

Abstract
Potent mechanism-based inactivators can be rationally designed against pyridoxal 5′-phosphate (PLP)-dependent drug targets, such as ornithine aminotransferase (OAT) or γ-amino-butyric acid aminotransferase (GABA-AT). An important challenge, however, is the lack of selectivity toward other PLP-dependent, off-target enzymes, because of similarities in mechanisms of all PLP-dependent aminotransferase reactions. On the basis of complex crystal structures, we investigate the inactivation mechanism of OAT, a hepatocellular carcinoma target, by (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid (FCP), a known inactivator of GABA-AT. A crystal structure of OAT and FCP showed the formation of a ternary adduct. This adduct can be rationalized as occurring via an enamine mechanism of inactivation, similar to that reported for GABA-AT. However, the crystal structure of an off-target, PLP-dependent enzyme, aspartate aminotransferase (Asp-AT), in complex with FCP, along with the results of attempted inhibition assays, suggests that FCP is not an inactivator of Asp-AT, but rather an alternate substrate. Turnover of FCP by Asp-AT is also supported by high-resolution mass spectrometry. Amid existing difficulties in achieving selectivity of inactivation among a large number of PLP-dependent enzymes, the obtained results provide evidence that a desirable selectivity could be achieved, taking advantage of subtle structural and mechanistic differences between a drug-target enzyme and an off-target enzyme, despite their largely similar substrate binding sites and catalytic mechanisms.
Graphical abstract
Pyridoxal 5′-phosphate (PLP), the active form of vitamin B6, is a coenzyme in a variety of enzymatic reactions. Aminotransferases make up a class of PLP-dependent enzymes that catalyze myriad biochemical reactions involving amino acid metabolism, amino acid-derived metabolites, and the biosynthesis of amine-containing compounds.1,2 The importance of these enzymes is further highlighted because some of them have been identified as drug targets. For example, inhibition of γ-aminobutyric acid aminotransferase (GABA-AT) was proven to be effective in the treatment of many neurological disorders,3 and inhibition of ornithine aminotransferase (OAT) was shown to suppress the growth of hepatocellular carcinoma (HCC).4,5 Although reactions catalyzed by PLP-dependent enzymes appear to be diverse, they all share a similar catalytic mechanism that makes it challenging to design specific inactivators for each of these drug-target enzymes without affecting one another or other PLP-dependent, off-target enzymes like aspartate aminotransferase (Asp-AT).
As an example of the general mechanism of amino-transferases, Asp-AT catalyzes a reversible transamination converting L-aspartate and α-ketoglutarate to oxaloacetate and L-glutamate (Scheme 1A).6 During the catalysis, the cofactor is interconverted between its PLP and pyridoxamine (PMP) forms, facilitating the aminotransferase reaction. The Asp-AT reaction is crucial in both amino acid degradation and biosynthesis.7 Therefore, as part of the design of an inactivator for PLP-dependent drug targets, any molecule that also inhibits and/or inactivates Asp-AT or similar off-target enzymes may not be promising for further pharmacological development because of possible side effects. OAT and GABA-AT are confirmed PLP-dependent, drug-target enzymes,3 and these two, as well as Asp-AT, catalyze an identical second-half “ping-pong” reaction, namely, the conversion of α-ketoglutarate to L-glutamate. OAT (EC 2.6.1.13) catalyzes the transfer of the δ-amino group of L-ornithine to α-ketoglutarate, forming glutamate γ-semialdehyde and L-glutamate (Scheme 1B). OAT is overexpressed in HCC cells, and its inhibition was shown to suppress their growth.8 GABA-AT (EC 2.6.1.19) catalyzes the transfer of the amino group of GABA to α-ketoglutarate, producing succinic semialdehyde and L-glutamate (Scheme 1C). GABA is the principal inhibitory neuro-transmitter in mammalian cells,9 and abnormally low levels of GABA in the brain have been associated with several neurological disorders, such as epilepsy, and several neuro-degenerative diseases, such as Parkinson’s, Huntington’s, and Alzheimer’s diseases.3 Inhibition of GABA-AT to increase the brain level of GABA has been proven to be beneficial in the treatment of epilepsy.10 Much of the drug design effort has focused on mechanism-based inactivators, which are unreactive compounds that require the catalytic activity of the enzyme to convert them into reactive species that then inactivate the enzyme.
Scheme 1.

General Mechanism of (A) Aspartate Aminotransferase, (B) Ornithine Aminotransferase, and (C) GABA Aminotransferase
In this study, we investigated the mechanism by which (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid (FCP), a known inactivator of GABA-AT11 and OAT,8 inactivates OAT, and the effects of FCP on the PLP-dependent, off-target enzyme Asp-AT. Our study serves as a proof of principle that new OAT inactivators could be developed from GABA analogues, without targeting other off-target, PLP-dependent enzymes. Previously, Storici et al.11 studied the inactivation of GABA-AT by FCP. It inactivated GABA-AT through an enamine mechanism (Figure 1A), with the crystal structure of the inactivated enzyme showing adduct 5 derived from this mechanism (Figure 1B). In this study, we obtained the crystal structure of inactivated OAT by FCP and examined the implications for the inactivation mechanism. We also report the crystal structures of the off-target enzyme Asp-AT in complex with FCP in the presence and absence of α-ketoglutarate during cocrystallization. The crystal structures of Asp-AT in complex with FCP, along with attempted inhibition assays and high-resolution mass spectrometry, support the turnover of FCP by Asp-AT and the proposed mechanism.
Figure 1.

Mechanism of inactivation of GABA-AT by FCP. (A) Scheme representing the mechanism of inactivation of GABA-AT by FCP. (B) Crystal structure of inactivated GABA-AT showing adduct 5 derived via an enamine mechanism.11
MATERIALS AND METHODS
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO), and all enzymes used for cloning were purchased from New England Biolabs (Beverly, MA).
