

Abstract
Biomarkers have transformed modern medicine but remain largely elusive in psychiatry, partly because there is a weak correspondence between diagnostic labels and their neurobiological substrates. Like other neuropsychiatric disorders, depression is not a unitary disease, but rather a heterogeneous syndrome that encompasses varied, co-occurring symptoms and divergent responses to treatment. By using functional magnetic resonance imaging (fMRI) in a large multisite sample (n = 1,188), we show here that patients with depression can be subdivided into four neurophysiological subtypes (‘biotypes’) defined by distinct patterns of dysfunctional connectivity in limbic and frontostriatal networks. Clustering patients on this basis enabled the development of diagnostic classifiers (biomarkers) with high (82–93%) sensitivity and specificity for depression subtypes in multisite validation (n = 711) and out-of-sample replication (n = 477) data sets. These biotypes cannot be differentiated solely on the basis of clinical features, but they are associated with differing clinical-symptom profiles. They also predict responsiveness to transcranial magnetic stimulation therapy (n = 154). Our results define novel subtypes of depression that transcend current diagnostic boundaries and may be useful for identifying the individuals who are most likely to benefit from targeted neurostimulation therapies.
Depression is a heterogeneous clinical syndrome that is diagnosed when a patient reports at least five of nine symptoms. This allows for several hundred unique combinations of changes in mood, appetite, sleep, energy, cognition and motor activity. Such remarkable heterogeneity reflects the consensus view that there are multiple forms of depression, but their neurobiological basis remains poorly understood1,2. So far, most efforts to characterize depression subtypes and develop diagnostic biomarkers have begun by identifying clusters of symptoms that tend to co-occur, and by then testing for neurophysiological correlates. These pioneering studies have defined atypical, melancholic, seasonal and agitated subtypes of depression associated with characteristic changes in neuroendocrine activity, circadian rhythms and other potential biomarkers3–5. Still, the association between clinical subtypes and their biological substrates is inconsistent and variable at the individual level, and unlike diagnostic biomarkers in other areas of medicine, they have not yet proven useful for differentiating individual patients from healthy controls or for reliably predicting treatment response at the individual level.
An alternative to subtyping patients on the basis of co-occurring clinical symptoms is to identify neurophysiological subtypes, or biotypes, by clustering subjects according to shared signatures of brain dysfunction6. This type of approach has already begun to yield insights into how differing biological mechanisms may give rise to overlapping, heterogeneous clinical presentations of psychotic disorders6,7. Neuroimaging biomarkers of abnormal brain function have proven utility in the assessment of pain8 and have also shown promise for depression, for both the prediction of treatment response9–13 and treatment selection14. Resting-state fMRI (rsfMRI) is an especially useful modality because it can be used easily in diverse patient populations to quantify functional network connectivity in terms of correlated, spontaneous MR signal fluctuations. Depression is associated with dysfunction and abnormal functional connectivity in frontostriatal and limbic brain networks15–20, in accordance with morphological and synaptic changes in chronic stress models in rodents21–24. These studies raise the intriguing possibility that fMRI measures of connectivity could be leveraged to identify novel subtypes of depression with stronger neurobiological correlates that predict treatment responsiveness.
To this end, we developed a method for defining depression subtypes by clustering subjects according to distinct, whole-brain patterns of abnormal functional connectivity in resting-state networks, unbiased by assumptions about the involvement of particular brain regions, and tested it in a large, multisite data set. Our analyses revealed four biotypes that were defined by homogeneous patterns of dysfunctional connectivity in frontostriatal and limbic networks, and that could be diagnosed with high sensitivity and specificity in individual subjects. Importantly, these biotypes were also prognostically informative, predicting which patients responded to repetitive transcranial magnetic stimulation (TMS), a targeted neurostimulation therapy.
RESULTS
Frontostriatal and limbic connectivity define four depression biotypes
We began by designing and implementing a preprocessing procedure (Online Methods) to control for motion-, scanner- and age-related effects in a multisite data set that comprised rsfMRI scans for 711 subjects (the ‘training data set’, n = 333 patients with depression; n = 378 healthy controls). No subjects had comorbid substance-abuse disorders, and patients and controls were matched for age and sex. Data that support our approach to controlling for motion-related Blood-oxygen-level dependent (BOLD) signal effects, a particularly important source of rsfMRI artifact25–27, are presented in Supplementary Figure 1. After co-registering the functional volumes to a common (Montreal Neurological Institute (MNI)) space, we applied an extensively validated parcellation system28 to delineate 258 functional network nodes that spanned the whole brain and had stable signals across all sites and scans in this data set (Fig. 1a). Next, we extracted BOLD signal residual time series and calculated correlation matrices between each node, which provided an unbiased estimate of the whole-brain architecture of functional connectivity in each subject (Fig. 1b).
Figure 1.
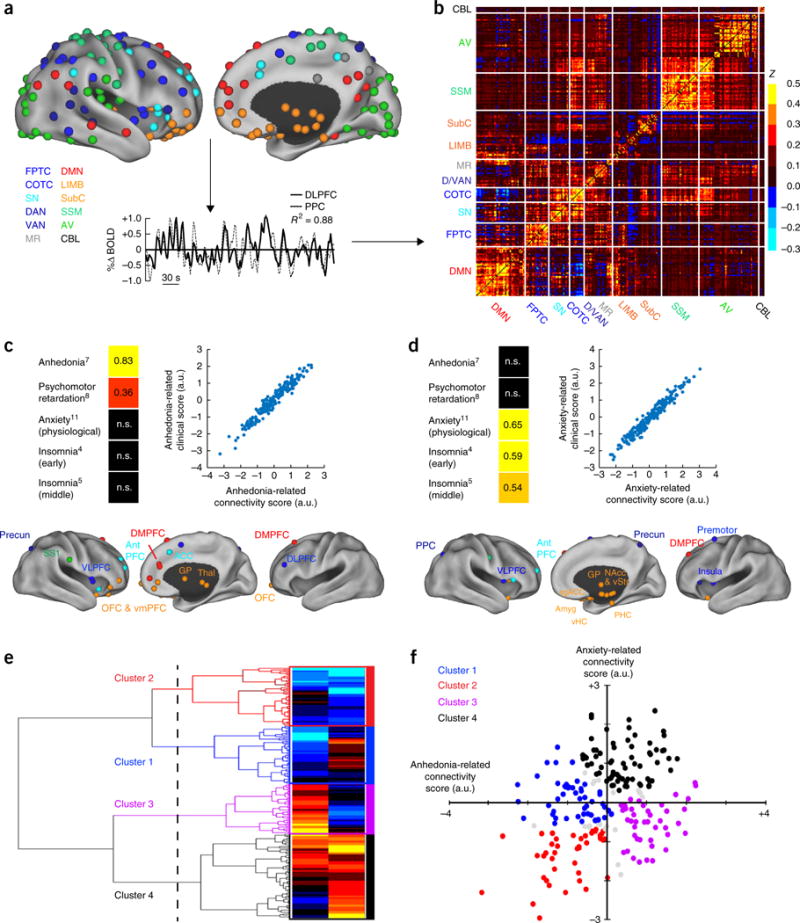
Canonical correlation analysis (CCA) and hierarchical clustering define four connectivity-based biotypes of depression. (a) Data analysis schematic and workflow. After preprocessing, BOLD signal time series were extracted from 258 spherical regions of interest (ROIs) distributed across the cortex and subcortical structures. The schematics (top) show lateral (left) and medial (right) views of right-hemisphere ROIs projected onto an inflated cortical surface and colored by functional network (lower left). Left-hemisphere ROIs (data not shown) were similar. For each subject, whole-brain functional-connectivity matrices were generated by calculating pairwise BOLD signal correlations between all ROIs, as in this example of correlated signals (r2 = 0.88) for DLPFC (solid line) and PPC (dashed line) nodes of the FPTC network in a representative subject. (b) Whole-brain, 258 × 258 functional-connectivity matrix averaged across all healthy controls (n = 378 subjects). z = Fischer transformed correlation coefficient. (c,d) CCA was used to define a low-dimensional representation of depression-related connectivity features and identified an “anhedonia-related” component (canonical variate; c) and an “anxiety-related” component (d), represented by linear combinations of connectivity features that were correlated with linear combinations of symptoms. The scatterplots in c and d illustrate the correlation between low-dimensional connectivity scores and low-dimensional clinical scores for the anhedonia-related (r2 = 0.91) and anxiety-related components (r2 = 0.95), respectively (P < 0.00001, n = 220 patients with depression). To the left of each scatterplot, clinical score loadings (i.e., the Pearson correlation coefficients between specific symptoms and the anhedonia- or anxiety-related clinical score (canonical variate)) are depicted for those symptoms with the strongest loadings (HAMD item #, indicated by numbers in superscript; for all loadings on all symptoms, see Supplementary Fig. 2). Below each scatterplot, connectivity score loadings are summarized by depicting the neuroanatomical distribution of the 25 ROIs (top 10%) that were most highly correlated with each component (summed across all significantly correlated connectivity features for a given ROI), colored by network, as in a. Projections to the medial wall map are for both left- and right-hemisphere ROIs. (e) Hierarchical clustering analysis. The height of each linkage in the dendrogram represents the distance between the clusters joined by that link. For reference, the dashed line denotes 20 times the mean distance between pairs of subjects within a cluster. For analyses of additional cluster solutions and further discussion, see Supplementary Figure 3. (f) Scatterplot for four clusters of subjects along dimensions of anhedonia- and anxiety-related connectivity. Gray data points indicate subjects with ambiguous cluster identities (edge cases, cluster silhouette values < 0; n = 15, or 6.8% of all subjects). ACC, anterior cingulate cortex; amyg, amygdala; antPFC, anterior prefrontal cortex; a.u., arbitrary units; AV, auditory/visual networks; CBL, cerebellum; COTC, cingulo-opercular task-control network; D/VAN, dorsal/ventral attention network; DLPFC, dorsolateral prefrontal cortex; DMN, default-mode network; DMPFC, dorsomedial prefrontal cortex; FPTC, frontoparietal task-control network; GP, globus pallidus; LIMB, limbic; MR, memory retrieval network; NAcc, nucleus accumbens; OFC, orbitofrontal cortex; PPC, posterior parietal cortex; precun, precuneus; sgACC, subgenual anterior cingulate cortex; SS1, primary somatosensory cortex; SN, salience network; SSM, somatosensory/motor networks; subC, subcortical; thal, thalamus; vHC, ventral hippocampus; VLPFC, ventrolateral prefrontal cortex; VMPFC, ventromedial prefrontal cortex; vStr, ventral striatum; n.s., not significant. See Supplementary Table 4 for MNI coordinates for ROIs in b and c.
Each correlation matrix comprised 33,154 unique connectivity features, which thus necessitated a protocol for selecting a subset of relevant, nonredundant connectivity features for use in clustering. We reasoned that biologically meaningful depression subtypes would be best characterized by a subset of connectivity features that were significantly correlated with depressive symptoms. Therefore, to select connectivity features for use in clustering, we used canonical correlation analysis (Online Methods) to define a low-dimensional representation of connectivity features that were associated with weighted combinations of clinical symptoms, as quantified by the 17-item Hamilton Depression Rating Scale (HAMD), a commonly used, clinician-rated assessment. To ensure that cluster discovery was not confounded by site-related differences in subject recruitment criteria or by other unidentified variables, the cluster-discovery analysis was restricted to a subset of patients (the ‘cluster-discovery subset’, n = 220 of the 333 patients with depression) from two sites with identical inclusion and exclusion criteria and statistically equivalent depression-symptom scores (see Supplementary Tables 1–3 for details). This analysis identified linear combinations of connectivity features (analogous to principal components) that predicted two distinct sets of depressive symptoms (Fig. 1c,d). The first connectivity component (canonical variate) defined a combination of predominantly frontostriatal and orbitofrontal connectivity features that were correlated with anhedonia and psychomotor retardation (Fig. 1c, Supplementary Fig. 2 and Supplementary Table 4). The second component defined a distinct set of predominantly limbic connectivity features involving the amygdala, ventral hippocampus, ventral striatum, subgenual cingulate and lateral prefrontal control areas, and that was correlated with anxiety and insomnia (Fig. 1d). Thus, this empirical, data-driven approach to feature selection and dimensionality reduction identified two sets of functional connectivity features that were correlated with distinct clinical-symptom combinations.
