN4 (Table 1). In MEN4, there are no reported cases of insulinoma, VIPoma, glucaconoma, ectopic-ACTH secreting NET or malignant transformation of pNETs. In MEN1, NETs are usually multiple with an uncertain behavior. It appears that there is a decreased penetrance of pNETs in MEN4 when compared to MEN1. The diagnosis and management of pNETs in MEN4 is similar to that in MEN1 (Thakker et al. 2012).
4. Adrenal neoplasia
Adrenal neoplasia is a frequent finding in MEN1 but figures for MEN4 are not available. MEN1 also predisposes to primary aldosteronism (Beckers, et al. 1992), bilateral adrenal non-functional nodular hyperplasia (Gatta-Cherifi, et al. 2012), primary bilateral adrenocortical hyperplasia (PBMAH) (Stratakis and Boikos 2007), and adrenocortical cancer (Gatta-Cherifi et al. 2012), which has not been reported in MEN4. While adrenal tumors are found in mouse MENX, only one case of non-functional bilateral adrenal nodules was reported in MEN4 (Table 2). Routine surveillance for the development of ACTH-independent Cushing syndrome, primary aldosteronism or pheochromocytoma [only reported in rats with MENX (Molatore, et al. 2010)] in patients with MEN4 should be performed on a case-by-case basis.
5. Other
Testicular cancer and neuroendocrine cervical carcinoma have been reported in MEN1 but not in MEN4. Likewise, skin manifestations that are commonly reported in MEN1, such as lipomas, angiofibromas, collagenomas have not been reported in MEN4. Finally, CDKN1B has been implicated in primary ovarian insufficiency: one study found two nonsynonymous variants of CDKN1B; p.V109G, a polymorphism, and p.I119T, a mutation with a potential deleterious effect requiring functional studies for confirmation (Ojeda, et al. 2011).
CDKI and Human Neoplasia
Endocrine tumors have been associated with over-expression of cyclins and/or loss of function of CDKI. Importantly, CDKN1B and CDKN2C are transcriptional gene targets of menin (Milne, et al. 2005). In a study of 196 patients with MEN1 (but no MEN1 gene mutations), the relative frequency of the various CDKI mutations were 1%, 0.5%, 0.5% and 1.5% for p15 (CDKN2B), p18 (CDKN2C), p21 (CDKN1A) and p27, respectively (Agarwal et al. 2009b). This report was the first to document a missense variant (p.V31L) in CDKN2C in an endocrinopathy. Other authors have confirmed the very rare or non-existent association between CDKN2C and parathyroid neoplasia (Tahara, et al. 1997) and a frequent promoter methylation of CDKN2C in pituitary adenomas (Kirsch, et al. 2009). Moreover, CDKN2C has been found significantly under-expressed in various pituitary tumors, including corticotropinomas, prolactinomas, and somatotropinomas (Hossain, et al. 2009; Kirsch et al. 2009; Morris, et al. 2005). In most patients with atypical manifestations of MEN1, the disease seems to occur due to genetic causes other than CDKN1B, implicating other genetic alternations that are yet to be identified (Ozawa, et al. 2007).
Somatic changes in CDKN1B without mutations in the remaining wild-type allele, as well as the reduced expression of p27 protein without CDKN1B mutations in various human tumors support the concept that CDKN1B is a likely tumor suppressor gene that confers tumorigenicity in haploinsufficiency. Nonsense mutations were discovered in adult T-cell leukemia/lymphoma (p.W76X) (Morosetti, et al. 1995) and breast cancer (p.Q104X) (Spirin, et al. 1996), while a missense change (p.I119T) was found in a myeloproliferative disorder (Pappa, et al. 2005). Somatic alterations in CDKN1B represent the second most common mutated gene in hairy-cell leukemia (Dietrich, et al. 2015). Other studies have identified recurrent somatic frameshift mutations and deletions in CDKN1B of small intestinal NET with inter and intratumor heterogeneity (Crona et al. 2015; Francis, et al. 2013), supporting its role as a haploinsufficient tumor suppressor gene. Several potentially functional single nucleotide polymorphisms (SNPs; −838C>A, −79C>T and 326T>G) were associated with a variety of human cancers including prostate, breast and thyroid cancer (Landa, et al. 2010). One study found that the CDKN1B rs2066827 polymorphism may be associated with decreased susceptibility to ovarian cancer (Lu, et al. 2015).