Cloning, Expression, and Purification of Human OAT
A shuttle vector (pCMV-SPORT6) carrying the OAT cDNA gene from Homo sapiens was purchased from plasmID (Harvard Medical School, Boston, MA). The coding sequence for OAT was cloned in a pMAL-C5X vector for expression in Escherichia coli. A sequence encoding the cleavage site for tobacco etch virus (TEV) protease (ENLYFQG) was inserted at the 5′ end of the OAT coding region using forward primer 5′GCGCT-CGGGGAAAACCTGTATTTTCAGGGCGCCTCTGCTAC-ATCTGTTGCAAC3′ and reverse primer 5′GCGGAATTCT-CAGAAAGACAAGATGGTC3′; restriction sites AvaI and EcoRI were added to the 5′ and 3′ ends, respectively. A commercial protein expression vector, pMAL-C5X (New England Biolabs), was digested using restriction enzymes EcoRI and AvaI, and the resulting polymerase chain reaction insert containing the OAT coding region was ligated into the expression vector using T4 DNA ligase (New England Biolabs) to yield a protein expression plasmid encoding an N-terminal maltose binding protein (MBP) linked through a TEV cleavage sequence to the full length OAT enzyme. The resulting vector (pMAL-t-OAT) was used to transform E. coli DH5α cells for plasmid storage and amplification. The entire coding region in pMAL-t-OAT was verified by DNA sequencing (CRC DNA Sequencing, University of Chicago, Chicago, IL).
For protein production and purification, pMAL-t-OAT was used to transform E. coli BL21(DE3) cells. The resulting E. coli BL21(DE3) (pMAL-t-OAT) cells were incubated at 37 °C while being shaken in Luria-Bertani (LB) medium supplemented with 100 μg/mL ampicillin. When the culture OD600 value reached 0.6–0.8 absorbance unit, expression of the MBP–t-OAT fusion protein was induced by addition of 0.3 mM isopropyl β-D-1-thiogalactopyranoside, and the expression was continued for an additional 16–18 h at 25 °C after induction. Cells were harvested by centrifugation at 12400g, washed with wash buffer [20 mM Tris-HCl buffer with 200 mM NaCl and 100 μM PLP (pH 7.4)], and stored at −80 °C after being flash-frozen in liquid nitrogen. The frozen cell pellet was thawed, sonicated in wash buffer, and centrifuged at 40000g to pellet cell debris, which was discarded. The resulting supernatant was loaded onto an amylose affinity column (16 mm × 25 mm Dextrin Sepharose-MBL Trap HP, GE LifeSciences, pre-equilibrated with wash buffer). The column was washed with wash buffer, and the MBP–t-OAT fusion protein was eluted from the column using wash buffer supplemented with maltose (10 mM). Fractions were evaluated using Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and those fractions containing the MBP–t-OAT fusion protein were combined and treated batchwise with TEV protease using previously published protocols.12 Fractions containing untagged OAT protein were combined, concentrated using a 30000 molecular weight cutoff (MWCO) Amicon (Millipore, MA) Ultra centrifugal filter device, and further purified by size exclusion chromatography using a HiLoad Superdex-200PG column (16 mm × 600 mm) (GE Lifesciences). The column was equilibrated, and the protein was purified using size exclusion buffer [50 mM HEPES buffer and 300 mM NaCl (pH 7.5)]. During the purification, fractions were assayed for the presence of the MBP–t-OAT fusion protein or OAT at each step by using 12% SDS–PAGE, followed by staining with EZ-Run Gel Staining Solution (Fisher BioReagents) to detect bands at ~92 or ~49 kDa, respectively. The final purified untagged OAT protein appeared to be homogeneous upon being characterized on a Coomassie-stained 12% SDS–PAGE gel. Protein concentrations in solution were measured using bovine serum albumin (BSA) standards and the Bradford assay (Bio-Rad). This purification procedure typically results in a yield of 30 mg of purified untagged OAT/L culture medium.
Purification of E. coli Aspartate Aminotransferase
The pJO2 plasmid (provided by J. F. Kirsch) containing the wild-type aspC gene was transformed into the DH5α E. coli strain. Wild-type Asp-AT was overexpressed and purified by following a published protocol.13 Accordingly, 0.1% PLP was added to the expression culture. After the harvested cells were broken via sonication and the cell extract was centrifuged at 12400g, the supernatant was removed and mixed with PEG 8000 that was slowly added to a final concentration of 20% of the total volume of the supernatant. The resulting suspension was again centrifuged at 12400g. The supernatant was diluted in a DEAE running buffer and run through a DEAE column and a hydroxyapatite column on an FPLC instrument consecutively. For crystallization, an additional purification step was added using a HiLoad Superdex-200PG column (16 mm × 600 mm) (GE Lifesciences). The final yield of purified Asp-AT was 80–100 mg/L of culture. For crystallization, the purified protein was exchanged into a 25 mM potassium phosphate buffer at pH 6.5 at a final concentration of 10 mg/mL. A 1 mM PLP solution was added to the protein solution to stabilize the enzyme. All purification procedures were performed at or near 4 °C. During the purification, fractions were assayed for the presence of Asp-AT at each step using 12% SDS–PAGE, followed by staining with EZ-Run Gel Staining Solution (Fisher BioReagents) to detect bands at ~47 kDa.
Attempted Inhibition Assays of FCP against Asp-AT
Microtiter plate wells were loaded with 90 μL of an assay mixture containing potassium phosphate (100 mM, pH 7.4), α-ketoglutarate (5.55 mM), NADH (1.11 mM), L-aspartate (5.55 mM), malate dehydrogenase (5.5 units), and various concentrations of FCP. After the mixture was incubated at room temperature for a few minutes, 10 μL of Asp-AT [2.0 units/mL in 100 mM potassium phosphate (pH 7.4)] was added. The plate was shaken at room temperature for 1 min, and the absorbance was measured at 340 nm every 10 s for 60 min. All assays were performed in duplicate.
Crystallization of OAT and FCP
Potassium pyrophosphate buffer (500 μL, 100 mM, pH 8.0), containing human OAT (2.2 mg, 49 nmol), α-ketoglutarate (10 mM), β-mercaptoethanol (10 mM), and FCP (50 mM), was protected from light and incubated at room temperature for 14 h until the human OAT was inactivated (activity of ≲0.1%). The inactivated OAT was then concentrated to 10 mg/mL using a 30000 MWCO Amicon Ultra centrifugal filter device, and the concentrated protein was exchanged into a crystallization buffer [50 mM Tricine HCl (pH 7.8) and 10 μM PLP]. Crystals were grown using previously published conditions.14 Crystallization was performed in 24-well Cryschem Plates (Hampton Research). Crystals with the best morphology appeared in the well solution containing NaCl (175 mM) and 8–10% (v/v) PEG 6000 after incubation for 1 week at room temperature. OAT crystals with the best morphology were transferred into a cryo-protecting solution [well solution supplemented with 25% (v/v) glycerol] before being flash-cooled in liquid nitrogen.