We then tested whether abnormalities in these connectivity feature sets tended to cluster in patient subgroups. Multiple statistical learning approaches are available for discovering notable structure in large data sets (‘unsupervised learning’). Here we chose to use hierarchical clustering—a standard approach that has been used extensively in the biological sciences29,30—to discover clusters of patients, by assigning them to nested subgroups with similar patterns of connectivity (Online Methods). This analysis revealed four patient clusters defined by distinct and relatively homogeneous patterns of connectivity along these two dimensions (Fig. 1e,f) and comprising 23.6%, 22.7%, 20.0% and 33.6% of the 220 patients with depression, respectively. This four-cluster solution was optimal for defining relatively homogeneous subgroups that were maximally dissimilar from each other (maximizing the ratio of between-cluster to within-cluster variance), while ensuring individual cluster sample sizes that provided sufficient statistical power to detect biologically meaningful differences (Supplementary Fig. 3). Therefore, we focused our subsequent analyses on characterizing and validating these four putative subtypes of depression.
Biotype-specific clinical profiles predicted by frontostriatal and limbic network dysfunction
To understand the neurobiological basis of these biotypes, we began by testing for differences in the whole-brain architecture of functional connectivity between patients (n = 220) and age-, sex- and site-matched healthy controls (n = 378) and for connectivity features that differed between patient subgroups. We observed a common neuroanatomical core of pathology underlying all four biotypes and encompassing areas spanning the insula, orbitofrontal cortex, ventromedial prefrontal cortex and multiple subcortical areas (Fig. 2a,b and Supplementary Table 5)—all of which have been implicated in depression previously15–20. This led us to ask whether these connectivity features predicted the severity of ‘core’ symptoms that were present in almost all patients, regardless of biotype. We found that, of the 17 symptoms quantified by the HAMD, three were present in almost all patients with depression (>90%): mood (“feelings of sadness, hopelessness, helplessness,” 97.1%), anhedonia (96.7%) and anergia or fatigue (93.9%). Across subjects, regardless of biotype, abnormal connectivity in this shared neuroanatomical core (as indexed by the first principal component in a principal-component analysis (PCA)) was correlated with severity scores on these three symptoms (Fig. 2c; r = 0.72–0.82).
Figure 2.
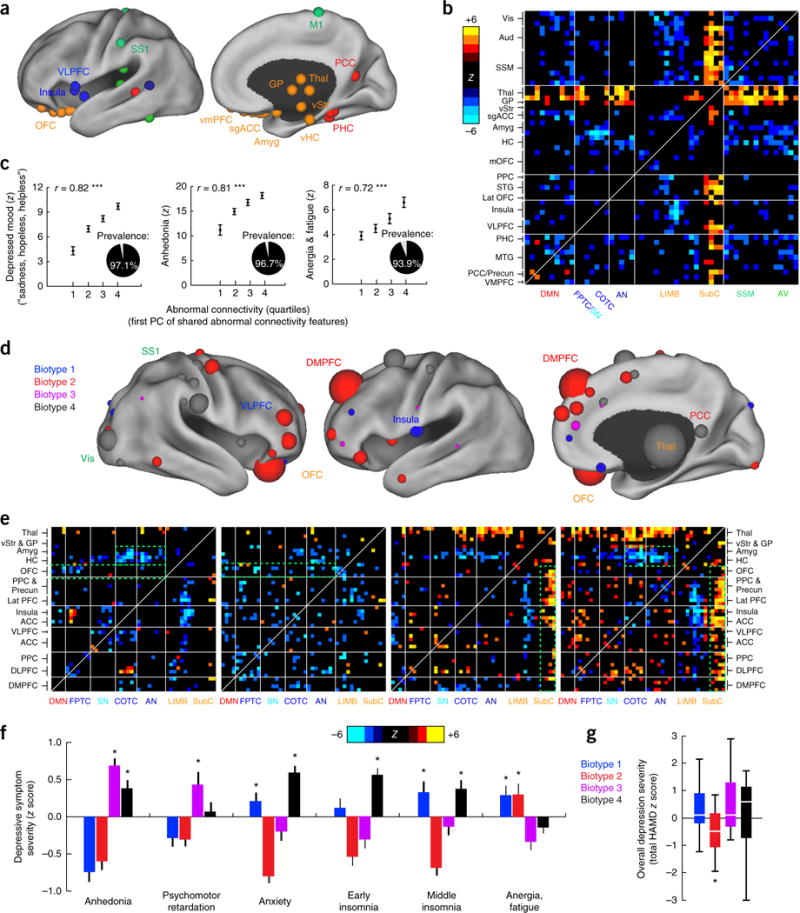
Connectivity biomarkers define depression biotypes with distinct clinical profiles. (a) Neuroanatomical distribution of the 25 ROIs (top 10%) with the most abnormal connectivity features shared by all four biotypes (summed across all connectivity features for a given ROI), identified using Wilcoxon rank–sum tests to test for connectivity features that were significantly abnormal in all four biotypes relative to healthy controls in data set 1 (n = 378). ROIs are colored by network, as in Figure 1a. (b) Heat maps depicting a pattern of abnormal connectivity (P < 0.05, false-discovery rate (FDR) corrected) shared by all four biotypes for the top 50 most abnormal ROIs, colored on the basis of Wilcoxon rank–sum tests comparing patients and controls, as in a. Warm colors represent increase and cool colors decrease in depression as compared to controls. (c) Correlations (r = 0.72–0.82, ***P < 0.001, Spearman) between shared abnormal connectivity features (as indexed by the first principal component (PC) of the features depicted in b and the severity of the core depressive symptoms. Insets depict the prevalence of each symptom. Symptom severity measures are z-scored with respect to controls and plotted as the mean for each quartile, ± s.e.m. (d) Neuroanatomical distribution of dysfunctional connectivity features that differed by biotype, as identified by Kruskal–Wallis analysis of variance (ANOVA) (P < 0.05, FDR corrected), summarized for the 50 ROIs (top ~20%) with the most biotype-specific connectivity features (i.e., the 50 ROIs with the largest test statistic summed across all connectivity features, showing cluster specificity at a threshold of P < 0.05, FDR corrected). Nodes (ROIs) are colored to indicate the biotype with the most abnormal connectivity features and scaled to indicate how many connectivity features exhibited significant effects of biotype. (e) Heat maps depicting biotype-specific patterns of abnormal connectivity for the functional nodes illustrated in d, plus selected limbic areas, colored as in b. Green boxes highlight corresponding areas in each matrix discussed in the main text. (f) Biotype-specific clinical profiles for the six depressive symptoms that varied most significantly by cluster (P < 0.005, Kruskal–Wallis ANOVA). Symptom severities (HAMD) are z-scored with respect to the mean for all patients in the cluster-discovery set. See Supplementary Figure 4 for all 17 HAMD items and for replication in data from subjects left out of the cluster-discovery set. (g) Boxplot of biotype differences in overall depression severity (total HAMD score), in which boxes denote the median and interquartile range (IQR) and whiskers the minimum and maximum values. In f and g, asterisk (*) indicates significant difference from mean symptom severity rating for all patients (z = 0) at P < 0.05; error bars depict s.e.m.; n.s., not significant. Aud, auditory cortex; HC, hippocampus; lat PFC, lateral prefrontal cortex; lat OFC, lateral orbitofrontal cortex; MTG, middle temporal gyrus; PHC, parahippocampal cortex; PCC, posterior cingulate cortex; SSM, primary sensorimotor cortex (M1 or S1); STG, superior temporal gyrus; vis, visual cortex. Other abbreviations are as in Figure 1. See Supplementary Table 5 for Montreal Neurological Institute coordinates for ROIs in a and d.
In addition, we found that, superimposed on this shared pathological core, distinct patterns of abnormal functional connectivity differentiated the four biotypes (Fig. 2d,e) and were associated with specific clinical-symptom profiles (Fig. 2f). For example, as compared to controls, reduced connectivity in frontoamygdala networks, which regulate fear-related behavior and reappraisal of negative emotional stimuli31–33, was most severe in biotypes 1 and 4, which were characterized in part by increased anxiety. By contrast, hyperconnectivity in thalamic and frontostriatal networks, which support reward processing, adaptive motor control and action initiation20,34–37, were especially pronounced in biotypes 3 and 4 and were associated with increased anhedonia and psychomotor retardation. And reduced connectivity in anterior cingulate and orbitofrontal areas supporting motivation and incentive-salience evaluation38–40 was most severe in biotypes 1 and 2, which were characterized partly by increased anergia and fatigue.
Importantly, although the connectivity-based biotypes revealed in our analysis were associated with differences in clinical symptoms, they did not simply reflect differences in overall depression severity. Although overall depression severity scores were modestly but significantly decreased in biotype 2 as compared to the other three groups (by 15–16%), there were no significant differences in severity between biotypes 1, 3 and 4 (Fig. 2g; see Supplementary Fig. 4 for convergent findings in independent data acquired from subjects not included in the cluster-discovery analysis). Furthermore, they did not simply recapitulate subtypes derived strictly from clinical-symptom measures; whereas clustering according to functional connectivity features in random patient subsamples yielded stable clustering outcomes, clustering according to clinical symptoms yielded unstable outcomes with relatively low longitudinal stability over time (Supplementary Fig. 5).
Functional connectivity biomarkers for diagnosing depression biotypes
By reducing diagnostic heterogeneity, we reasoned that clustering could be leveraged to develop classifiers for the diagnosis of depression biotypes solely on the basis of fMRI measures of functional connectivity, which have shown promise in smaller-scale, single-site studies of depression41–43 and other neuropsychiatric disorders44,45, but that have not performed as well when tested in multisite data sets44. To this end, we developed classifiers for each depression biotype, testing and optimizing standard, extensively used methods for brain parcellation, subject clustering, feature selection and classification to identify empirically the most successful approach to clustering and classification (Fig. 3a and Online Methods). Throughout, clustering analysis was performed in the same cluster-discovery sample (n = 220), whereas classification of patients versus controls was optimized in the full training data set (n = 333 patients; n = 378 controls), and leave-one-out cross-validation and permutation testing were used to assess performance and significance (Supplementary Fig. 6; for additional analysis confirming the stability of cluster assignments, see Supplementary Fig. 3d–f). The optimization process yielded progressive improvements in classifier performance (Fig. 3b). Support-vector machine (SVM) classifiers (using linear kernel functions) performed best, yielding overall accuracy rates of up to 89.2% for the clusters characterized above, on the basis of connectivity features associated with the neuroanatomical areas summarized in Figure 3c–f. In cross-validation (leave-one-out), individual patients and healthy controls were diagnosed correctly with sensitivities of 84.1–90.9% and specificities of 84.1–92.5% (Fig. 3g).