It is known that downregulation of p27, through a mechanism that enhances proteasome-mediated degradation, is associated with tumor progression, aggressiveness, poor clinical outcome and decreased survival in these malignancies (Chu, et al. 2008; Lloyd, et al. 1999). Oncogenic activation of phosphatidylinositol 3-kinase (PI3K) (Liang and Slingerland 2003), proto-oncogene tyrosine-protein kinase Src, or MAPKs inactivate p27 or accelerate its proteolysis in human cancers (Busse, et al. 2000; Donovan et al. 2001). These findings highlight the potential role of tyrosine kinase inhibitors for the management of aggressive NETs associated with MEN4 or somatic p27 mutations.
Genetic Testing and Counseling for MEN4
The identification of the genes responsible for MEN1, MEN2 and MEN4 has enabled the genetic diagnosis and, thus, early detection of patients with suspected endocrine tumor syndromes. Genetic testing in clinical practice for affected patients and their families with MENs has now become routine. In addition, sequencing for MEN1 and RET is now included in clinical exome and genome sequencing of other cases and reported as secondary findings (Kalia, et al. 2017).
Genetic screening is useful in identifying carriers or at risk family members who can then be monitored for the clinical manifestations of the respective syndrome(s). Negative genetic testing offers reassurance to those who do not carry the mutation and prevents unnecessary screening. Preimplantation and/or prenatal screening may also be offered. Patient and family counseling should incorporate patient’s values and attitudes toward their disease, underscoring the risks and benefits of genetic screening and counseling, psychosocial interventions, and service delivery. An experienced genetic counselor and team should provide a comprehensive assessment, including education and discussions on preventing and screening options.
As we mentioned above, since the identification of mutations in CDKN1B as causative for MEN4, only 19 cases have been reported in the medical literature (Table 1). Thus, guidelines and recommendations for MEN4 are lacking and difficult to formulate given the paucity of cases described in the literature. Moreover, the limited analysis of relatives with CDKN1B that do not manifest with any signs or symptoms suggestive of MEN4 is consistent with an incomplete penetrance of the disease.
In clinical practice, if a clinician encounters patients with asymptomatic or symptomatic PHPT that are young (typically <30 years old), with multigland disease, parathyroid carcinoma, or atypical adenoma, or those with a family history or evidence of syndromic disease and negative for MEN1 or MEN2, genetic testing for CDKN1B should be pursued. However, no guidelines exist. As we already know from MEN2, the genetic status of suspected pre-symptomatic patients provides survival benefits based on preemptive management of the potential morbidities (Brandi et al. 2001). On the other hand, MEN1-related tumors have no effective prevention except for prophylactic thymectomy for thymic NET (Brandi et al. 2001; Thakker et al. 2012).
An approach to screening in MEN4 is outlined in Figure 3. Index cases, or individuals with MEN1-like features and negative MEN1 testing should be offered genetic counseling and testing for MEN4 (CDKN1B) in accredited laboratories. Screening should also be offered to a first-degree relative with or without MEN1 features. The identification of a germline CDKN1B mutation should prompt periodic clinical, biochemical, and radiological screening for MEN4. Mutations leading to the MEN4 phenotype are transmitted in an autosomal dominant fashion, and each sibling has a 50% risk of having the mutation. If neither parent carries the mutation, the risk to siblings is low, but the possibility of germline mosaicism or de novo mutations exists. It is advisable to refer the patient and/or family members to a tertiary center with expertise in these rare conditions. It is also important for the proband or at risk family members to receive genetic counseling and testing for risk stratification of affected and unaffected individuals.
Figure 3.