Cocrystallization of Asp-AT and FCP in the Presence and Absence of α-Ketoglutarate
The cocrystallization of Asp-AT and FCP in the absence of α-ketoglutarate was conducted using the hanging-drop method. The well solutions contained 25 mM potassium phosphate and 43% saturated ammonium sulfate. Hanging drops were set up by mixing 4 μL of the protein solution (10 mg/mL), 2 μL of FCP (50 mM, stock solution), and 4 μL of the well solution. The pH values of the solutions were adjusted to 6.5. The crystals appeared overnight. The crystals were then transferred to a cryo-protectant solution that consisted of 48% saturated ammonium sulfate, 25 mM potassium phosphate, 20% glycerol, and 1 mM PLP and was adjusted to the pH value at which the crystals were formed. The crystals were frozen in liquid nitrogen after being dipped into the cryo-protectant solution. For the cocrystallization of Asp-AT and FCP in the presence of α-ketoglutarate, the protein buffer was substituted with a solution containing α-ketoglutarate (10 mM); other crystallization conditions remained the same.
Data Collection and Processing
Monochromatic data sets were collected at the LS-CAT, Advanced Photon Source (APS), at Argonne National Laboratory (ANL, Argonne, IL). Diffraction data were collected at a wavelength of 0.98 Å at 100 K using a Mar300 Charge Coupled Device (CCD) detector. All collected data sets were indexed and integrated using the HKL2000 suite.15 Data collection statistics are summarized in Table 1.
Table 1.
Crystallographic Data for OAT–FCP, Asp-AT–FCP, and Asp-AT–FCP–KG Complexes
| OAT–FCP | Asp-AT–FCP | Asp-AT–FCP–KG | |
|---|---|---|---|
| Data Processinga | |||
| space group | P3221 | P1 | C2221 |
| no. of molecules per asymmetric unit | 3 | 4 | 1 |
| cell dimensions | |||
| α, β, γ(deg) | 115.3, 115.3, 187.9 | 118.8, 90.1, 89.6 | 84.4, 155.5, 77.4 |
| a, b, c (Å) | 90, 90, 120 | 78.9, 84.9, 88.0 | 90.0, 90.0, 90.0 |
| resolution (Å) | 1.78 | 1.80 | 1.72 |
| resolution at I/σ(I) = 2 | 1.84 | 1.80 | 1.75 |
| Rmergeb (%) | 9.3 (0.0) | 6.1 (40.0) | 4.0 (98.0) |
| Rpimc (%) | 3.9 (79.3) | – | 2.7 (53.0) |
| I/σ(I) | 26.2 (1.5) | 20.0 (2.8) | 25.3 (1.9) |
| CC1/2d | 1.00 (0.57) | – | 1.00 (0.70) |
| completeness (%) | 99.8 (99.0) | 98.2 (83.4) | 99.7 (99.5) |
| multiplicity | 9.4 (7.5) | 3.8 (3.3) | 4.3 (4.0) |
| no. of reflections | 1315591 | 676385 | 240049 |
| no. of unique reflections | 139552 | 178845 | 55946 |
| Refinement | |||
| Rwork,e Rfreef(%) | 17.8, 21.0 | 16.2, 19.2 | 16.7, 19.04 |
| no. of atoms | |||
| protein | 9626 | 13737 | 3105 |
| ligand | 32 | 64 | 25 |
| water | 1309 | 1434 | 366 |
| B factor (Å2) | |||
| protein | 26.4 | 30.3 | 25.2 |
| ligand | 16.7–32.9 | 17.72–32.7 | 11.9–32.0 |
| RMSDg | |||
| bond lengths (Å) | 0.01 | 0.01 | 0.01 |
| bond angles (deg) | 1.11 | 1.49 | 1.14 |
| Ramachandran plot (%) | |||
| most favored | 96.41 | 96.47 | 95.77 |
| allowed | 3.27 | 3.04 | 4.23 |
| outliers | 0.33 | 0.50 | 0.00 |
The values for the highest-resolution bin are in parentheses.
Rmerge = Σ|Iobs − Iavg|/ΣIavg.
Precision-indicating merging R.
Pearson correlation coefficient of two “half” data sets.
Rwork = Σ|Fobs − Fcalc|/ΣFobs.
Five percent of the reflection data were selected at random as a test set, and only these data were used to calculate Rfree.
Root-mean-square deviation.
Model Building and Refinement
The OAT structure and Asp-AT structures were determined by molecular replacement using PHASER in the Phenix software suit.16 The initial search model was based on a previously published structure of OAT [Protein Data Bank (PDB) entry 1OAT] and Asp-AT (PDB entry 2AAT). Models were rebuilt using COOT,17 refined using Phenix,16 and analyzed using COOT17 and UCSF Chimera.18 All ligands were built and regularized using JLigand in the CCP419 program suit. Final refinement statistics are listed in Table 1. Structural figures were made using UCSF Chimera.
Mass Spectrometry Study on a Single Turnover of FCP
Ammonium bicarbonate buffer (200 μL, 50 mM, pH 7.4) containing Asp-AT (39.5 μM) and FCP (15.8 mM) was protected from light and incubated at room temperature for 12 h. A similar control solution that did not contain FCP was also incubated. After 12 h, formic acid (5 μL) was added to each solution, and both were centrifuged in a 0.5 mL 10 kDa MWCO centrifuge tube at 13400g for 20 min or until most of the solution had passed through. Triplicate deproteinated reaction mixtures from incubation of Asp-AT with FCP, or the control, were analyzed by high-performance liquid chromatography with electrospray ionization mass spectrometry (HPLC–ESI-MS). Ten microliters of each mixture was injected on a Phenomenex Luna C18(2) column (2.1 mm × 150 mm) and analyzed by a linear gradient at 200 μL/min from 10 to 90% buffer B over 15 min, where buffer A was water with 0.1% formic acid and buffer B was acetonitrile with 0.1% formic acid. The eluent from LC was split 1:4 so that 20% of the eluent (40 μL/min) flowed directly into a Thermo Ion Max ESI source connected to a Thermo Q-Exactive. Data were collected in negative ion mode through a data-dependent top five method where the first duty cycle scan was a global scan from 100 to 1000 Th, followed by MS2 fragmentation of the five most abundant ions with the higher-energy collisional dissociation cell set to a normalized collision energy of 45 arbitrary units. Extracted ion chromatograms for ions of interest were integrated for total peak area and normalized relative to the total ion current for each run. Normalized values were then scaled to represent normalized absolute ion abundances. Values from triplicate samples were averaged, and error bars were generated on the basis of the standard deviation.