Figure 3.

Functional connectivity biomarkers for diagnosing neurophysiological biotypes of depression. (a) Data analysis schematic and workflow (Online Methods for additional details). (b) Optimization of diagnostic-classifier performance (accuracy) across the indicated combinations of methods for parcellation, clustering and classification. *P < 0.005, as estimated by permutation testing (Online Methods). Double asterisk (**) indicate the best performing protocol for parcellation, clustering and classification, and the focus of all subsequent analyses. (c–f) The neuroanatomical locations of the nodes with the most discriminating connectivity features are illustrated for each biotype for the four-cluster solution denoted by the double asterisk in b, colored and scaled by summing the results of Wilcoxon rank–sum tests of patients as compared to controls across all connectivity features associated with that node. Red represents increased and blue decreased functional connectivity in depression. (g) Sensitivity and specificity by biotype for the most successful classifiers identified in b (**). Error bars depict 95% confidence interval for the mean accuracy across all iterations of leave-one-out cross-validation. (h) Reproducibility of cluster assignments in a second fMRI scan (n = 50) obtained 4–5 weeks after the initial scan (χ2 = 112.7, P < 0.00001). (i) Classifier performance in an independent, out-of-sample replication data set (n = 125 patients, 352 healthy controls). Cross-hatched bars depict classifier accuracy with more stringent data quality controls (Online Methods) and excluding equivocal classification outcomes (the 10% of subjects with the lowest absolute SVM classification scores). Error bars depict 95% confidence intervals.
To further validate the biotypes, we asked whether biotype diagnosis (cluster membership) was stable over time by testing these classifiers on a subset of patients (n = 50) who received a second fMRI scan while they were actively experiencing depression, 4–6 weeks after the first scanning session. We found that, overall, 90.0% of subjects were assigned to the same biotype in both scans (Fig. 3h; χ2 = 84.6, P < 0.0001). There were no significant between-group differences in age, medication usage or head motion during scanning, variables that may affect rsfMRI connectivity measures (Supplementary Fig. 7).
It is well established in the machine-learning literature that iterative training and cross-validation on the same data overestimate classifier performance46, and other studies have raised questions about the capacity for classifiers trained on one data set at a single site to generalize to data collected at multiple sites44. Therefore, we tested the most successful classifier for each depression biotype in an independent replication data set that consisted of 125 patients and 352 healthy controls acquired from 13 sites, including five sites that were not included in the original training data set (Supplementary Table 3). To avoid overestimating diagnostic sensitivity, only one classifier—the classifier for the best-fitting biotype—was tested on each subject (Online Methods). Overall, 86.2% of subjects in this independent, out-of-sample replication data set were correctly diagnosed, including >90% of patients in biotypes 3 and 4 (Fig. 3i; Supplementary Table 6). By implementing stricter data quality controls and by treating subjects with ambiguous classification outcomes (the lowest absolute SVM classification scores; Online Methods) as equivocal test results, as is common practice for biomarkers in other areas of medicine, these accuracy rates exceeded 95%.
Connectivity biomarkers predict responsiveness to rTMS
Treatment-response prediction is an important element of validating biomarkers and establishing potential for clinical actionability, and neuroimaging measures have already shown promise for predicting treatment response in depression9–14. Repetitive transcranial magnetic stimulation (rTMS) is a noninvasive neurostimulation treatment for medication-resistant depression that modulates functional connectivity in cortical networks47–49. Although the left dorsolateral prefrontal cortex is the most common target for stimulation48, recent studies have demonstrated efficacy for a dorsomedial prefrontal (DMPFC) target13, which raises the intriguing possibility that biotype differences in dysfunctional connectivity at the DMPFC target (Fig. 2d) site may give rise to differing treatment outcomes. To test this, we asked first whether the four depression biotypes were differentially responsive to rTMS in 124 subjects who received repetitive high-frequency stimulation of the dorsomedial prefrontal cortex for 5 weeks, beginning shortly after their fMRI scan (Online Methods). Treatment response varied significantly with cluster membership (χ2 = 25.7, P = 1.1 × 10−5). rTMS was most effective for patients in biotype 1, 82.5% of whom (n = 33/40) improved significantly (>25% HAMD reduction), as compared to 61.0% for biotype 3 (n = 25/41) and only 25.0% and 29.6% for biotypes 2 (n = 4/16) and 4 (n = 8/27), respectively (see Fig. 4a,b full response rates (>50% reduction) and percentage change in depression severity by total HAMD score).
Figure 4.

Connectivity biomarkers predict differential antidepressant response to rTMS. (a) Differing response rates to repetitive transcranial magnetic stimulation (rTMS) of the dorsomedial prefrontal cortex across patient biotypes (clusters) in n = 124 subjects. Response rate indicates percentage of subjects showing at least a partial clinical response to rTMS (χ2 = 25.7, P = 1.1 × 10−5), defined conventionally as >25% reduction in symptom severity by HAMD. Full response rates (>50% reduction by HAMD, cross-hatched bars) also varied by biotype (χ2 = 22.9, P = 4.3 × 10−5). (b) Boxplot of percent improvement in depression severity by biotype (P = 1.79 × 10−6, Kruskal–Wallis ANOVA), in which boxes denote the median and interquartile range and whiskers the minimum and maximum up to 1.5 × the IQR, beyond which outliers are plotted individually. Percent improvement = total HAMD score before treatment – total HAMD score after treatment/total HAMD score before treatment. **P = 0.00001–0.002 (Mann–Whitney), indicating significantly increased versus biotypes 2–4; *P = 0.007 (Mann–Whitney), indicating significantly increased versus biotype 4. (c) Functional connectivity differences in the DMPFC stimulation target in treatment responders versus nonresponders (Wilcoxon rank–sum tests, thresholded at P < 0.005). Warm colors represent increased and cool colors decreased functional connectivity in treatment responders as compared to nonresponders. The 12 ROIs depicted here were located within 3 cm of the putative DMPFC target site, estimated in a previously published report to be located at Talairach coordinates, x = 0, y = +30, z = +30 (ref.13). (d) The neuroanatomical distribution of the most discriminating connectivity features for the comparison of rTMS responders versus non-responders, summarized by illustrating the locations of the 25 (top 10%) most discriminating ROIs indexed by summing across all significantly discriminating connectivity features and colored by functional network as in Figure 1a. The red arrows denote the rTMS target site in the two (lower) medial panels. (e) Heat maps depicting differences in functional connectivity in patients who subsequently improved after receiving rTMS (n = 70), as compared to those who did not (n = 54). (f–i) Confusion matrices depicting the performance of classifiers trained to identify subsequent treatment responders on the basis of the most discriminating connectivity features (f), connectivity features plus biotype diagnosis (g), clinical symptoms alone (h) or connectivity features plus biotype diagnosis in an independent replication set (i, n = 30 patients with depression). NR, nonresponder; R, responder. (j) Summary of performance (overall accuracy) for classifiers in f–i. **significantly greater than clinical features alone (P < 0.001) and connectivity features alone (P = 0.003) by permutation testing; *P = 0.04 (significantly greater than clinical features alone by permutation testing). Cross-hatched bars depict classifier accuracy with more stringent data quality controls (Online Methods) and excluding equivocal classification outcomes (the 10% of subjects with the lowest absolute SVM classification scores). Error bars depict s.e.m. in a and 95% confidence intervals in j. All abbreviations as in Figures 1 and 2. See Supplementary Table 7 for MNI coordinates for ROIs in d.
Next, we tested whether connectivity-based biotypes could be used to predict treatment response more effectively than clinical symptoms alone. To this end, we trained classifiers to differentiate responders and nonresponders using the same approach to feature selection, training and leave-one-out cross-validation. The most discriminating connectivity features involved the dorsomedial prefrontal stimulation target and the left amygdala, left dorsolateral prefrontal cortex, bilateral orbitofrontal cortex and posterior cingulate cortex (Fig. 4c; Supplementary Table 7). Connectivity between other neuroanatomical areas that were not directly stimulated by the rTMS protocol—including the ventromedial prefrontal cortex, thalamus, nucleus accumbens and globus pallidus—also predicted treatment response (Fig. 4d,e). Connectivity features predicted individual differences in the rTMS responsiveness with 78.3% accuracy in leave-one-out cross-validation (Fig. 4f,j). Classification according to connectivity features plus biotype diagnosis yielded the highest predictive accuracy (89.6%; Fig. 4g,j).
By contrast, clinical symptoms alone were not strong predictors of rTMS treatment responsiveness at an individual level. To test this, we trained classifiers to differentiate responders and nonresponders solely on the basis of clinical data. We found that clinical features (insomnia, anhedonia and psychomotor retardation by HAMD) were only modestly (62.6%) predictive of treatment responsiveness (Fig. 4h,j). Overall, classifiers based on connectivity features and biotype diagnosis significantly outperformed those based on clinical features alone (Fig. 4j; P < 0.005). Furthermore, just as we observed for diagnostic classifiers in Figure 3, accuracy rates could be improved further (>94%, Fig. 4j) by implementing stricter data quality controls and treating subjects with ambiguous classification outcomes as equivocal test results (Online Methods). Finally, to further evaluate predictive validity, we tested the best-performing classifier, which used a combination of connectivity features and biotype diagnosis, in an independent replication set (n = 30 subjects) and obtained comparable accuracy rates (87.5–92.6%; Fig. 4i,j). By contrast, subtyping subjects on the basis of clinical symptoms yielded highly variable, longitudinally unstable clustering outcomes that failed to predict treatment response (Supplementary Fig. 5).
Depression biotypes transcend conventional diagnostic boundaries
Collectively, these findings show that our current diagnostic system merges groups of patients with at least four distinct patterns of abnormal connectivity under a single diagnostic label—major depressive disorder. We concluded our study by testing whether the converse also occurs: that is, does our diagnostic system assign different diagnostic labels to patients who exhibit the same connectivity biotype? Motivated by studies identifying common neuroanatomical and functional changes that are shared across mood and anxiety disorders50–53, we first asked whether patients diagnosed with generalized anxiety disorder (GAD; n = 39) shared similar patterns of abnormal connectivity with one or more of the depression biotypes identified above. GAD was associated with widespread connectivity differences in resting-state networks (Fig. 5a–c) that overlapped significantly with those in depression (χ2 = 5,457; P < 0.0001; Fig. 5a–c). Next, to test whether subsets of patients with GAD resemble one or more depression biotypes, we applied the optimized classifiers developed above to the GAD cohort (Online Methods). Although none of the patients with GAD in this analysis met clinical criteria for a diagnosis of depression, 69.2% of them were nevertheless classified as belonging to one of the depression biotypes, and a majority of these (59.3%) were assigned to the anxiety-associated biotype 4 (Fig. 5d).
Figure 5.