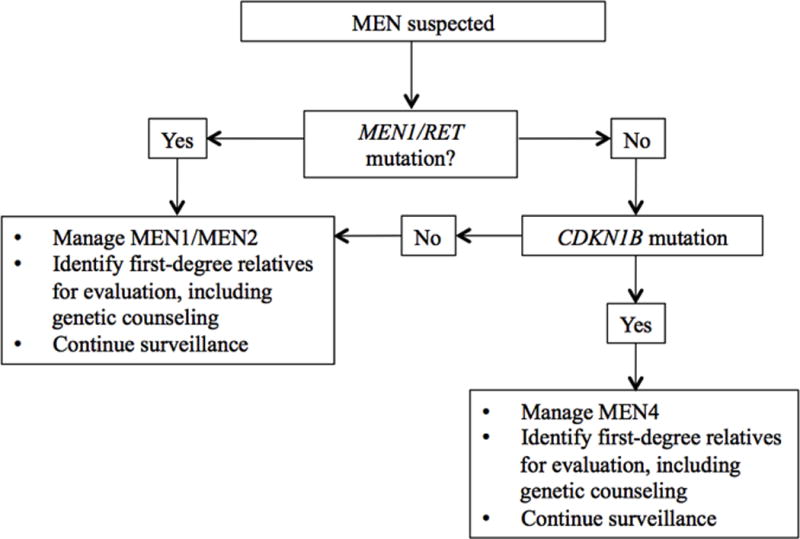
An approach to screening in MEN4. Index cases, or individuals with MEN1-like features and negative MEN1 testing should be offered genetic counseling and testing for MEN4 (CDKN1B). Screening should also be offered to a first-degree relative with or without MEN1 features. The identification of a germline CDKN1B mutation should prompt periodic clinical, biochemical, and radiological screening for MEN4.
Summary
Over the last two decades, significant progress has been made in the understanding of the molecular and genetic mechanisms of tumor pathogenesis in MENs. In this review, we presented the genetic and clinical features of MEN4, being the newest member to join the MEN family of conditions. MEN4 is a rare syndrome with clinical features that overlap with the other MENs. The discovery of CDKN1B mutations that cause MEN4 enabled personalized approaches to diagnosis, risk stratification, and appropriate treatment for individuals with MEN and other sporadic tumors affecting various organs and systems. Index cases, or individuals with MEN1-like features and negative MEN1 testing should be offered genetic counseling and testing for MEN4. Screening should also be offered to a first-degree relative with or without MEN1 features. The identification of a germline CDKN1B mutation should prompt periodic clinical, biochemical, and radiological screening for MEN4. CDKN1B somatic mutations are frequent in NETs and other non-endocrine neoplasms pointing to potential use of this knowledge in molecularly targeted therapies for these lesions.
Selected Accredited Laboratories for MEN Molecular analysis.
United States
-
Esoterix Molecular Endocrinology, Calabasas Hills, California
-
Molecular Genetics Laboratory, Emory, Atlanta, Georgia
-
Molecular Genetics Laboratory, Mayo Clinic, Rochester, Minnesota
-
PreventionGenetics, Marshfield, Wisconsin
-
Quest Diagnostics, Nichols Institute, San Juan Capistrano, California
-
GeneDx, Gaithersburg, Maryland
Europe
-
Reference Laboratory Genetics, Spain
-
Service Hormonologie Métabolisme Nutrition Oncologie, Institut de biochimie et biologie moléculaire, CHRU de Lille - Centre de Biologie Pathologie Génétique, France
-
Pränatal-Medizin München, Germany
-
Molecular Genetics Laboratory, Oxford Medical Genetics Laboratories, The Churchill Hospital, England
www.ouh.nhs.uk/services/referrals/genetics/genetics-laboratories
Acknowledgments
Funding statement:
Funded by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health (project number Z1A HD008920).
Footnotes
Conflict of Interest Statement: The authors declare that the research was conducted in absence of any potential conflict of interest.
Disclosure
The authors have no multiplicity of interest to disclose.