RESULTS AND DISCUSSION
Cloning and Protein Purification
The cloning results were confirmed by DNA sequencing. Heterologous expression of OAT and Asp-AT in E. coli BL21(DE3) cells led to good yields of purified proteins (~10 and ~30 mg/L of culture, respectively). The purified enzymes were used for structural and biochemical studies.
Inactivation of OAT by FCP
Crystallographic Studies of OAT Inactivated by FCP (OAT–FCP) Yielded a Complex Structure with a Ternary Adduct
FCP was previously shown to inactivate GABA-AT and OAT.8 On the basis of a reported complex structure with GABA-AT,11 FCP inactivates GABA-AT through an enamine mechanism (Figure 1A). In this work, the obtained OAT–FCP complex structure was determined by molecular replacement using a monomer of a known OAT structure as a search model (PDB entry 1OAT) after deletion of all water and ligand molecules. In space group P3221, three monomers were found in one asymmetric unit. Two of the three monomers form a biological assembly that is a homodimer; the other one forms a homodimer with a monomer from another asymmetric unit. The final model was refined to a resolution of 1.8 Å with Rfree and Rwork values of 21.3 and 18.4%, respectively. Final refinement statistics are listed in Table 1.
No major conformational changes were observed when the known structure of OAT (PDB entry 1OAT) was compared to that of the OAT–FCP complex (root-mean-square deviation of 0.4 Å using SSM superposition20 in COOT). The active site of the OAT–FCP complex (Figure 2A) displays a ternary adduct formed by Lys292, PLP, and FCP. Atom C4 of PLP, at which three parts of the ternary adduct are joined covalently, was refined with an R configuration and tetrahedral geometry. This ternary adduct is similar to the adduct observed in the active site of the crystal structure of inactivated GABA-AT by FCP11 (Figure 1B). In the OAT–FCP complex, Arg180 forms a salt bridge with the carboxyl group of FCP; this noncovalent interaction has been reported to be important for substrate binding at the active site.3 Figure 2B shows a simulated annealing omit map (Fo – Fc) superimposed with the ternary adduct.
Figure 2.

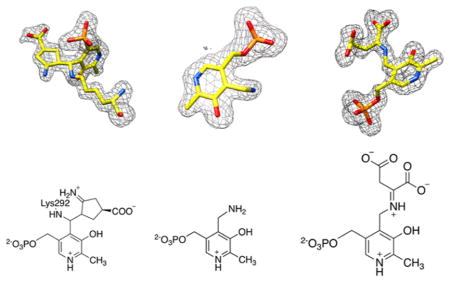
Structure of the adduct formed after the inactivation of OAT by FCP. Protein atoms are shown as sticks, with oxygen colored red, nitrogen colored blue, and phosphorus colored orange. (A) Active site of OAT with the ternary adduct formed by K292, FCP, and PLP, which is colored yellow (PDB: 5VWO). The dashed lines represent hydrogen bonds or electrostatic interactions between the active site residues of OAT (green) and the adduct. (B) Simulated annealing omit map (Fo – Fc) of the adduct. The omit map is shown as a gray mesh at 3σ. The omit map was generated after FCP, PLP, and K292 had been omitted from the coordinates. (C) Chemical structure of the adduct formed by FCP, PLP, and K292.
The formation of the ternary adduct implies that the inactivation of OAT by FCP proceeds via an enamine mechanism, similar to that of the inactivation of GABA-AT by FCP. This mechanism is initiated by the formation of the Schiff base between FCP and PLP (1), followed by γ-proton removal and tautomerization that lead to the release of the fluoride ion and enamine 7 (Scheme 2). Subsequent nucleophilic addition of 7 to the lysine-bound PLP on OAT gives rise to ternary adduct 8, which is similar to 5 in the mechanism of inactivation of GABA-AT by FCP (Figure 1A).
Scheme 2.

Proposed Mechanism of Inactivation of OAT by FCP
FCP Acts as an Alternative Substrate, Not an Inactivator of Asp-AT
FCP was tested for its potential inhibitory capacity on Asp-AT using the coupled enzyme assay described in Materials and Methods. The results showed FCP did not inactivate or inhibit the activity of Asp-AT at a concentration of ≤10 mM (Figure S1). Cocrystallization of Asp-AT and FCP in the absence of α-ketoglutarate (Asp-AT–FCP) yielded crystals that diffracted to 1.8 Å. The data set was processed in space group P1, and the crystal structure was determined by molecular replacement using a known structure of Asp-AT (PDB entry 2AAT) as a search model. The resulting structure has four monomers per asymmetric unit, and the model was refined with Rfree and Rwork values of 18.7 and 16.4%, respectively. Final refinement statistics are listed in Table 1.
PMP was identified in the crystal structure of the Asp-AT–FCP complex. The active site of the Asp-AT-–FCP complex (Figure 3A) displays the electron density that is not consistent with the formation of a ternary adduct as being observed in the crystal structures of FCP-inactivated GABA-AT or OAT. Instead, this electron density fits well with PMP. The discontinuity in the electron density between Lys246 and the putative pyridoxamine moiety is consistent with the turnover of PLP to PMP. In addition, the electron density around C7 of the putative PMP fits well with an sp3 hybridized carbon, providing further evidence that the coenzyme is in the pyridoxamine rather than the pyridoxal form (Figure 3C). Figure 3B shows a simulated annealing omit map (Fo – Fc) superimposed with the atomic model of PMP. The interactions between the active site residues and PMP (Figure 3A) in this crystal structure are the same as those in a previously reported structure of the Asp-AT_PMP holoenzyme.21,22
Figure 3.