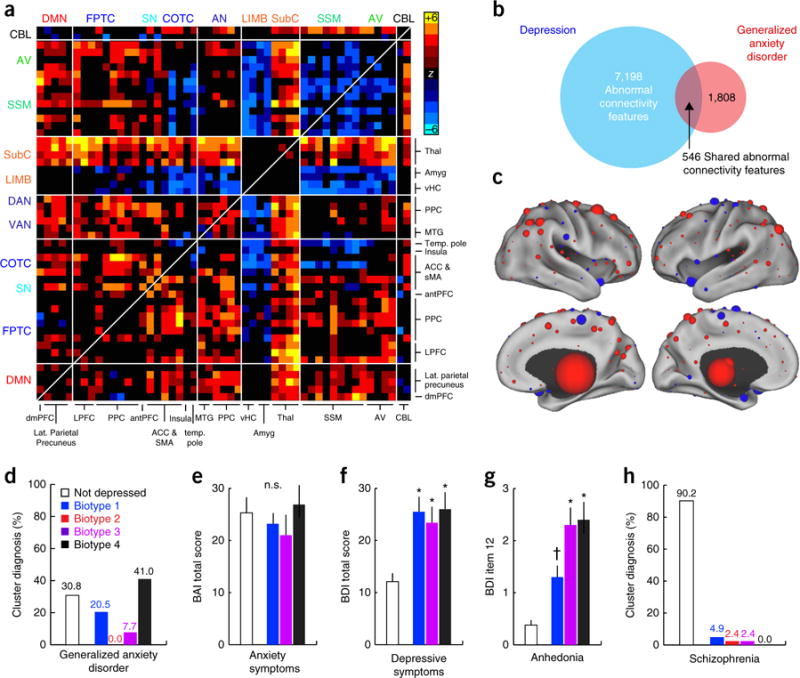
Connectivity biomarkers of depression biotypes transcend diagnostic boundaries. (a) Abnormal connectivity features in patients with generalized anxiety disorder (GAD, n = 39) relative to healthy controls (n = 378). In this matrix depicting the 50 neuroanatomical nodes with the most significantly different connectivity features (Wilcoxon rank–sum tests, summed across all 258 features), elements in warm and cool colors depict connectivity features that are significantly increased or decreased in GAD, respectively. (b) 30.2% of connectivity features that were significantly abnormal in GAD (threshold of P < 0.001 versus controls, Wilcoxon) were also abnormal in depression (χ2 = 5,457, P < 0.0001). (c) The neuroanatomical distribution of the most discriminating connectivity features for the comparison of GAD patients versus controls. The nodes are colored and scaled by summing across all significantly abnormal connectivity features associated with that node. Red represents increased and blue decreased functional connectivity in GAD. (d) Distribution of biotype diagnoses in patients with GAD. (e) No significant biotypes differences in anxiety symptom severity (P = 0.692; Kruskal–Wallis ANOVA). BAI, Beck anxiety inventory. (f,g) Significantly (P < 0.005, Kruskal–Wallis) elevated total depressive-symptom severity (f; BDI, Beck depression inventory) and anhedonia severity (g; BDI item 12) in GAD patients who tested positive for a depression biotype as compared to those who did not. *P < 0.01, †P = 0.064 in post hoc Mann–Whitney tests relative to “not depressed” group. (h) Distribution of biotype diagnoses in patients with schizophrenia (n = 41). Error bars depict s.e.m. throughout. All abbreviations as in Figures 1 and 2.
Although anxiety symptom severity did not vary significantly by biotype classification (Fig. 5e), depressive symptom severity (Fig. 5f) and anhedonia (Fig. 5g) were significantly increased in patients with GAD who tested positive for one of the depression biotypes, as compared to patients with GAD who did not test positive. Furthermore, just as anhedonia was increased in patients with depression in biotypes 3 and 4, patients with GAD showed a similar trend (Fig. 5g; P < 0.05). Finally, to understand whether these classifiers were detecting pathological connectivity related specifically to mood and anxiety as opposed to nonspecific differences associated with psychiatric illness in general, we tested them on patients with schizophrenia (n = 41), a disorder that is not thought to be closely related to unipolar depression. Just 9.8% of patients with schizophrenia tested positive for a depression biotype (Fig. 5h).
DISCUSSION
Increasingly, diagnostic heterogeneity has emerged as a major obstacle to understanding the pathophysiology of mental illnesses and, in particular, depression. Although major depressive disorder—especially highly recurrent depression—is up to 45% heritable54, identifying genetic risk factors has proven challenging, even in extremely large genome-wide association studies55. Likewise, efforts to develop new treatments have slowed, owing in part to a lack of physiological targets for the assessment of treatment efficacy and the selection of individuals who are most likely to benefit56. All of these challenges have been attributed in part to the fact that our diagnostic system assigns a single label to a syndrome that is not unitary and that might be caused by distinct pathological processes, which would thus require different treatments. Here we have defined four subtypes of depression associated with differing patterns of abnormal functional connectivity and distinct clinical-symptom profiles that transcend conventional diagnostic boundaries, and we have shown how neuroimaging biomarkers can be used to diagnose them. Our sample size, cross-validation in strictly independent samples and replication in independent data sets support these results.
However, this is to our knowledge the first effort to apply this type of statistical clustering for the purpose of defining depression subtypes and diagnosing them in individual patients, so caution is warranted. Replication of our findings in additional, independent, prospectively acquired data sets will be crucial for addressing some of the limitations inherent in our retrospective, multisite sample. We designed a preprocessing scheme specifically to control for site- and scanner-related artifacts, and we performed our clustering analysis on data from just two sites with nearly identical acquisition protocols and recruitment criteria. Still, it will be essential to replicate these findings in an equally large sample acquired from a single site. Furthermore, more extensive and uniform clinical phenotyping—especially within the relatively broad domains of anhedonia and anxiety—will be crucial for further understanding how connectivity-based biotypes relate to distinct symptoms and behaviors.
Importantly, we regard the four biotypes identified here as just one, initial solution to the problem of diagnostic heterogeneity in a system that relies primarily on the reporting of clinical symptoms. This solution is capable of predicting treatment response in a controlled, laboratory setting and advances our understanding of how heterogeneous symptom profiles in depression might be related to clustered patterns of dysfunctional connectivity. But alternative solutions to the problem of depression subtyping also exist, even in our 220-subject hierarchical clustering analysis, which was suggestive of additional subtypes nested within these four clusters. It is likely that relatively restrictive patient-recruitment criteria, the size of our cluster-discovery data set, and the ordinal nature of our clinical-symptom assessments were also limiting factors. For these reasons, clinical and neuroimaging data acquired from much larger populations will be useful for characterizing more complex associations between connectivity features and symptoms; for defining robust low-dimensional representations of this connectivity feature space; and for optimizing the mapping between diagnostic subtypes and their underlying neurobiology. It will also be crucial to evaluate how these biomarkers perform in real-world, clinical settings, in which clinical assessments and treatments might be administered with varying fidelity, which could potentially diminish diagnostic and prognostic performance.
These caveats notwithstanding, our results have several potential applications. They may inform recent initiatives to rethink our system for diagnosing psychiatric disorders and investigating their neuro-physiological and genetic basis, by stratifying subjects into subgroups defined by shared neurobiological substrates1. They might also guide optogenetic and other circuit neuroscience approaches to investigating how dysfunction in specific circuits contributes to depression-and anxiety-related behaviors in experimentally tractable animal models57–59. Finally, these biomarkers also have prognostic potential. Patients in biotype 1 were approximately three times more likely to benefit from TMS of the dorsomedial prefrontal cortex than those in biotypes 2 or 4, and together, biotype diagnosis and functional connectivity features could be leveraged to accurately differentiate treatment responders from nonresponders on an individual basis. Validating and adapting them for use in naturalistic clinical settings will be a key challenge, but our data are also consistent with other recent reports that highlight the potential of neuroimaging tools to predict treatment response9–14, a major priority for a condition in which most treatments are effective only after several months. Biomarkers have already transformed the diagnosis and management of cancer, diabetes, heart disease and even pain syndromes8, but they have proven more elusive for psychiatry. Our results define one approach for using neuroimaging biomarkers to delineate and diagnose novel subtypes of mental illness characterized by uniform neurobiological substrates.
ONLINE METHODS
Subjects
All analyses were conducted in one of two data sets, unless otherwise noted (see also ‘Statistical analysis’ section below for subject details for each analysis, organized by figure panel). Data set 1 (n = 711 subjects, 333 patients and 378 controls) was used for all analyses, except those depicted in Figures 3i, 4i and 5. That is, data set 1 was used to identify clusters (biotypes) of patients with distinct patterns of dysfunctional connectivity in resting-state networks, testing for neurobiological and clinical correlates of these biotypes, and for training and testing classifiers to diagnose them. To ensure that cluster discovery was not confounded by site-related differences in subject recruitment criteria or other unidentified variables, the cluster-discovery analysis (Fig. 1) was restricted to a subset of patients in data set 1, the ‘cluster-discovery set’ (n = 220 of the 333 patients), who were recruited and scanned from just two sites with identical inclusion and exclusion criteria. Subjects in the cluster-discovery set were adult patients meeting Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria for (unipolar) major depressive disorder and seeking treatment for a currently active, nonpsychotic major depressive episode. They had a history of failure to respond to at least two antidepressant medication trials at adequate doses, including at least one during the current episode. Patients in the cluster-discovery set were excluded from enrollment if they had a currently active substance-use disorder, a psychotic disorder, bipolar depression, a history of seizures, unstable medical conditions, current pregnancy or other contraindications to MRI (for example, implanted devices, claustrophobia or head injury with loss of consciousness). As described in Supplementary Table 1, subjects from the two sites included in the cluster-discovery set were matched for age, sex and depression severity (HAMD-17 total score). Supplementary Table 1 also describes medication status, comorbid diagnoses and additional details about the scanning protocols for data acquired at these two sites.
Classifier training, cross-validation and optimization was performed in the full data set 1, i.e., the ‘training data set,’ which included patients diagnosed with unipolar major depressive disorder and a currently active major depressive episode (n = 333, 59.2% female, mean age = 40.6 years) and healthy control subjects without any history of a psychiatric condition (n = 378, 57.7% female, mean age = 38.0 years). The patient and control groups did not differ significantly in age (P = 0.189, Mann-Whitney) or sex (χ2 = 0.61, P = 0.688). The patient scans were acquired at separate sites by five principal investigators (the two sites from the cluster-discovery set plus three additional sites). The control scans were acquired at these same five sites, as well as from seven additional sites that have provided unrestricted public access to their data through the 1000 Functional Connectomes Project (http://fcon_1000.projects.nitrc.org). Inclusion and exclusion criteria were generally similar to those described above for the two sites in the cluster-discovery set, except that a history of treatment resistance was not a requirement. Exclusion criteria common to all sites were contraindications for MRI and a recent history of substance abuse or dependence. Other inclusion and exclusion criteria—and consequently, the presence of psychiatric co-morbidities and use of psychiatric medications—varied by site and are detailed in Supplementary Table 2. Clustering into connectivity biotypes was not related to medication history, age or head motion (Supplementary Fig. 7). Additional demographic information for all sites in data set 1 is reported in Supplementary Table 3.
Data set 2 (n = 477)—the ‘replication data set’—was used to test the most successful classifier of each depression biotype in patients with active depression (n = 125 from seven sites) and healthy controls (n = 352 from 13 sites). Scans in data set 2 were acquired in separate studies, at a later date or were not initially available to us, and they were not used in any step of the cluster identification or classifier training procedure. Furthermore, five sites were unique to data set 2. Patients with depression at all sites in both data sets met DSM-IV criteria for a current major depressive episode (n = 109 unipolar; n = 16 bipolar 2), and healthy controls were subjects without any current or past history of a psychiatric or neurological condition.