References
- Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR, Skarulis MC, Doppman JL, Kim YS, Lubensky IA, Zhuang Z, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet. 1997;6:1169–1175. doi: 10.1093/hmg/6.7.1169. [DOI] [PubMed] [Google Scholar]
- Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009a;94 doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009b;94:1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alevizaki M, Stratakis CA. Multiple endocrine neoplasias: advances and challenges for the future. J Intern Med. 2009;266:1–4. doi: 10.1111/j.1365-2796.2009.02108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu EJ, Lledo E, Poch E, Ivorra C, Albero MP, Martinez-Climent JA, Montiel-Duarte C, Rifon J, Perez-Calvo J, Arbona C, et al. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res. 2005;65:3264–3272. doi: 10.1158/0008-5472.CAN-04-1357. [DOI] [PubMed] [Google Scholar]
- Beckers A, Abs R, Willems PJ, van der Auwera B, Kovacs K, Reznik M, Stevenaert A. Aldosterone-secreting adrenal adenoma as part of multiple endocrine neoplasia type 1 (MEN1): loss of heterozygosity for polymorphic chromosome 11 deoxyribonucleic acid markers, including the MEN1 locus. J Clin Endocrinol Metab. 1992;75:564–570. doi: 10.1210/jcem.75.2.1639957. [DOI] [PubMed] [Google Scholar]
- Belar O, De La Hoz C, Perez-Nanclares G, Castano L, Gaztambide S, Spanish MENG. Novel mutations in MEN1, CDKN1B and AIP genes in patients with multiple endocrine neoplasia type 1 syndrome in Spain. Clin Endocrinol (Oxf) 2012;76:719–724. doi: 10.1111/j.1365-2265.2011.04269.x. [DOI] [PubMed] [Google Scholar]
- Borsari S, Pardi E, Pellegata NS, Lee M, Saponaro F, Torregrossa L, Basolo F, Paltrinieri E, Zatelli MC, Materazzi G, et al. Loss of p27 expression is associated with MEN1 gene mutations in sporadic parathyroid adenomas. Endocrine. 2017;55:386–397. doi: 10.1007/s12020-016-0941-6. [DOI] [PubMed] [Google Scholar]
- Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- Busse D, Doughty RS, Ramsey TT, Russell WE, Price JO, Flanagan WM, Shawver LK, Arteaga CL. Reversible G (1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J Biol Chem. 2000;275:6987–6995. doi: 10.1074/jbc.275.10.6987. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chiappetta G, De Marco C, Quintiero A, Califano D, Gherardi S, Malanga D, Scrima M, Montero-Conde C, Cito L, Monaco M, et al. Overexpression of the S-phase kinase-associated protein 2 in thyroid cancer. Endocr Relat Cancer. 2007;14:405–420. doi: 10.1677/ERC-06-0030. [DOI] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Costa-Guda J, Marinoni I, Molatore S, Pellegata NS, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2011;96:E701–706. doi: 10.1210/jc.2010-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crona J, Gustavsson T, Norlen O, Edfeldt K, Akerstrom T, Westin G, Hellman P, Bjorklund P, Stalberg P. Somatic Mutations and Genetic Heterogeneity at the CDKN1B Locus in Small Intestinal Neuroendocrine Tumors. Ann Surg Oncol. 2015;22(Suppl 3):S1428–1435. doi: 10.1245/s10434-014-4351-9. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Aguiar RC, Honegger J, Fahlbush R, Jordan S, Lowe DG, Lu X, Clayton RN, Besser GM, Grossman AB. Mutation and expression analysis of the p27/kip1 gene in corticotrophin-secreting tumours. Oncogene. 1998;16:69–76. doi: 10.1038/sj.onc.1201516. [DOI] [PubMed] [Google Scholar]
- de Laat JM, van der Luijt RB, Pieterman CR, Oostveen MP, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML, Bisschop PH, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med. 2016;14:182. doi: 10.1186/s12916-016-0708-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich S, Hullein J, Lee SC, Hutter B, Gonzalez D, Jayne S, Dyer MJ, Oles M, Else M, Liu X, et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood. 2015;126:1005–1008. doi: 10.1182/blood-2015-04-643361. [DOI] [PubMed] [Google Scholar]
- Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276:40888–40895. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- El-Maouche D, Welch J, Agarwal SK, Weinstein LS, Simonds WF, Marx SJ. A patient with MEN1 typical features and MEN2-like features. Int J Endocr Oncol. 2016;3:89–95. doi: 10.2217/ije-2015-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston MS, Meyer-Rochow GY, Dray M, Swarbrick M, Conaglen JV. Early Onset Primary Hyperparathyroidism Associated with a Novel Germline Mutation in CDKN1B. Case Rep Endocrinol. 20152015:510985. doi: 10.1155/2015/510985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, Banck MS, Kanwar R, Kulkarni AA, Karpathakis A, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013;45:1483–1486. doi: 10.1038/ng.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000;20:6147–6158. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz A, Walch A, Piotrowska K, Rosemann M, Schaffer E, Weber K, Timper A, Wildner G, Graw J, Hofler H, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 2002;62:3048–3051. [PubMed] [Google Scholar]
- Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, Young J, Delemer B, Du Boullay H, Verger MF, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d’etude des Tumeurs Endocrines database. Eur J Endocrinol. 2012;166:269–279. doi: 10.1530/EJE-11-0679. [DOI] [PubMed] [Google Scholar]
- Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, Vierimaa O, Makinen MJ, Tuppurainen K, Paschke R, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. 2007;92:3321–3325. doi: 10.1210/jc.2006-2843. [DOI] [PubMed] [Google Scholar]
- Gopfert U, Kullmann M, Hengst L. Cell cycle-dependent translation of p27involves a responsive element in its 5′-UTR that overlaps with a uORF. Hum Mol Genet. 2003;12:1767–1779. doi: 10.1093/hmg/ddg177. [DOI] [PubMed] [Google Scholar]
- Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994;91:5291–5295. doi: 10.1073/pnas.91.12.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- Hossain MG, Iwata T, Mizusawa N, Qian ZR, Shima SW, Okutsu T, Yamada S, Sano T, Yoshimoto K. Expression of p18(INK4C) is down-regulated in human pituitary adenomas. Endocr Pathol. 2009;20:114–121. doi: 10.1007/s12022-009-9076-0. [DOI] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Yoshimoto T, Shida N. Molecular analysis of p21 and p27 genes in human pituitary adenomas. Br J Cancer. 1997;76:1119–1123. doi: 10.1038/bjc.1997.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasturi K, Fernandes L, Quezado M, Eid M, Marcus L, Chittiboina P, Rappaport M, Stratakis CA, Widemann B, Lodish M. Cushing Disease in a patient with Multiple Endocrine Neoplasia type 2B. J Clin Transl Endocrinol Case Rep. 2017;4:1–4. doi: 10.1016/j.jecr.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S, et al. Molecular analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in human malignancies. Cancer Res. 1995;55:2266–2269. [PubMed] [Google Scholar]
- Kirsch M, Morz M, Pinzer T, Schackert HK, Schackert G. Frequent loss of the CDKN2C (p18INK4c) gene product in pituitary adenomas. Genes Chromosomes Cancer. 2009;48:143–154. doi: 10.1002/gcc.20621. [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Landa I, Montero-Conde C, Malanga D, De Gisi S, Pita G, Leandro-Garcia LJ, Inglada-Perez L, Leton R, De Marco C, Rodriguez-Antona C, et al. Allelic variant at −79 (C>T) in CDKN1B (p27Kip1) confers an increased risk of thyroid cancer and alters mRNA levels. Endocr Relat Cancer. 2010;17:317–328. doi: 10.1677/ERC-09-0016. [DOI] [PubMed] [Google Scholar]
- Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- Lee M, Pellegata NS. Multiple endocrine neoplasia type 4. Front Horm Res. 2013;41:63–78. doi: 10.1159/000345670. [DOI] [PubMed] [Google Scholar]
- Liang J, Slingerland JM. Multiple roles of the pI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- Lindberg D, Akerstrom G, Westin G. Mutational analysis of p27(CDKN1B) and p18(CDKN2C) in sporadic pancreatic endocrine tumors argues against tumor-suppressor function. Neoplasia. 2007;9:533–535. doi: 10.1593/neo.07328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RV, Erickson LA, Jin L, Kulig E, Qian X, Cheville JC, Scheithauer BW. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol. 1999;154:313–323. doi: 10.1016/S0002-9440(10)65277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longuini VC, Lourenco DM, Jr, Sekiya T, Meirelles O, Goncalves TD, Coutinho FL, Francisco G, Osaki LH, Chammas R, Alves VA, et al. Association between the p27rs2066827 variant and tumor multiplicity in patients harboring MEN1 germline mutations. Eur J Endocrinol. 2014;171:335–342. doi: 10.1530/EJE-14-0130. [DOI] [PubMed] [Google Scholar]