Structure of Asp-AT and PMP. Protein atoms are shown as pink sticks, with oxygen colored red, nitrogen colored blue, and phosphorus colored orange. (A) Active site of Asp-AT and PMP (colored yellow, PDB: 5VWQ). The dashed green lines represent the hydrogen bonds or electrostatic interactions between PMP and the active site residues of Asp-AT. (B) Simulated annealing omit map (Fo – Fc) of the adduct. The omit map is shown as a gray mesh at 2.5σ. The omit map was generated after PMP had been omitted from the coordinates. (C) Chemical structure of PMP.
The conversion of PLP to PMP indicates that off-target Asp-AT can turn over FCP as an alternative substrate. E. coli Asp-AT is similar to H. sapiens (human) Asp-AT with >40% sequence identity and is a model enzyme among Asp-ATs, which are known to catalyze reactions via the same mechanism.2,6 Overexpressed and purified E. coli Asp-AT has been used as the off-target model enzyme to design inactivators against PLP-dependent enzymes.23,24 The lack of inactivation by FCP and the observation of PMP bound to Asp-AT in the presence of FCP suggest that Asp-AT catalyzes the conversion of the amino group on FCP to a keto group through the same mechanism by which it converts aspartate to oxaloacetate, and this reaction stops at the generation of PMP after the first half of the “ping-pong” catalytic cycle.
Trapping a Ketimine Intermediate in the Asp-AT Crystal Structure Further Supports the FCP Turnover
On the basis of the interpretation of the turnover of FCP by Asp-AT and the conversion of PLP to PMP, it is expected that cocrystallization of Asp-AT in the presence of FCP and its second substrate, α-ketoglutarate, should reveal a structure representing regeneration of the enzyme back to the PLP form, as proposed in the second half of the Asp-AT catalytic cycle.
Cocrystallization of Asp-AT and FCP in the presence of α-ketoglutarate yielded crystals that diffracted to 1.7 Å. Interestingly, the obtained complex structure (Asp-AT–FCP–α-ketoglutarate) revealed electron density at the active site that was interpreted as a ketimine intermediate (Figure 4A). After refinement, the B factors of the built ligand are between 11.9 and 33.5 Å2 (Table 1). Figure 4B shows a simulated annealing omit map (Fo – Fc) superimposed with the atomic model of this ketimine intermediate. The carbon–nitrogen double bond formed between N2 on the pyridoxamine moiety and C2 on α-ketoglutarate is the key structural characteristic for this ketimine intermediate (Figure 4C). There is no electron density between Lys246 and the pyridoxal moiety, consistent with the measured distance of ~3.2 Å. The carboxyl group on the α-ketoglutarate makes a salt bridge with Arg374. The phosphate group on the pyridoxal moiety interacts with Arg254 and Thr104 through hydrogen bonds. These electrostatic interactions are consistent with the previous understanding that they hold the substrate and cofactor in the active site during the catalytic transformation.6 This ketimine intermediate has long been proposed as a transient intermediate in the Asp-AT catalytic mechanism before the generation of the final products, glutamate and the cofactor PLP (Scheme S1). Ketimine intermediates have been previously trapped in the crystal structures of DesV from Streptococcus pneumoniae (PLP-dependent transaminase)25 and ColD from E. coli (dehydratase),26,27 but to the best of our knowledge, they have not reportedly been trapped in Asp-AT.
Figure 4.

Structure of Asp-AT with a ketimine intermediate. Protein atoms are shown as pink sticks, with oxygen colored red, nitrogen colored blue, and phosphorus colored orange. (A) Active site of Asp-AT and a ketimine intermediate formed between PMP and α-ketoglutarate, which is colored yellow (PDB: 5VWR). The dashed green lines represent the electrostatic interactions or hydrogen bonds between the ketimine and the active site residues of Asp-AT. (B) Simulated annealing omit map (Fo – Fc) of the adduct. The omit map is shown as a gray mesh at 2.5σ. The omit map was generated after the ketimine had been omitted from the coordinates. (C) Chemical structure of the ketimine intermediate.
In the Asp-AT active site, PMP, which is a product in the reaction of FCP with Asp-AT, can react with α-ketoglutarate to form a ketimine intermediate, which undergoes fast tautomerization and then hydrolysis to produce L-glutamate and PLP in solution. The trapping of this ketimine intermediate in the crystal may have occurred serendipitously during cocrystallization, providing evidence of the occurrence of the second half of the catalytic cycle, and further suggesting that FCP is an alternate substrate of Asp-AT.
Identification of the Products and Proposed Mechanism of the Turnover of FCP by Asp-AT
To test for the turnover of FCP by Asp-AT and probe its turnover mechanism, we incubated Asp-AT with FCP in the absence of α-ketoglutarate, allowing the putative first half-reaction of FCP turnover to occur in a single-turnover fashion. After being desalted and deproteinated, FCP turnover samples were analyzed by high-mass accuracy HPLC–ESI-MS/MS, using a Q-Exactive mass spectrometer in negative ion mode to detect reaction products. The relative abundance of ions with molecular formulas corresponding to several putative FCP turnover products was observed changing significantly when FCP and Asp-AT were incubated together (Table 2 and Figure 5).
Table 2.
Proposed FCP Turnover Products and Intermediates Detected by Mass Spectrometry
| compound | change | observed m/z | theoretical m/z | Δppm | isotopic envelope supported | MS2 supported | synthetic standard supported |
|---|---|---|---|---|---|---|---|
| FCP | no change | 146.0615 | 146.0623 | −5 | yes | yes | yes |
| 9/10 | increase | 375.077 | 375.076 | 4 | NDa | NDa | NAb |
| 3 | increase | 355.072 | 355.070 | 5 | NDa | NDa | NAb |
| PLP | decreasec | 246.017 | 246.017 | 1 | yes | yes | yes |
| PMP | decreasec | 247.049 | 247.049 | 0 | yes | NDa | yes |
| 12 | increase | 127.039 | 127.040 | −7 | yes | yes | NAb |
| 11 | increase | 145.030 | 145.031 | 4 | NDa | NDa | NAb |
| 7 | no change | 126.055 | 126.056 | −7 | yes | yes | NAb |
The compound was below the limit of detection for MS2 or isotopic envelope.