To test whether patterns of abnormal connectivity that were evident in clusters of patients with depression were also present in subsets of patients with other psychiatric disorders (Fig. 5), we tested the same classifiers on patients meeting DSM-IV criteria for a diagnosis of generalized anxiety disorder (GAD, n = 39, 69.2% female, mean age = 32.4 years) or schizophrenia (n = 41, 78.0% male, mean age = 38.2 years; no co-morbid mood disorders and no schizoaffective disorder). Data for the GAD subjects were acquired by one of the co-authors of this report (A.E.), and inclusion and exclusion criteria are described in Supplementary Table 2 (site: Stanford 1; PI: A. Etkin). Data for the schizophrenia subjects were obtained through the 1000 Functional Connectomes Project (http://fcon_1000.projects.nitrc.org), made publicly available by the Center of Biomedical Research Excellence in Brain Function and Mental Illness (PIs: J. Sui, J. Liu, C. Harenski, R. Thoma and C. Abbott). Inclusion criteria were a diagnosis of schizophrenia (but not schizoaffective disorder), as confirmed by the Structured Clinical Interview for DSM Disorders (SCID), and exclusion criteria were a history of neurological disorder, mental retardation, head trauma with loss of consciousness or substance abuse or dependence within the past 12 months. All subjects in all data sets provided informed consent, and all recruitment procedures and experimental protocols were approved by the Institutional Review Boards of the principal investigators’ respective institutions (Weill Cornell Medical College, Stanford University, Toronto Western Hospital, Emory University and Harvard Medical School).
Clinical measures
At all sites, initial screening interviews were conducted to determine eligibility to participate, and a trained clinician conducted a structured clinical interview (MINI or SCID) to confirm all psychiatric diagnoses and rule out exclusionary co-morbid conditions as defined in Supplementary Table 2. In addition, specific clinical symptoms were evaluated using the Hamilton Rating Scale for Depression (HAMD; n = 312 patients; n = 65 healthy controls), the Beck depression inventory (BDI, n = 39 patients with GAD) and the Beck anxiety inventory (BAI; n = 39 patients with GAD). These assessments were used to test the depression biotypes that were associated with specific clinical symptom profiles. For details, see ‘Clinical data analysis’ section below.
Magnetic resonance imaging (MRI) data acquisition
A resting-state functional MRI scan was obtained by using a T2*-weighted gradient echo spiral in-out sequence or a Z-SAGA sequence, yielding whole-brain coverage in all subjects. A high-resolution T1-weighted anatomical scan (MP-RAGE or SPGR) was obtained for brain parcellation and co-registration purposes. Specific scanning parameters varied by site. Most used a TR of ~2 s, in-plane resolution of ~3.5 mm, and obtained 150–180 volumes in ~5–6 min. Detailed scanning parameters for each site are reported in Supplementary Table 1 and Supplementary Table 3.
fMRI data analysis: preprocessing
All data sets were preprocessed using the Analysis of Functional Neuroimages (AFNI) software package. Prior to other preprocessing steps, framewise motion parameters were calculated by using AFNI’s 3dvolreg function, owing to concerns that slice-time correction might lead to systematic underestimates of motion when this step is performed first. After estimating framewise motion parameters, preprocessing included standard procedures for slice-timing correction, spatial smoothing (with a 4-mm-full-width, half-maximum Gaussian kernel), temporal bandpass filtering (0.01–0.1 Hz), linear and quadratic detrending and removal of nuisance signals related to head motion, physiological variables and local and global hardware artifacts. Functional data sets were co-registered to the corresponding high-resolution T1 anatomical images, and T1 anatomicals were transformed into the Montreal Neurological Institute (MNI) common space by using AFNI’s 3dQwarp function to calculate and optimize a nonlinear transformation. To reduce the number of interpolations performed on resting-state data, we combined motion-correcting, anatomical-to-structural and structural-to-MNI template alignments and applied them to functional scans in a single step.
Motion correction was achieved using AFNI’s 3dvolreg function. Motion artifact is increasingly recognized as an important potential confound in resting-state fMRI studies, especially those involving clinical populations, and can introduce systematic shifts in signal correlations that vary as a function of the distance separating two brain regions25–27. To balance the demands of noise reduction and data preservation, we censored volumes preceding or following any movement (framewise displacement (FD)) greater than 0.3 mm. These volumes were excluded from all further analysis steps, including nuisance regression. A small number of subjects (8.9%) were excluded from further analysis if the number of remaining volumes was insufficient for performing simultaneous nuisance signal regression and band-pass filtering as described below. (Note that descriptions of the number of subjects comprising each data set in the ‘Subjects’ section above and in the main text refer to subjects that were actually used in each analysis, after excluding scans because of motion contamination or poor signal quality, as defined below.)
Next, nuisance signal regression and band-pass filtering were performed simultaneously, only on volumes that survived motion censoring, and excluding high-motion volumes. This is because noise from high-motion volumes has been shown to contaminate other volumes, even if they are eventually omitted from final analyses60,61. Accordingly, the regression step included 12 motion parameters (roll, pitch, yaw, translation in three dimensions and their first derivatives); non-neuronal signals from eroded white matter and CSF masks; and regressors for temporal filtering. Finally, we used AFNI’s ANATICOR function to eliminate local and global hardware artifacts62–63. After preprocessing, the residual time series files, co-registered to MNI space, were used for all subsequent analyses.
A note on motion artifact
We selected a censoring threshold (FD > 0.3 mm) empirically based on analyses showing that it was sufficient to exclude the majority of excursions from so-called floor values in single-subject FD traces (Supplementary Fig. 1), which have been associated with significant motion artifact, while preserving enough data to allow for stable estimates of signal cor-relations25–27. It is also worth noting that this threshold resembles commonly used thresholds (0.2–0.5 mm) in recently published reports (reviewed in ref. 64). However, we found that a small number of RSFC features (just 0.7% of the connectivity features that differentiated patients and controls, at a liberal threshold of P < 0.005, uncorrected) were significantly different in low- versus high-motion subjects after ANATICOR regression and censoring at 0.3 mm (Supplementary Fig. 1d). To further evaluate whether motion artifact affected cluster discovery and biotype diagnoses, we repeated the hierarchical clustering analysis depicted in Figure 1 after excluding the 0.7% of RSFC features that varied with motion at this liberal threshold (P < 0.005). 99.1% of all subjects were assigned to the same cluster (Supplementary Fig. 1h). To rule out the possibility that multivariate classifiers may been influenced by the aggregation of subtle between-group differences in motion artifact that were undetectable by the mass univariate approach implemented in ref. 64, we conducted additional analyses reported in Supplementary Figure 1i,j. The results indicate that our clustering and classification results were not biased substantially by motion.
fMRI data analysis: parcellation and whole-brain connectivity estimation
The objective of this analysis was to extend conventional seed-based approaches to generate a whole-brain correlation matrix for each subject, quantifying functional connectivity in regions of interest spanning the entire brain in terms of correlated, spontaneous fluctuations in the resting-state BOLD signal. Most data sets were acquired in a native grid space of ~3.5 × 3.5 × 5 mm, yielding ~30,000 brain voxels and up to ~4.5 × 108 unique, potential pairwise correlations. To increase computational tractability and biological interpretability, all analyses reported in the main text used an established and extensively validated functional parcellation system28 to delineate functional network nodes (10-mm diameter spheres) spanning most cortical, subcortical and cerebellar areas. The originally published parcellation identified 264 nodes (ROIs). Here 13 ROIs that have hypothesized roles in depression-related pathology, but that are not represented in this 264-node parcellation, were added, including the left and right nucleus accumbens, subgenual anterior cingulate, head of the caudate nucleus, amygdala, ventral hippocampus, locus coeruleus, ventral tegmental area and raphe nucleus, for a total of 264 + 13 = 277 nodes. However, 19 of the 277 nodes—mostly cerebellar and inferior temporal areas—were excluded from further analyses owing to incomplete MRI volume coverage or because of inadequate signal (SNR < 100), as discussed in more detail below. Thus, the primary parcellation used in all analyses included 264 +13 − 19 = 258 functional nodes. In addition, when optimizing the biomarkers developed in Figure 3, we tested four strategies for parcellation: (i) The primary functional parcellation of Power and colleagues that is described above and is the focus of the analyses in the main text28; (ii) a ‘coarse voxelwise’ parcellation strategy, a standard anatomical template brain (1 × 1 × 1–mm resolution in MNI space) was resampled to a 10 × 10 × 15–mm grid space. After excluding voxels (or portions of voxels) corresponding to white matter or CSF using masks derived from a segmentation of the original template brain into tissue classes (via AFNI’s 3dSeg function), we were left with 945 ROIs spanning all cortical, subcortical and cerebellar gray matter; (iii) an anatomical parcellation used the Freesurfer atlas developed by Desikan, Killiany and colleagues that segments the brain into 68 gyral-based cortical ROIs and an additional 22 subcortical and cerebellar areas for a total of 90 anatomical regions of interest65; (iv) finally, a second functional parcellation (in addition to the used 90 cortical and subcortical ROIs defined by Shirer, Greicius and colleagues using independent-components analysis to identify brain voxels that exhibit correlated activity in association with one or more cognitive states (rest, episodic-memory retrieval, serial calculations or singing lyrics; see ref.66 for details). The best results were obtained from the primary functional parcellation devised by Power and colleagues28, which was the focus of all other analyses.
After preprocessing the resting-state fMRI data and parcellating the brain as described above, BOLD signal time series were extracted from each ROI by averaging across all voxels in that ROI, and a correlation matrix was calculated for each subject by using AFNI’s 3dNetCorr function. However, before doing so, we took additional steps to control for scanner- and site-related differences that could potentially confound analyses of data pooled across multiple sites. First, we controlled for site-related differences in signal quality or scan coverage by excluding ROIs if the signal-to-noise ratio (SNR, the voxelwise mean of the magnetic resonance signal over time divided by the s.d. of the time series) was less than 100 in >5% of subjects. On this basis, we excluded 13 of the 277 ROIs in the primary functional parcellation, leaving 264 ROIs for further analysis. Most excluded ROIs were located in the inferior cerebellum, which did not have consistent coverage across all sites, or on the ventral surface of the temporal lobe or the orbital surface of the frontal lobe, which tended to have lower SNR in some scans, likely owing to artifact at the interface with air sinuses. Second, for each subject, only voxels with SNR > 100 were used to calculate the mean BOLD signal time series for each ROI, to further control for local differences in signal quality on a per subject basis. And third, a small number of subjects (2.9%) was excluded from further analysis if the signal quality was low (SNR < 100) in any of the remaining 258 ROIs.
Thus, after excluding 13 ROIs with low-quality signal and a small number of subjects with excessive head motion (8.9%) or poor signal quality (2.9%), we calculated 258 × 258–element correlation matrices for each of the remaining subjects (n = 711 for data set 1; n = 477 for data set 2; see ‘Subjects’ above). To enable us to test hypotheses about functional connectivity differences in the depressed and control populations, we applied the Fisher z-transformation to each correlation coefficient. Next, we used multiple linear regression to further control for site- and age-related effects on functional connectivity by regressing the Fisher z-transformed correlation coefficients for each matrix element on subjects’ ages and dummy variables for each site. The resulting residuals— comprising a 258 × 258–element matrix for each subject—were an estimate of the functional connectivity between each ROI and every other ROI, controlling for age effects and relative to other subjects whose data were acquired on the same scanner. Henceforth, we refer to these matrices of residuals as functional connectivity matrices.
fMRI data analysis: canonical correlation analysis and clustering
To ensure that cluster discovery was not confounded by site-related differences in subject recruitment criteria or other unidentified variables, the cluster-discovery analysis was restricted to a subset of patients (the ‘cluster-discovery set,’ n = 220 of the 333 patients) from two sites with identical inclusion and exclusion criteria (see Supplementary Tables 1–3 for details). Each subject’s 258 × 258–element correlation matrix contained 33,154 unique functional connectivity features, necessitating a protocol for selecting a subset of relevant, nonredundant connectivity features for use in clustering. We reasoned that biologically meaningful depression subtypes would be best characterized by a low-dimensional representation of a subset of those 33,154 connectivity features that were significantly correlated with depressive symptoms. Therefore, to select a set of connectivity features for use in clustering, we (i) used Spearman’s rank correlation coefficients to identify connectivity features that were significantly correlated (P < 0.005) with severity scores for one or more of the 17 depressive symptoms, as indexed by individual item responses on the Hamilton Depression Rating Scale (HAMD-17), and then (ii) used canonical correlation analysis to define a low-dimensional representation of those connectivity features, in terms of linear combinations of connectivity features that were correlated with linear combinations of clinical symptoms. This empirical, data-driven approach to feature selection and dimensionality reduction identified two linear combinations of functional connectivity features (canonical variates) that were correlated with distinct clinical-symptom combinations, which we term “anhedonia-related connectivity features” and “anxiety-related connectivity features.” The results are depicted in Figure 1, with additional details in Supplementary Figure 2.