- Lu Y, Gao K, Zhang M, Zhou A, Zhou X, Guan Z, Shi X, Ge S. Genetic Association Between CDKN1B rs2066827 Polymorphism and Susceptibility to Cancer. Medicine (Baltimore) 2015;94:e1217. doi: 10.1097/MD.0000000000001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanga D, De Gisi S, Riccardi M, Scrima M, De Marco C, Robledo M, Viglietto G. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol. 2012;166:551–560. doi: 10.1530/EJE-11-0929. [DOI] [PubMed] [Google Scholar]
- Millard SS, Vidal A, Markus M, Koff A. A U-rich element in the 5′ untranslated region is necessary for the translation of p27 mRNA. Mol Cell Biol. 2000;20:5947–5959. doi: 10.1128/mcb.20.16.5947-5959.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS, Ko YW, Lee MH. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004;64:5225–5231. doi: 10.1158/0008-5472.CAN-04-0174. [DOI] [PubMed] [Google Scholar]
- Moeller SJ, Head ED, Sheaff RJ. p27Kip1 inhibition of GRB2-SOS formation can regulate Ras activation. Mol Cell Biol. 2003;23:3735–3752. doi: 10.1128/MCB.23.11.3735-3752.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molatore S, Marinoni I, Lee M, Pulz E, Ambrosio MR, degli Uberti EC, Zatelli MC, Pellegata NS. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat. 2010;31:E1825–1835. doi: 10.1002/humu.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morosetti R, Kawamata N, Gombart AF, Miller CW, Hatta Y, Hirama T, Said JW, Tomonaga M, Koeffler HP. Alterations of the p27KiP1 gene in non-Hodgkin’s lymphomas and adult T-cell leukemia/lymphoma. Blood. 1995;86:1924–1930. [PubMed] [Google Scholar]
- Morris DG, Musat M, Czirjak S, Hanzely Z, Lillington DM, Korbonits M, Grossman AB. Differential gene expression in pituitary adenomas by oligonucleotide array analysis. Eur J Endocrinol. 2005;153:143–151. doi: 10.1530/eje.1.01937. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Namihira H, Sato M, Matsubara S, Ohye H, Bhuiyan M, Murao K, Takahara J. No evidence of germline mutation or somatic deletion of the MEN1 gene in a case of familial multiple endocrine neoplasia type 1 (MEN1) Endocr J. 1999;46:811–816. doi: 10.1507/endocrj.46.811. [DOI] [PubMed] [Google Scholar]
- Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, Ferasin S, Cetani F, Pardi E, Korbonits M, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. 2013;9:e1003350. doi: 10.1371/journal.pgen.1003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda D, Lakhal B, Fonseca DJ, Braham R, Landolsi H, Mateus HE, Restrepo CM, Elghezal H, Saad A, Laissue P. Sequence analysis of the CDKN1B gene in patients with premature ovarian failure reveals a novel mutation potentially related to the phenotype. Fertil Steril. 2011;95:2658–2660 e2651. doi: 10.1016/j.fertnstert.2011.04.045. [DOI] [PubMed] [Google Scholar]
- Ozawa A, Agarwal SK, Mateo CM, Burns AL, Rice TS, Kennedy PA, Quigley CM, Simonds WF, Weinstein LS, Chandrasekharappa SC, et al. The parathyroid/pituitary variant of multiple endocrine neoplasia type 1 usually has causes other than p27Kip1 mutations. J Clin Endocrinol Metab. 2007;92:1948–1951. doi: 10.1210/jc.2006-2563. [DOI] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Pappa V, Papageorgiou S, Papageorgiou E, Panani A, Boutou E, Tsirigotis P, Dervenoulas J, Economopoulos T, Raptis S. A novel p27 gene mutation in a case of unclassified myeloproliferative disorder. Leuk Res. 2005;29:229–231. doi: 10.1016/j.leukres.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Pardi E, Mariotti S, Pellegata NS, Benfini K, Borsari S, Saponaro F, Torregrossa L, Cappai A, Satta C, Mastinu M, et al. Functional characterization of a CDKN1B mutation in a Sardinian kindred with multiple endocrine neoplasia type 4 (MEN4) Endocr Connect. 2015;4:1–8. doi: 10.1530/EC-14-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103:15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001;264:148–168. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Bohlander SK, Sato Y, Papadopoulos N, Liu B, Friedman C, Trask BJ, Roberts JM, Kinzler KW, Rowley JD, et al. Assignment of the human p27Kip1 gene to 12p13 and its analysis in leukemias. Cancer Res. 1995;55:1206–1210. [PubMed] [Google Scholar]