Synthetic standard not available.
Probably because of an ion suppression that resulted from the co-elution with FCP, and because of the sequestration of some PLP and PMP molecules in the formation of intermediates 1–3, 9, and 10.
Figure 5.

Relative levels of proposed FCP turnover products and intermediates. The relative abundance of each compound was determined by LC–MS and normalized relative to the total ion current. The level of each compound increased in the incubated solution of FCP and Asp-AT, except for those of PLP and PMP, which were detected at lower levels probably because of an ion suppression that resulted from their co-elution with FCP, and because of the sequestration of some PLP and PMP molecules in the formation of intermediates 1–3, 9, and 10. Abbreviations: Blank, acetonitrile; Buffer, reaction buffer; FCP, FCP at 15.8 mM; Enz., Asp-AT at 39.5 μM; Rxn., Asp-AT (39.5 μM) and FCP (15.8 mM). Molecular anions of compounds 1, 9, and 10 have the same mass and cannot be distinguished.
Ions were putatively identified on the basis of intact mass, and, when possible, isotopic envelopes, MS2 fragment assignment, and comparison to synthetic standards. Figures S2–S5 present MS2 and/or isotopic envelope evidence for the identification of 3-oxocyclopentane-1-carboxylate (12), PLP, PMP, and 7. In some cases, analysis of isotopic envelopes and MS2 was not possible because of the low overall abundance (Table 2). The relative level of each of these metabolites is shown in Figure 5. Notably, the level of each metabolite increased when FCP and enzyme were mixed, relative to negative controls, with the exception of PLP and PMP, which decreased, and 7 and FCP itself, which did not change significantly. FCP levels did not change significantly because of the large molar excess of FCP used in these experiments, and 7 (in addition to being a product of pathway b) is an in-source fragmentation product of FCP, which explains why its relative levels mirror those of FCP. Altogether, this is consistent with the mechanism proposed in Figure 6, where FCP turnover is partitioned between the two separate pathways.
Figure 6.

Proposed turnover mechanism of FCP by Asp-AT based on mass spectrometry analysis of a single turnover of FCP. Species detected to be increased are boxed in green. Both pathways a and b are proposed to occur concurrently.
The resulting Schiff base from the reaction of FCP and the lysine-bound PLP on Asp-AT is subjected to γ-proton removal, followed by tautomerization that leads to the formation of intermediates 9 and 10 (Figure 6, path a) and the release of the fluoride ion (Figure 6, path b). In path a, hydrolysis of intermediate 10 gives 3-fluoro-4-oxocyclopentane-1-carboxylate 11 and releases PMP. In path b, hydrolysis of 3, the intermediate formed after the release of the fluoride ion, gives enamine 7 and releases PLP; hydrolysis of 7 generates 3-oxocyclopentane-1-carboxylate 12. Pathways a and b occur concurrently. Levels of intermediates 9 and 10, 3-fluoro-4-oxocyclopentane-1-carboxylate (11), 3-oxocyclopentane-1-carboxylate (12), and 7 increased in the incubated solution of FCP and Asp-AT. The observed decrease in the levels of PLP and PMP is likely due to ion suppression effects of FCP, because PLP and PMP both co-elute with FCP, as well as due to the fact that some PLP and PMP cofactors are sequestered in the formation of intermediates 1–3, 9, and 10. Notably, the PMP:PLP ratio increases significantly upon the addition of FCP to OAT, indicating that PLP is converted to PMP through the FCP turnover mechanism (Figure S6).
Comparison of Structures of OAT–FCP, GABA–FCP, and Asp-AT–FCP Complexes
OAT, GABA-AT, and Asp-AT (Figure S7) belong to the type 1 fold class of amino-transferases.28 Within this type I fold class, Asp-AT belongs to subclass I (AT-I), while OAT and GABA-AT belong to subclass II (AT-II).25,28 A structural alignment of GABA-AT and OAT (root-mean-square deviation of 2.2 Å using SSM superposition20 in COOT) shows no major differences in the positions of the conserved key catalytic residues (Figure S8A), including the catalytic Lys and an Arg that is involved in substrate binding. Lee et al.3 previously proposed that inactivators of GABA-AT could also bind to OAT because the distances between the catalytic Lys residue and the Arg residues are similar (Figure S8A). Therefore, it is reasonable that FCP inactivates both GABA-AT and OAT via a similar enamine mechanism. Despite the presence of the catalytic Lys and PLP, the active site residues in Asp-AT do not align well with those of OAT and GABA-AT (Figure S8B), displaying subtle differences (Tyr65 from subunit b and the position of R254 for substrate recognition)6 that could contribute to the difference in reaction mechanisms with FCP.
In the mechanisms of inactivation of OAT and GABA-AT by FCP (Figure 1A and Scheme 2), 7 is held in position in the active site via a salt bridge between a conserved Arg and the carboxyl group on FCP, allowing 7 to irreversibly form a covalent adduct with the catalytic Lys and PLP, which leads to the inactivation of the enzyme. In the mechanism of FCP with Asp-AT, this ternary adduct is not formed; instead, FCP is turned over to a keto product, which could be the result of the hydrolysis of 3 that occurs before the ternary adduct could be formed (Figure 6, path b). Examination of the active site pockets reveals that, in GABA-AT and OAT, the pyridine ring of the cofactor is buried in the enzymes’ active sites and is barely exposed to the solvent (Figure 7A,B), which likely helps to retain 7 in the active site cavities long enough for the formation of the final ternary adducts to occur. In Asp-AT (Figure 7C), the pyridine ring of the cofactor is more exposed and closer to the surface of the protein, which may account for the availability of a water molecule that performs the rapid hydrolysis of 3.
Figure 7.