Next, to assess whether these abnormalities were evenly distributed across patients or tended to cluster in subgroups, we used hierarchical clustering to assign subjects to nested subgroups with similar patterns of abnormal connectivity along these two dimensions. We calculated a dissimilarity matrix describing the Euclidean distance between every pair of subjects in this two-dimensional feature space, and then used Ward’s minimum variance method to iteratively link pairs of subjects in closest proximity, forming progressively larger clusters in a hierarchical tree. These methods were implemented by using MATLAB’s pdist, linkage, cluster and clusterdata functions. The height of each link in the resulting dendrogram (Fig. 1d) represents the distance between the clusters being linked. On this basis, we conservatively identified at least four clusters for which the distance between cluster centroids was at least 20 times the mean distance between pairs of subjects within a cluster. Additional potential clustering solutions were also evident, nested within these subgroups. However, this four-cluster solution was optimal for defining relatively homogeneous subgroups that were maximally dissimilar from each other (maximizing the ratio of between-cluster to within-cluster variance), while ensuring individual cluster sample sizes that provided sufficient statistical power to detect biologically meaningful differences between biotypes (Supplementary Fig. 3). To construct the heat maps depicted in Figure 2, we used Wilcoxon rank-sum tests to identify connectivity features that were significantly different in patients with depression from each cluster, as compared to all controls, and Kruskal-Wallis ANOVA to identify connectivity features that differed most between clusters.
As described in the following section, we also investigated whether abnormal resting-state connectivity features could be used to diagnose these putative depression subtypes in individual subjects by training classifiers to detect them (Fig. 3). In our efforts to optimize classifier performance, we compared the hierarchical clustering method described above with k-means clustering, as implemented by MATLAB’s kmeans function, which assigns each subject to exactly one of k clusters on the basis of their squared Euclidean distance from the centroid of each cluster, iteratively assigning and reassigning subjects to a cluster to minimize the sum of the within-cluster sum-of-squares subject-to-centroid distances.
Classification: training and cross-validation of diagnostic classifiers for depression biotypes
In analyses depicted in Figure 3, we developed classifiers for diagnosing depression in subgroups of patients with similar patterns of abnormal functional connectivity in resting-state networks, testing and optimizing methods for brain parcellation and feature extraction, subject clustering, feature selection and classification to identify empirically the most successful approach. This optimization process was conducted exclusively in subjects from data set 1 (n = 711). As depicted in Figure 3a and in greater detail in Supplementary Figure 6, each optimization trial tested a combination of one of four methods for parcellation and feature extraction (coarse voxelwise parcellation, anatomical parcellation and two functional parcellations; see ‘Parcellation’ above); one of three methods for clustering (no clustering, k-means clustering or hierarchical clustering; see ‘Clustering’ above); and one of three methods for classification: logistic regression, support vector (SVM) classification or linear discriminant analysis (LDA).
On each optimization trial, a given combination of methods was evaluated by iteratively training classifiers on a subset (the ‘training subset’) of the subjects in data set 1 and then testing them on the remaining subjects (the ‘test subset’) through leave-one-out cross validation (LOOCV). As above, only the 220 patients in the two-site cluster-discovery set were used in the clustering analysis, whereas all 333 patients and 378 controls in data set 1 were eligible to be used in classification.
Assigning left-out subjects to clusters
The 133 patients (n = 333 − 220 = 133) left out of the cluster-discovery set were assigned to one of the four clusters in a two-step process. First, the canonical coefficients estimated in the cluster-discovery set were used to calculate canonical variate (component) scores for the left-out subjects. Second, LDA classifiers trained on the cluster-discovery sample were used to assign left-out subjects to one of the four clusters. The same two-step process was used to assign test subjects to the best-fitting cluster for the leave-one-out cross-validation analyses described below.
Classifier training
Classifier training was performed using the libsvm classification package67, the SPSS Statistics package (IBM: http://www.ibm.com/software/analytics/spss/products/statistics), or MATLAB classification functions (see schematic in Supplementary Fig. 6). Classifiers were trained to discriminate between patients with depression and healthy controls on the basis of a set of the most abnormal connectivity features, which were selected from the full set of all possible connectivity features (33,154 for the primary functional parcellation used in all other figures; 337,431 for the voxelwise parcellation; ~4,000 for the anatomical and second functional parcellations). In preliminary analyses (data not shown), we found that the optimal number of features depended on the parcellation strategy and classifier method. Simple logistic-regression classifiers could be trained only on a small set of features constrained by the number of subjects in each group; optimal performance was obtained in most cases with the top 20 features. SVM and LDA classifiers performed best when trained on the top ~5–10% of the most abnormal features for the primary functional and voxelwise parcellations (~1.5–3,000 and 10,000–25,000 features, respectively) and the top 25% for the coarser anatomical and functional parcellations (1,000 features). Thus, in Figure 3b, simple logistic-regression classifiers were trained on the top 20 features, whereas LDA and SVM classifiers were trained on the top ~2,000 features for the primary functional parcellation, ~1,000 features for the anatomical and secondary functional parcellations or ~10,000 features for voxelwise parcellation.
After being trained on subjects in the training subset, the resulting classifiers were tested on subjects in the test subset. Importantly, subjects in the test subset were left out of all aspects of the optimization procedure, including dimension reduction by canonical correlation analysis, clustering, feature selection and classifier training. This is crucial, because including members of the test subset in the clustering or feature-selection procedures will yield biased, inflated estimates of classifier accuracy. Trials that did not use clustering yielded one classifier on each iteration, which was then applied to subjects in the test subset, and the accuracy rates in Figure 3b represent the percentage of patients and healthy controls correctly classified as patients and healthy controls, respectively, averaged over all iterations. Trials that used clustering yielded three, four or five classifiers as indicated in Figure 3b. Testing each of them on every subject would tend to overestimate accuracy for patients and underestimate accuracy for healthy controls. Therefore, we tested only one of the biotype classifiers on each subject, on the basis of proximity to the cluster centroid or (in the case of the best performing classifiers depicted in Fig. 3g), by using the LDA classifiers for cluster assignment described above. For the purposes of defining a cluster’s centroid in order to make new cluster assignments, we excluded a small number of subjects (n = 15, or 6.8% of all subjects in the cluster-discovery set) with ambiguous cluster identities. These ‘edge cases’ were defined as cases with cluster silhouette values <0, indicating a case that was poorly matched to its own cluster and possibly better matched to a neighboring cluster. (We found that for small clusters, these edge cases could distort the calculation of the cluster’s centroid location, resulting in unstable cluster assignments across iterations.) In Figure 3c–f, the neuroanatomical locations of the most discriminating nodes were plotted by selecting connectivity features that were significantly different from controls (by Wilcoxon rank-sum tests) across each round of training and cross-validation. The nodes were colored and scaled by summing across all connectivity features associated with that node, as described in ref.68.
Permutation testing
By systematically testing various combinations of methods for parcellation, clustering, and classification, we found that the most successful classifier used our primary functional parcellation28, hierarchical clustering and SVM classification with linear kernel functions, and correctly identified healthy controls and patients with sensitivities of 84.1–90.9% and specificities of 84.1–92.5% (Fig. 3g). The statistical significance of these results was estimated by permutation testing, randomly permuting the diagnostic labels for each subject and applying the exact same procedure for clustering, feature selection and classifier training and repeating this procedure 200 times. Permutation testing was used to assess the statistical significance of the most successful classifier derived from each of the three classification methods (logistic regression, SVM and LDA). For all three methods, the reported accuracy rates exceeded those obtained on all 200 permutation tests, indicating a statistical significance of P < 0.005.
Classification: testing classifiers in an independent replication data set
It is well established in the machine-learning literature that iterative training and cross-validation on the same data overestimate classifier performance, and other studies have raised questions about the capacity for classifiers trained on one data set at a single site to generalize to data collected at multiple sites44,46. To address these issues, we tested the most successful classifier for each depression biotype (primary functional parcellation, hierarchical clustering and SVM classification) in an independent replication data set (data set 2; n = 477 subjects), comprising 125 patients and 352 healthy controls acquired from 13 sites, including five sites that were not included in the original training data set. This analysis was essentially identical to the analysis of test subjects in cross-validation described above. After preprocessing, parcellation and BOLD signal time-series extraction, we calculated correlation matrices, and the Fisher z-transformed correlation coefficients were corrected for age and site effects. For subjects in data set 2 who were scanned at a site that was included in data set 1, we corrected for age and site effects using the beta weights calculated for subjects in data set 1 to calculate residuals as described above. For subjects in data set 2 who were scanned at new sites that were not included in data set 1 (all healthy controls), we used multiple linear regression to estimate beta weights for these new sites. Next, the classifier for one depression biotype was tested on each subject by using the two-step procedure for cluster/biotype assignment described above (‘Assigning left-out subjects to clusters’). The overall accuracy rates and accuracies by cluster are reported in Figure 3i. To better understand the potential for further improvements in classifier performance in future, prospective data sets, we also calculated accuracy rates separately after implementing stricter data quality controls and by treating subjects with ambiguous classification outcomes as equivocal test results, as is common practice for biomarkers in other areas of medicine. These calculations excluded subjects with <300 s of data after censoring, motivated by reports that the stability of low-frequency BOLD signal-correlation estimates is higher for longer-duration scans;69 subjects with FD motion estimates exceeding 0.18 mm, i.e., the 95th percentile in our training set, motivated by our finding in Supplementary Figure 1 that classification rates in cross-validation (i.e., in data set 1) were slightly lower in the 5% of subjects with the highest levels of motion (χ2 = 5.096, P = 0.024); and the 10% of subjects with the lowest absolute SVM classification scores, i.e., equivocal classification outcomes. The results of these analyses are depicted in the cross-hatched bars in Figures 3i and 4j.
We also tested whether cluster assignments were stable over time, reasoning that if these clusters represent biologically meaningful depression subtypes, then a patient diagnosed with one of these subtypes should be diagnosed with the same subtype when re-tested at a later date. To assess this, we tested for reproducibility in a subset of subjects (n = 48) who were re-scanned 4–6 weeks after the initial scan and remained actively depressed (meeting DSM-IV criteria for a major depressive episode). As above, each subject was assigned to a cluster using the two-step procedure for biotype assignment described above (‘Assigning left-out subjects to clusters’), and we assessed the stability of cluster assignments across scans (Fig. 3h). A chi-squared test was used to assess the statistical significance of the longitudinal-stability results.
