

Abstract
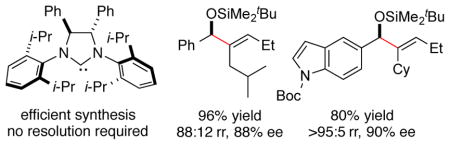
An exceptionally hindered class of enantiopure NHC ligands has been developed. While racemic forms had previously been utilized, a scalable and practical route to the enantiopure form of this ligand class is described utilizing a two-directional Buchwald-Hartwig amination in a highly sterically demanding environment. Using this newly accessible ligand class, nickel-catalyzed enantioselective reductive coupling reactions of aldehydes and alkynes has been developed. These studies illustrate that the newly available NHC ligands are well suited for simultaneous control of regio- and enantioselectivity, even in cases with internal alkynes possessing only very subtle steric differences in two aliphatic substituents. The steric demand of the new ligand class enables a complementary regiochemical outcome compared with previously described enantioselective processes. Using this method, a number of allylic alcohol derivatives were efficiently obtained with high regioselectivity (up to >95:5) and high enantioselectivity (up to 94% ee). The reaction conditions can also be extended to the reaction of aldehydes and allenes, providing silyl-protected allylic alcohol derivatives possessing a terminal methylene substituent. Computational studies have explained the origin of the exceptional steric demand of this ligand class, the basis for enantioselectivity, and the cooperative relationship of the aldehyde, alkyne, and ligand in influencing enantioselectivity.
Keywords: regioselective, enantioselective, N-heterocyclic carbene, allylic alcohol, addition
Graphical Abstract
Introduction
Tremendous strides have been made in the development of regioselective and enantioselective C-C bond-forming processes. However, the optimization of ligand structures becomes complex when high levels of both enantioselectivity and regioselectivity are desired with a substrate class that does not possess electronic or steric biases that effectively control regiochemistry. Ligand changes that enable high degrees of regioselectivity for a challenging substrate class often compromise the ability to simultaneously tune enantioselectivity. Similarly, the most broadly useful classes of chiral ligands are typically not amenable to modifications that enable access to elusive regiochemical outcomes.
Recent studies from our laboratory have focused on the development of classes of N-heterocyclic carbene (NHC) ligands that enable regiodivergence (access to either regioisomer from a common substrate) in catalytic, reductive coupling processes.1–3 The high levels of regiodivergence that are now possible have not yet been paired with an effective strategy for enabling enantioselectivity. Numerous important advances in enantiocontrol in reductive couplings have been illustrated in cases where regioselectivity is governed by substrate control.4 In these cases, access to only one of the two possible regioisomers is typically possible. Therefore, the ability to tune regioselectivity through catalyst control while obtaining high levels of enantioselectivity remains as an unsolved problem in this group of reactions.
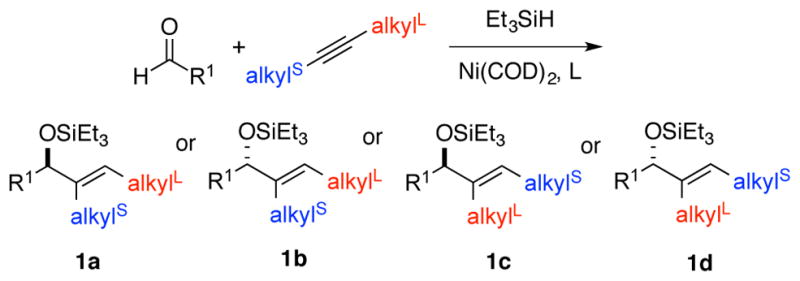
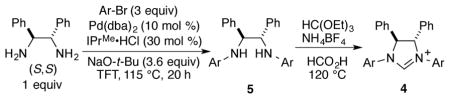
In the reductive coupling of aldehydes and alkynes to prepare allylic alcohols, a benchmark challenge in regiocontrol is presented by internal alkynes that possess two different alkyl substituents. This class of alkynes leads to the unselective production of two possible regioisomers in nearly all catalytic bond-forming processes of internal alkynes.5 The previous advances in enantioselective aldehyde-alkyne reductive couplings with internal alkynes that lack conjugation or directing functionality typically favor the production of products 1a and 1b where the least hindered alkyne terminus (alkylS) undergoes addition to the aldehyde (Scheme 1).4h–i However, the products 1c and 1d derived from coupling at the more hindered alkyne terminus (alkylL) can only be accessed in racemic form using current methods.2a–c Building on the insights provided by our previous efforts in establishing racemic regiodivergent processes,2,6 in this study, we have now focused on the development of processes that enable the highly selective production of the more hindered allylic alcohol regioisomers 1c and 1d, while simultaneously providing high levels of enantioselectivity.
Scheme 1.
Challenge of Simultaneous Regio- and Enantio-control.