Cofactor binding pockets. (A) A surface-coated GABA-AT is colored green and purple to represent the amino acids from two different subunits. PLP is represented as yellow sticks, with oxygen colored red, nitrogen colored blue, and phosphorus colored orange. (B) A surface-coated OAT is colored green and brown to represent the amino acids from two different subunits. PLP is represented as yellow sticks, with oxygen colored red, nitrogen colored blue, and phosphorus colored orange. (C) A surface-coated Asp-AT is colored pink and salmon to represent the amino acids from two different subunits. PMP is represented as yellow sticks, with oxygen colored red, nitrogen colored blue, and phosphorus colored orange.
To further analyze the possible hydrolysis in Asp-AT, structures of the Asp-AT–PMP and OAT–PLP–FCP complexes are manually superimposed by overlapping the atom positions of the atoms in the PLP moieties (Figure 8). Assuming that FCP reacts with PLP and the catalytic Lys in a similar fashion in the active sites of both enzymes, this superimposition reveals a hypothetical position of bound FCP in Asp-AT. In Asp-AT, Tyr214, instead of a Thr in OAT, is close to the PLP and places a water molecule via hydrogen bonding close to the hypothetical binding position of FCP, presenting an estimated distance of ~3.3 Å to the point of the nucleophilic attack. Thus, this water could be the nucleophile that performs the hydrolysis to complete the alternative turnover of FCP, instead of inactivation, in Asp-AT.
Figure 8.
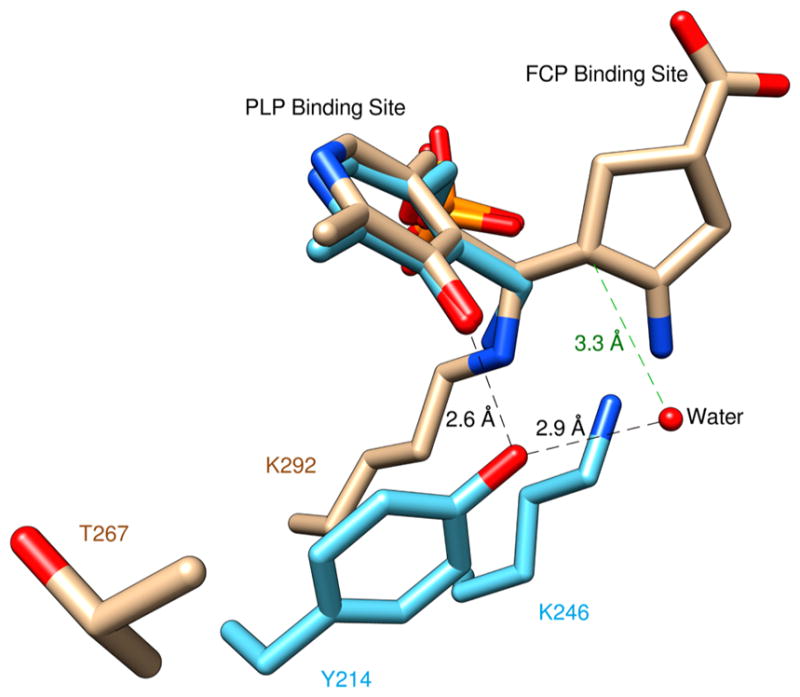
Superposition of the structures of the OAT–PLP–FCP and Asp-AT–PMP complexes by manually overlapping the atom positions of PLP moieties. The carbon atoms in the OAT–PLP–FCP complex are colored brown. The carbon atoms in the Asp-AT–PMP complex are colored cyan. The estimated distance from the water molecule that potentially performs the nucleophilic attack on the hypothetical FCP position in Asp-AT is colored green.
Selectivity of Mechanism-Based Inactivators for PLP-Dependent Enzymes
Although FCP is not selective for OAT or GABA-AT, the active sites of GABA-AT and OAT display certain structural differences. Comparison of their substrates shows that ornithine is one carbon longer than GABA, and it has an additional amino group at the α-carbon. Previous studies of mechanism-based inactivators of these two enzymes showed that vigabatrin is specific for GABA-AT10 and (2S,5S)-5-fluoromethylornithine (5FMOrn)29 is specific for OAT. 5FMOrn is one carbon longer than vigabatrin, and it has an additional amino group at the α-carbon. An overlap of the active sites of GABA-AT and OAT (Figure S8A), though very similar, shows differences in some amino acid residues that could account for their selectivity for one substrate over the other. It has been suggested that residues Tyr55 and Tyr85 make the active site of OAT more flexible to accommodate substrates that are larger and have an amino group at the α-carbon, when compared to the hydrophobic environment in the active site of GABA-AT, which is created by residues Ile72 and Phe351.3 These subtle differences can be exploited when designing mechanism-based inactivators that are specific for either OAT or GABA-AT.
CONCLUSION
FCP inactivates a drug-target OAT via an enamine mechanism similar to that of another drug-target GABA-AT, but it is an alternate substrate for an apparent PLP-dependent off-target Asp-AT. The proposed mechanisms of inactivation and alternative turnover in all three enzymes are complex, and all contain multiple reaction pathways. Although FCP is still too toxic to become a drug itself,11,29 it displays a desirable selectivity in the context of drug development as a mechanism-based inactivator. The reason for this desirable selectivity results from the subtle differences in the shapes of the active site cavities for retaining an intermediate, as well as the availability of a water molecule for a proposed hydrolysis in a complex mechanism with multiple reaction pathways. In general, the reactive intermediates generated during the complex reactions of these mechanism-based inactivators in the active sites could be capable of displaying desirable specificity of inactivation, discerning drug-target and off-target enzymes, even with similar structures and reaction mechanisms.
Supplementary Material
Acknowledgments
Funding
We gratefully acknowledge the National Institutes of Health (R01 DA030604 to R.B.S., R01 AT009143 to N.L.K., and 1R15GM113229-01 to D.L.) for financial support. We also thank Loyola University Chicago for funding support (to D.L.) and the Arthur J. Schmitt Foundation (fellowship to R.M.).
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) OFFce of Science User Facility operated for the DOE OFFce of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Footnotes
Notes
The authors declare no competing financial interest.
Accession Codes
Coordinates and structure factors have been submitted to the Protein Data Bank as entries 5VWO, 5VWQ, and 5VWR.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.7b00499.