- Piotrowska K, Pellegata NS, Rosemann M, Fritz A, Graw J, Atkinson MJ. Mapping of a novel MEN-like syndrome locus to rat chromosome 4. Mamm Genome. 2004;15:135–141. doi: 10.1007/s00335-003-3027-8. [DOI] [PubMed] [Google Scholar]
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- Ponce-Castaneda MV, Lee MH, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew S, Krauter K, Sheinfeld J, Massague J, et al. p27Kip1: chromosomal mapping to 12p12–12p13.1 and absence of mutations in human tumors. Cancer Res. 1995;55:1211–1214. [PubMed] [Google Scholar]
- Sambugaro S, Di Ruvo M, Ambrosio MR, Pellegata NS, Bellio M, Guerra A, Buratto M, Foschini MP, Tagliati F, degli Uberti E, et al. Early onset acromegaly associated with a novel deletion in CDKN1B 5′UTR region. Endocrine. 2015;49:58–64. doi: 10.1007/s12020-015-0540-y. [DOI] [PubMed] [Google Scholar]
- Schernthaner-Reiter MH, Trivellin G, Stratakis CA. MEN1, MEN4, and Carney Complex: Pathology and Molecular Genetics. Neuroendocrinology. 2016;103:18–31. doi: 10.1159/000371819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T, Bronstein MD, Benfini K, Longuini VC, Jallad RS, Machado MC, Goncalves TD, Osaki LH, Higashi L, Viana J, Jr, et al. p27 variant and corticotropinoma susceptibility: a genetic and in vitro study. Endocr Relat Cancer. 2014;21:395–404. doi: 10.1530/ERC-13-0486. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler HP. p27/Kip1 mutation found in breast cancer. Cancer Res. 1996;56:2400–2404. [PubMed] [Google Scholar]
- Stegmaier K, Pendse S, Barker GF, Bray-Ward P, Ward DC, Montgomery KT, Krauter KS, Reynolds C, Sklar J, Donnelly M, et al. Frequent loss of heterozygosity at the TEL gene locus in acute lymphoblastic leukemia of childhood. Blood. 1995;86:38–44. [PubMed] [Google Scholar]
- Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab. 2007;3:748–757. doi: 10.1038/ncpendmet0648. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, Verma S, Daly AF, Raygada M, Keil MF, et al. The role of germline AIP, MEN1, pRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet. 2010;78:457–463. doi: 10.1111/j.1399-0004.2010.01406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara H, Smith AP, Gaz RD, Zariwala M, Xiong Y, Arnold A. Parathyroid tumor suppressor on 1p: analysis of the p18 cyclin-dependent kinase inhibitor gene as a candidate. J Bone Miner Res. 1997;12:1330–1334. doi: 10.1359/jbmr.1997.12.9.1330. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Koeffler HP, Hinton DR, Miyoshi I, Melmed S, Shimon I. Mutation and expression analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in pituitary tumors. J Endocrinol. 1998;157:337–341. doi: 10.1677/joe.0.1570337. [DOI] [PubMed] [Google Scholar]
- Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97:2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- Tichomirowa MA, Lee M, Barlier A, Daly AF, Marinoni I, Jaffrain-Rea ML, Naves LA, Rodien P, Rohmer V, Faucz FR, et al. Cyclin-dependent kinase inhibitor 1B (CDKN1B) gene variants in AIP mutation-negative familial isolated pituitary adenoma kindreds. Endocr Relat Cancer. 2012;19:233–241. doi: 10.1530/ERC-11-0362. [DOI] [PubMed] [Google Scholar]
- Tonelli F, Giudici F, Giusti F, Marini F, Cianferotti L, Nesi G, Brandi ML. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. 2014;171:K7–K17. doi: 10.1530/EJE-14-0080. [DOI] [PubMed] [Google Scholar]
- Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, Cougard P, Chambe B, Montvernay C, Calender A. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab. 2002;87:457–465. doi: 10.1210/jcem.87.2.8145. [DOI] [PubMed] [Google Scholar]