Clinical-data analysis
To assess whether biotypes of depression defined by unique patterns of resting state functional connectivity were associated with specific clinical profiles (Fig. 2f), we used Kruskal-Wallis analysis of variance to test for biotype differences in the severity of depressive symptoms in the cluster-discovery set (n = 220), as indexed by the HAMD. The six symptoms reported in Figure 2f showed the largest main effects of biotype (see Supplementary Fig. 4a for results for all 17 HAMD items). In Supplementary Figure 4c, we also tested for differences in these same six measures in clinical data acquired from subjects that were not included in the clustering analysis (n = 92).
In Figure 2c, we tested whether abnormal connectivity features that were shared across all four biotypes predicted the severity of ‘core’ symptoms that were present in almost all patients, regardless of biotype. We found that of the 17 symptoms quantified by the HAMD, three were present in almost all patients with depression (>90%); these included depressed mood (“feelings of sadness, hopelessness, helplessness”, 97.1%), anhedonia (96.7%) and anergia or fatigue (93.9%). We used principal-components analysis to define a low-dimensional representation of these shared, abnormal connectivity features and correlated the first component with severity scores for these three symptoms. The results are depicted in quartile plots in Figure 2c.
Repetitive transcranial magnetic stimulation and related analyses
In Figure 4, we tested whether depression biotypes defined by unique patterns of abnormal functionally connectivity were differentially responsive to rTMS in a subset of subjects (n = 154 in total) who received a course of excitatory repetitive TMS (10 Hz or intermittent theta burst stimulation) targeting the dorsomedial prefrontal cortex, beginning the week after their fMRI scan. The left dorsolateral prefrontal cortex is the most common target for stimulation in rTMS clinical trials48, but recent studies have demonstrated efficacy for the dorsomedial prefrontal cortical (DMPFC) target used here13,70. Of note, DMPFC was among the most important neuroanatomical areas differentiating the four biotypes in Figure 2d, which suggested to us that biotype differences in dysfunctional connectivity at the DMPFC target site may give rise to differing treatment outcomes.
The treatment parameters and scanning parameters for this sample have been previously described in detail elsewhere13,71. To summarize, all subjects received five sessions of TMS per week for 4–6 weeks (20–30 sessions total), delivered using a MagPro R30 rTMS device (MagVenture, Farum, Denmark) and a Cool-DB80 stimulation coil. For subjects who received 10-Hz stimulation (n = 86), each session included 3,000 pulses per hemisphere, delivered to the dorsomedial prefrontal cortex at 120% of resting motor threshold at a frequency of 10 Hz and with a duty cycle of 5 s on and 10 s off, for a total of 3,000 pulses in 60 trains per hemisphere per session (6,000 pulses total). For subjects who received intermittent theta burst stimulation (n = 68), each session included 600 pulses per hemisphere, delivered to the dorsomedial prefrontal cortex, at 120% of resting motor threshold, in 50 Hz triplet bursts, five bursts per second, with a duty cycle of 2 s on and 8 s off, for a total of 600 pulses in 20 trains per hemisphere per session (1,200 pulses total). To increase the tolerability of the DMPFC stimulation protocol, which has been associated with discomfort in some reports, all subjects also underwent a scalp-pain acclimatization protocol, as detailed in refs. 13,71. Depression severity was assessed using the 17-item HAMD before and after the course of treatment, and clinical improvements were measured in terms of changes in the total HAMD score.
To assess whether treatment response varied with depression biotype, subjects were classified as “treatment responders” or “treatment nonresponders”. Treatment responders were subjects who showed either a partial or full response to treatment, conventionally defined as a 25–50% or >50% reduction in HAMD scores, and “treatment nonresponders” were subjects who showed a <25% reduction in HAMD scores. A chi-squared test was used to assess whether treatment response rates varied with depression biotype, and Kruskal-Wallis analysis of variance was used to test whether change in HAMD varied with depression biotype (Fig. 4a,b).
In addition, we tested whether functional connectivity features and biotype diagnosis were predictive of treatment response in a training and cross-validation sample (~80% or n = 124 of the 154 patients; Fig. 4c–g) and then tested the best-performing classifier in an independent replication sample (~20%, n = 30 of the 154 patients). Using a procedure identical to the one described above, we used the primary functional parcellation, feature selection and SVM classification methods to iteratively train classifiers to prospectively identify TMS responders and nonresponders on the basis of connectivity features assessed before treatment, with leave-one-out cross validation (Fig. 4f). As above, the test subjects were left out of all aspects of feature selection and classifier training. We repeated this process using both connectivity features and biotype diagnosis, coded as four binary dummy variables (Fig. 4g). To understand whether clinical profiles were sufficient to predict treatment response without resting-state connectivity measures, we trained classifiers to differentiate responders and nonresponders solely on the basis of clinical data using an identical approach (Fig. 4h). Finally, we tested the best-performing classifier, which used both functional connectivity features and biotype diagnosis, in the independent replication sample (Fig. 4i).
Statistics
In Figure 1, canonical correlation analysis was used to define a low-dimensional representation of connectivity features (n = 220 patients from the “Toronto” and “Cornell 1” sites, Supplementary Table 1) that were predictive of two specific combinations of clinical symptoms (see above), and hierarchical clustering analysis (Fig. 1e–f) was used to delineate clusters of subjects in a two-dimensional space defined by these two canonical variates.
In Figure 2a–c, Wilcoxon rank-sum tests were used to test for differences in functional connectivity between all patients in the cluster-discovery set (n = 220) and all healthy controls (n = 378, Supplementary Table 3, training Data set), and Spearman rank correlations were used to test for associations with three clinical symptoms that were present in at least 90% of patients (n = 220). In Figure 2d,e, Kruskal-Wallis ANOVA (n = 220) was used to test for connectivity features that varied by biotype, and Wilcoxon rank-sum tests were used to assess whether these connectivity features were increased or decreased in depression (n = 220) as compared to controls (n = 378). In Figure 2f,g, Kruskal-Wallis ANOVA (n = 220) was used to test for differences in clinical-symptom severity by biotype.
In Figure 3b,g, classifier accuracy was assessed in leave-one-out cross validation in the full training data set (n = 333 patients, n = 378 healthy controls; Supplementary Table 3, training data set), with the test subject strictly excluded from all aspects of the clustering and classification optimization process, and statistical significance was assessed by establishing a null hypothesis distribution by randomly permuting diagnostic labels 500 times (see ‘Classification’ and ‘Permutation testing’ sections above). In Figure 3h, the longitudinal stability of biotype assignments was assessed in a subset of subjects from the cluster-discovery set (n = 50 patients with depression from “Cornell 1” site) who received a second fMRI scan obtained 4–5 weeks after the initial scan, and a chi-squared test (n = 50) was used to assess for a statistical dependence between biotype ID on scans 1 and 2. In Figure 3i, the most successful classifier identified in Figure 3b was tested in an independent replication data set (n = 125 patients, n = 352 healthy controls; Supplementary Table 3, replication data set). In Figures 3h and 3i, the scans used for testing longitudinal stability and for replicating classifier performance were not used in any aspect of the cluster-discovery process or classifier optimization.
In Figure 4a,b, chi-squared tests (a) and Kruskal-Wallis ANOVA (b) were used to test for biotype differences in response rates and improvements in depression severity (change in total HAMD), respectively, in patients after treatment with TMS (n = 124 patients with depression from training data set, “Toronto” site). In Figure 4c–e, Wilcoxon rank-sum tests were used to test for functional connectivity differences in TMS partial responders (n = 70) versus nonresponders (n = 54). In Figure 4f–i, classifier accuracy for differentiating responders (n = 70) and nonresponders (n = 54) was assessed by using leave-one-out cross validation and permutation testing, as in Figure 3, and the best-performing classifier was tested in an independent replication set (n = 30 patients with depression from replication data set, “Toronto” site) in Figure 4j.
In Figure 5a–c, Wilcoxon rank-sum tests were used to test for functional connectivity differences in patients with generalized anxiety disorder (n = 39 patients with GAD from “Cornell 1” and “Stanford 1” sites) versus healthy controls (n = 378, training data set; Supplementary Table 3), and a chi-squared test was used to test for significant overlap in depression- and GAD-related connectivity features (Fig. 5b). In Figure 5d and h, we applied the biotype classifiers developed in Figure 3 to the patients with GAD (n = 39) and to a separate cohort of patients diagnosed with schizophrenia (n = 41 patients with rsfMRI scans shared through the 1000 Functional Connectomes Project and the Center of Biomedical Research Excellence in Brain Function and Mental Illness (COBRE)). In Figure 5e–g, Kruskal-Wallis ANOVA was used to test for biotype differences in clinical symptom severity in the same patients with GAD (n = 39). Throughout, all P values are two-tailed, and all error bars are either s.e.m. or 95% confidence intervals, as defined in the corresponding figure legends.
Data availability
Data from the following sites (Supplementary Tables 2 and 3) are publicly available for download through the 1000 Functional Connectomes Project International Data Sharing Initiative (http://fcon_1000.projects.nitrc.org/index.html): NKI, Atlanta, Cambridge, Cleveland, ICBM, New York, COBRE, Beijing, Milwaukee and Leipzig. Data from the remaining sites are available at the discretion of the respective principal investigators, listed in Supplementary Table 2.