Structural overlays, schemes, and mass spectrum (PDF)
References
- 1.Karmen BA, Wroblewski F, Ladue JS. Transaminase activity in Human blood. J Clin Invest. 1955;34:126–133. doi: 10.1172/JCI103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eliot AC, Kirsch JF. Pyridoxal Phosphate Enzymes: Mechanistic, Structural and Evolutionary Considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 3.Lee H, Juncosa JI, Silverman RB. Ornithine Aminotransferase versus GABA Aminotransferase: Implications for the Design of New Anticancer Drugs. Med Res Rev. 2015;35:286–305. doi: 10.1002/med.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herzfeld A, Knox WE. The Properties, Developmental Formation, and Estrogen Induction of Ornithine Aminotransferase and Estrogen in Rat Tissues. J Biol Chem. 1968;243:3327–3332. [PubMed] [Google Scholar]
- 5.Miyasaka Y, Enomoto N, Nagayama K, Izumi N, Marumo F, Watanabe M, Sato C. Analysis of differentially expressed genes in human hepatocellular carcinoma using suppression subtractive hybridization. Br J Cancer. 2001;85:228–234. doi: 10.1054/bjoc.2001.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirsch JF, Eichele G, Ford G, Vincent MG, Jansonius J, Gehring H, Christen P. Mechanism of Action of Aspartate Aminotransferase Proposed on the Basis of its Spatial Structure. J Mol Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 7.Mckenna MC, Stevenson JH, Huang X, Hopkins IB. Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem Int. 2000;37:229–241. doi: 10.1016/s0197-0186(00)00042-5. [DOI] [PubMed] [Google Scholar]
- 8.Zigmond E, Ya’acov A, Lee H, Lichtenstein Y, Shalev Z, Smith Y, Zolotarov L, Ziv E, Kalman R, Le HV, Lu H, Silverman RB, Ilan Y. Suppression of Hepatocellular Carcinoma by Inhibition of Overexpressed Ornithine Aminotransferase. ACS Med Chem Lett. 2015;6:840–844. doi: 10.1021/acsmedchemlett.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe M, Maemura K, Kanbara K, Tamayama T, Hayasaki H. GABA and GABA Receptors in the Central Nervous System and Other Organs. Int Rev Cytol. 2002;213:1–47. doi: 10.1016/s0074-7696(02)13011-7. [DOI] [PubMed] [Google Scholar]
- 10.Hawker DD, Silverman RB. Synthesis and Evaluation of Novel Heteroaromatic Substrates of GABA Aminotransferase. Bioorg Med Chem. 2012;20:5763–5773. doi: 10.1016/j.bmc.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Storici P, Qiu J, Schirmer T, Silverman RB. Mechanistic Crystallography. Mechanism of Inactivation of γ-Amino-butyric Acid Aminotransferase by (1R,3S,4S)-3-Amino-4-fluorocyclo-pentane-1-carboxylic Acid As Elucidated by Crystallography. Biochemistry. 2004;43:14057–14063. doi: 10.1021/bi0487185. [DOI] [PubMed] [Google Scholar]
- 12.Fast W. Quorum-quenching N-acyl-homoserine lactonase. In: Chang A, editor. Springer Handbook of Enzymes. Springer; Berlin: 2009. pp. 23–38. [Google Scholar]
- 13.Onuffer JJ, Kirsch JF. Redesign of the substrate specificity of Escherichia coli aspartate aminotransferase to that of Escherichia coli tyrosine aminotransferase by homology modeling and site-directed mutagenesis. Protein Sci. 1995;4:1750–1757. doi: 10.1002/pro.5560040910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen BW, Hennig M, Hohenester E, Jansonius JN, Schirmer T. Crystal Structure of Human Recombinant Ornithine Aminotransferase. J Mol Biol. 1998;277:81–102. doi: 10.1006/jmbi.1997.1583. [DOI] [PubMed] [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and Development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera: A visulization system for exploratory analysis and reseach. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 19.Winn M, et al. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 21.Smith DL, Almo JSC, Toney MD, Ringe D. 2.8-A-Resolution Crystal Structure of an Active-Site Mutant of Aspartate Aminotransferase from Escherichia coli. Biochemistry. 1989;28:8161–8167. doi: 10.1021/bi00446a030. [DOI] [PubMed] [Google Scholar]
- 22.Metzler DE, Christen P. Transaminases. 1985:133–134. [Google Scholar]
- 23.Liu D, Pozharski E, Lepore BW, Fu M, Silverman RB, Petsko GA, Ringe D. Inactivation of Escherichia coli L-aspartate aminotransferase by (S)-4-amino-4,5-dihydro-2-thiophene-carboxylic acid reveals “A tale of two mechanisms. Biochemistry. 2007;46:10517–10527. doi: 10.1021/bi700663n. [DOI] [PubMed] [Google Scholar]
- 24.Liu D, Pozharski E, Fu M, Silverman RB, Ringe D. Mechanism of inactivation of escherichia coli aspartate aminotransferase by (S)-4-amino-4,5-dihydro-2-furancarboxylic acid. Biochemistry. 2010;49:10507–10515. doi: 10.1021/bi101325z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgie ES, Thoden JB, Holden HM. Molecular architecture of DesV from Streptomyces venezuelae: a PLP-dependent transaminase involved in the biosynthesis of the unusual sugar desosamine. Protein Sci. 2007;16:887–896. doi: 10.1110/ps.062711007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cook PD, Holden HM. A structural study of GDP-4-keto-6-deoxy-D-mannose-3-dehydratase: Caught in the act of geminal diamine formation. Biochemistry. 2007;46:14215–14224. doi: 10.1021/bi701686s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook PD, Thoden JB, Holden HM. The structure of GDP-4-keto-6-deoxy- D -mannose-3- dehydratase: A unique coenzyme B 6 -dependent enzyme. Protein Sci. 2006;15:2093–2106. doi: 10.1110/ps.062328306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider G, Käck H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure. 2000;8:R1–R6. doi: 10.1016/s0969-2126(00)00085-x. [DOI] [PubMed] [Google Scholar]
- 29.Storici P, Capitani G, Müller R, Schirmer T, Jansonius JN. Crystal structure of Human Ornithine Aminotransferase Complexed with the Highly Specific and Potent Inhibitor 5-Fluoromethylornithine. J Mol Biol. 1999;285:297–309. doi: 10.1006/jmbi.1998.2289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.