Supplementary Material
Acknowledgments
We wish to thank all investigators who volunteered to share MRI data via the 1000 Functional Connectomes Project (http://fcon_1000.projects.nitrc.org/index.html), which was supported by grants from the NIMH, NIDA, Autism Speaks, NINDS and HHMI. Principal investigators from sites that provided data used here include: R.L. Buckner (Harvard–MGH), F.X. Castellanos (NYU), A.C. Evans (ICBM), B. Leventhal (Nathan Kline Institute), S.J. Li (Medical College of Wisconsin), M.J. Lowe (Cleveland Clinic), H.M. Mayberg (Emory), M.P. Milham (Nathan Kline Institute), V. Riedl (Munchen), C. Sorg (Munchen), A. Villringer (Leipzig) and Y.F. Zang (Beijing Normal University). We also thank the following investigators at the University of New Mexico who provided public access to MRI data from patients diagnosed with schizophrenia through the Center of Biomedical Research Excellence in Brain Function and Mental Illness (COBRE): C. Aine, V. Calhoun, J. Canive, F. Hanlon, R. Jung, K. Kiehl, A. Mayer, N. Perrone-Bizzozero, J. Stephen and C. Tesche, who were supported by NIH COBRE grant 1P20RR021938-01A2. We also thank D. Fair (OHSU) and J. Power (NIMH, Weill Cornell) for providing comments on the data analysis, as well as members of the Liston Lab and Sackler Institute, for their helpful comments on the manuscript. H.S.M. was supported by a grant from the NIMH (P50 MH077083). C.L. was supported by grants from the Dana Foundation, Hartwell Foundation, International Mental Health Research Organization, Klingenstein-Simons Foundations, NARSAD and NIMH (R00 MH097822, R01 MH109685).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
J.D., K.D., F.M., D.J.O., A.E., A.F.S., K.S., J.K., H.S.M., F.M.G., G.S.A., M.D.F., A.P.-L., H.U.V., B.J.C., M.J.D. and C.L. collected the data. L.G. consulted on all statistical analyses. C.L. designed the protocol for analyzing data pooled across multiple sites and identifying clusters. A.T.D., R.F. and C.L. designed and implemented the preprocessing pipeline and methods for validating clusters and optimizing classifiers, and C.L. developed and implemented the method for clustering and classification in a low-dimensional connectivity-feature space by using canonical correlation analysis (Figs. 1–3). J.D., K.D. and F.M. collected the TMS data. C.L. analyzed the TMS response data and other clinical data (Figs. 2 and 4) and tested the subtype classifiers on subjects with other diagnoses (Fig. 5). A.T.D., Y.M. and C.L. implemented the permutation testing. A.T.D., B.Z. and C.L. created the figures and wrote the manuscript. All authors discussed the results and conclusions and edited the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Insel TR, Cuthbert BN. Medicine. Brain disorders? Precisely. Science. 2015;348:499–500. doi: 10.1126/science.aab2358. [DOI] [PubMed] [Google Scholar]
- 2.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll BJ, et al. A specifc laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry. 1981;38:15–22. doi: 10.1001/archpsyc.1981.01780260017001. [DOI] [PubMed] [Google Scholar]
- 4.Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7:254–275. doi: 10.1038/sj.mp.4001032. [DOI] [PubMed] [Google Scholar]
- 5.Lewy AJ, Sack RL, Miller LS, Hoban TM. Antidepressant and circadian phase-shifting effects of light. Science. 1987;235:352–354. doi: 10.1126/science.3798117. [DOI] [PubMed] [Google Scholar]
- 6.Clementz BA, et al. Identifcation of distinct psychosis biotypes using brain-based biomarkers. Am J Psychiatry. 2016;173:373–384. doi: 10.1176/appi.ajp.2015.14091200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill SK, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: fndings from the Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170:1275–1284. doi: 10.1176/appi.ajp.2013.12101298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wager TD, et al. An fMRI-based neurologic signature of physical pain. N Engl J Med. 2013;368:1388–1397. doi: 10.1056/NEJMoa1204471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liston C, et al. Default mode network mechanisms of transcranial magnetic stimulation in depression. Biol Psychiatry. 2014;76:517–526. doi: 10.1016/j.biopsych.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CH, et al. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry. 2007;62:407–414. doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Salvadore G, et al. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65:289–295. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox MD, Buckner RL, White MP, Greicius MD, Pascual-Leone A. Effcacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol Psychiatry. 2012;72:595–603. doi: 10.1016/j.biopsych.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Downar J, et al. Anhedonia and reward-circuit connectivity distinguish nonresponders from responders to dorsomedial prefrontal repetitive transcranial magnetic stimulation in major depression. Biol Psychiatry. 2014;76:176–185. doi: 10.1016/j.biopsych.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 14.McGrath CL, et al. Toward a neuroimaging treatment selection biomarker for major depressive disorder. JAMA Psychiatry. 2013;70:821–829. doi: 10.1001/jamapsychiatry.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greicius MD, et al. Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry. 2007;62:429–437. doi: 10.1016/j.biopsych.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drevets WC, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 17.Pezawas L, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- 18.Mayberg HS, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 19.Sheline YI, et al. The default mode network and self-referential processes in depression. Proc Natl Acad Sci USA. 2009;106:1942–1947. doi: 10.1073/pnas.0812686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knutson B, Bhanji JP, Cooney RE, Atlas LY, Gotlib IH. Neural responses to monetary incentives in major depression. Biol Psychiatry. 2008;63:686–692. doi: 10.1016/j.biopsych.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60:236–248. doi: 10.1002/neu.20025. [DOI] [PubMed] [Google Scholar]
- 22.Liston C, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gourley SL, Swanson AM, Koleske AJ. Corticosteroid-induced neural remodeling predicts behavioral vulnerability and resilience. J Neurosci. 2013;33:3107–3112. doi: 10.1523/JNEUROSCI.2138-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dias-Ferreira E, et al. Chronic stress causes frontostriatal reorganization and affects decision-making. Science. 2009;325:621–625. doi: 10.1126/science.1171203. [DOI] [PubMed] [Google Scholar]
- 25.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage. 2012;59:2142–2154. doi: 10.1016/j.neuroimage.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satterthwaite TD, et al. Impact of in-scanner head motion on multiple measures of functional connectivity: relevance for studies of neurodevelopment in youth. Neuroimage. 2012;60:623–632. doi: 10.1016/j.neuroimage.2011.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Dijk KRA, Sabuncu MR, Buckner RL. The infuence of head motion on intrinsic functional connectivity MRI. Neuroimage. 2012;59:431–438. doi: 10.1016/j.neuroimage.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Power JD, et al. Functional network organization of the human brain. Neuron. 2011;72:665–678. doi: 10.1016/j.neuron.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 30.Rihel J, et al. Zebrafsh behavioral profling links drugs to biological targets and rest/wake regulation. Science. 2010;327:348–351. doi: 10.1126/science.1183090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wager TD, Davidson ML, Hughes BL, Lindquist MA, Ochsner KN. Prefrontal-subcortical pathways mediating successful emotion regulation. Neuron. 2008;59:1037–1050. doi: 10.1016/j.neuron.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 33.Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 34.Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- 35.Pizzagalli DA, et al. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry. 2009;166:702–710. doi: 10.1176/appi.ajp.2008.08081201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferenczi EA, et al. Prefrontal cortical regulation of brainwide circuit dynamics and reward-related behavior. Science. 2016;351:aac9698. doi: 10.1126/science.aac9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- 38.Cardinal RN, Parkinson JA, Hall J, Everitt BJ. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev. 2002;26:321–352. doi: 10.1016/s0149-7634(02)00007-6. [DOI] [PubMed] [Google Scholar]
- 39.Gottfried JA, O’Doherty J, Dolan RJ. Encoding predictive reward value in human amygdala and orbitofrontal cortex. Science. 2003;301:1104–1107. doi: 10.1126/science.1087919. [DOI] [PubMed] [Google Scholar]
- 40.Schultz W. Behavioral theories and the neurophysiology of reward. Annu Rev Psychol. 2006;57:87–115. doi: 10.1146/annurev.psych.56.091103.070229. [DOI] [PubMed] [Google Scholar]
- 41.Rosa MJ, et al. Sparse network-based models for patient classifcation using fMRI. Neuroimage. 2015;105:493–506. doi: 10.1016/j.neuroimage.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craddock RC, Holtzheimer PE, III, Hu XP, Mayberg HS. Disease state prediction from resting state functional connectivity. Magn Reson Med. 2009;62:1619–1628. doi: 10.1002/mrm.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng LL, et al. Identifying major depression using whole-brain functional connectivity: a multivariate pattern analysis. Brain. 2012;135:1498–1507. doi: 10.1093/brain/aws059. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen JA, et al. Multisite functional connectivity MRI classifcation of autism: ABIDE results. Front Hum Neurosci. 2013;7:599. doi: 10.3389/fnhum.2013.00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plitt M, Barnes KA, Martin A. Functional connectivity classifcation of autism identifes highly predictive brain features but falls short of biomarker standards. Neuroimage Clin. 2014;7:359–366. doi: 10.1016/j.nicl.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. Springer-Verlag; 2009. [Google Scholar]
- 47.George MS, et al. Daily repetitive transcranial magnetic stimulation (rTMS) improves mood in depression. Neuroreport. 1995;6:1853–1856. doi: 10.1097/00001756-199510020-00008. [DOI] [PubMed] [Google Scholar]
- 48.Pascual-Leone A, Rubio B, Pallardó F, Catalá MD. Rapid-rate transcranial magnetic stimulation of left dorsolateral prefrontal cortex in drug-resistant depression. Lancet. 1996;348:233–237. doi: 10.1016/s0140-6736(96)01219-6. [DOI] [PubMed] [Google Scholar]
- 49.Huang YZ, Rothwell JC, Edwards MJ, Chen RS. Effect of physiological activity on an NMDA-dependent form of cortical plasticity in human. Cereb Cortex. 2008;18:563–570. doi: 10.1093/cercor/bhm087. [DOI] [PubMed] [Google Scholar]
- 50.Davidson RJ, Pizzagalli D, Nitschke JB, Putnam K. Depression: perspectives from affective neuroscience. Annu Rev Psychol. 2002;53:545–574. doi: 10.1146/annurev.psych.53.100901.135148. [DOI] [PubMed] [Google Scholar]
- 51.Oathes DJ, Patenaude B, Schatzberg AF, Etkin A. Neurobiological signatures of anxiety and depression in resting-state functional magnetic resonance imaging. Biol Psychiatry. 2015;77:385–393. doi: 10.1016/j.biopsych.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodkind M, et al. Identifcation of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72:305–315. doi: 10.1001/jamapsychiatry.2014.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker JT, et al. Disruption of cortical association networks in schizophrenia and psychotic bipolar disorder. JAMA Psychiatry. 2014;71:109–118. doi: 10.1001/jamapsychiatry.2013.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 55.Ripke S, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pankevich DE, Altevogt BM, Dunlop J, Gage FH, Hyman SE. Improving and accelerating drug development for nervous system disorders. Neuron. 2014;84:546–553. doi: 10.1016/j.neuron.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krishnan V, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 58.Chaudhury D, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–536. doi: 10.1038/nature11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tye KM, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Steps toward optimizing motion artifact removal in functional connectivity MRI; a reply to Carp. Neuroimage. 2013;76:439–441. doi: 10.1016/j.neuroimage.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carp J. Optimizing the order of operations for movement scrubbing: Comment on Power et al. Neuroimage. 2013;76:436–438. doi: 10.1016/j.neuroimage.2011.12.061. [DOI] [PubMed] [Google Scholar]
- 62.Jo HJ, et al. Effective preprocessing procedures virtually eliminate distance-dependent motion artifacts in resting state fMRI. J Appl Math. 2013;2013:935154. doi: 10.1155/2013/935154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jo HJ, Saad ZS, Simmons WK, Milbury LA, Cox RW. Mapping sources of correlation in resting state FMRI, with artifact detection and removal. Neuroimage. 2010;52:571–582. doi: 10.1016/j.neuroimage.2010.04.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Power JD, Schlaggar BL, Petersen SE. Recent progress and outstanding issues in motion correction in resting state fMRI. Neuroimage. 2015;105:536–551. doi: 10.1016/j.neuroimage.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Desikan RS, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 66.Shirer WR, Ryali S, Rykhlevskaia E, Menon V, Greicius MD. Decoding subject-driven cognitive states with whole-brain connectivity patterns. Cereb Cortex. 2012;22:158–165. doi: 10.1093/cercor/bhr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang CC, Lin CJ. LIBSVM: A library for support vector machines. ACM Trans Intell Syst Technol. 2011;2:27. [Google Scholar]
- 68.Dosenbach NUF, et al. Prediction of individual brain maturity using fMRI. Science. 2010;329:1358–1361. doi: 10.1126/science.1194144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Dijk KRA, et al. Intrinsic functional connectivity as a tool for human connectomics: theory, properties, and optimization. J Neurophysiol. 2010;103:297–321. doi: 10.1152/jn.00783.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salomons TV, et al. Resting-state cortico-thalamic-striatal connectivity predicts response to dorsomedial prefrontal rTMS in major depressive disorder. Neuropsychopharmacology. 2014;39:488–498. doi: 10.1038/npp.2013.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bakker N, et al. rTMS of the dorsomedial prefrontal cortex for major depression: safety, tolerability, effectiveness, and outcome predictors for 10 Hz versus intermittent theta-burst stimulation. Brain Stimul. 2015;8:208–215. doi: 10.1016/j.brs.2014.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the following sites (Supplementary Tables 2 and 3) are publicly available for download through the 1000 Functional Connectomes Project International Data Sharing Initiative (http://fcon_1000.projects.nitrc.org/index.html): NKI, Atlanta, Cambridge, Cleveland, ICBM, New York, COBRE, Beijing, Milwaukee and Leipzig. Data from the remaining sites are available at the discretion of the respective principal investigators, listed in Supplementary Table 2.