Results and Discussion
Design of a Ligand Classes to Exert Regio- and Enantiocontrol
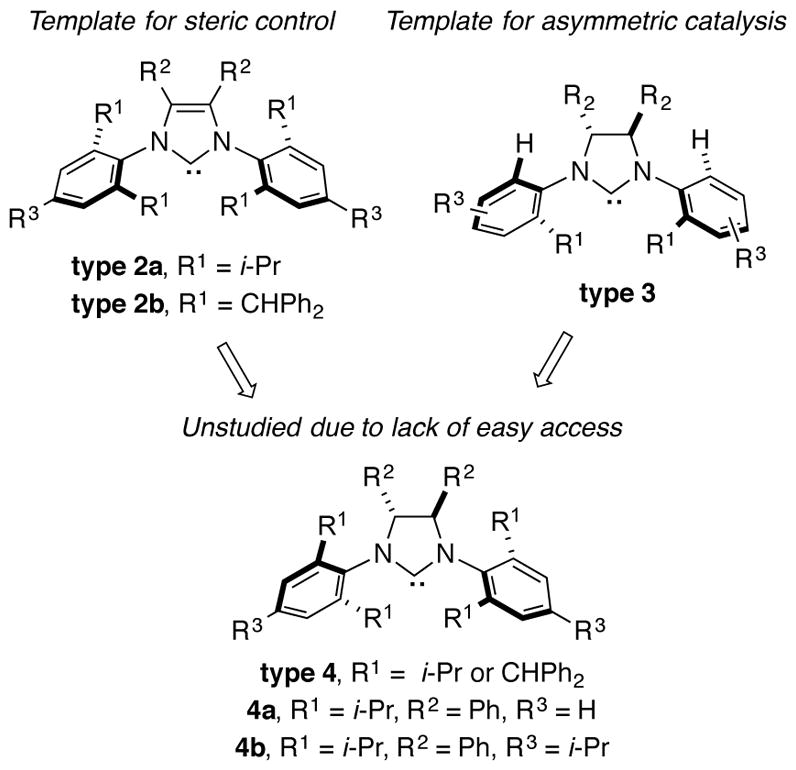
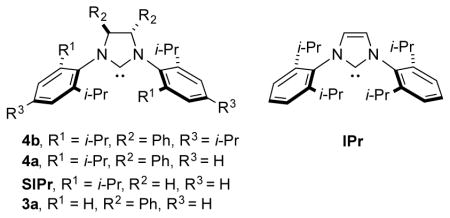
The most robust classes of NHC ligands in exerting steric control in catalytic processes are those that possess 2,6-disubstitution within an N,N-diaryl imidazolium framework. For example, the IPr and IPr*OMe classes of ligands 2a and 2b have been widely exploited in a vast array of catalytic processes that are governed by ligand steric control.1–2 Towards the goal of achieving enantioselectivity, multiple types of chiral NHC ligands exist for catalytic asymmetric processes.4,7 While many of these possess chirality within the N-substituent, these ligand classes designed for enantioselectivity typically lack a steric environment that is well positioned for regiocontrol. Installation of backbone chirality within the imidazolidine core enables a variety of N-aryl substituents to be utilized in ligand framework 3,7a,8 but none of the previously utilized chiral ligands of this class possess secondary branching of both substituents at the ortho positions. The ligand class 4 that possesses C-2 backbone chirality and N-aryl substituents that possess a bulky 2,6-disubstutution pattern seems an obvious choice, but this ligand type has only been exploited in racemic form in catalytic processes.
Chiral ligands of the type 4 have rarely been obtained in enantiopure form, and never by a route that avoids multistep synthesis including a resolution.9 The obvious synthetic route involving N,N-diarylation of a commercially available enantiopure diamine has not been previously been accomplished due to the low yields of the two-directional Buchwald-Hartwig coupling in an extremely sterically demanding environment. The original strategy to this ligand class from Sigman, involving a pinacol coupling of N-aryl imines followed by separation of rac- and meso isomers of the resulting diamine, has been employed in several contexts to prepare racemic ligand 4a.9a This ligand provided unique regiochemical outcomes in several different contexts in our own laboratory,2 and these promising outcomes motivated us to better explore the synthesis and utility of the enantiopure ligand class.
Ligand Synthesis Design and Optimization
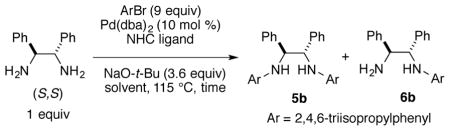
Initial studies in aldehyde-alkyne reductive couplings were conducted with ligand 4a (Scheme 2) obtained in enantiopure form by preparative chiral SFC chromatography.10 Given the promise of preliminary results in asymmetric couplings, the more straightforward route involving palladium-catalyzed C-N couplings was then explored. Considering the low cost of 1-bromo-2,4,6-triisopropylbenzene, we opted to explore this substrate in the synthesis of ligand 4b (Scheme 2). Under a variety of standard conditions for Buchwald-Hartwig amination, low yields of product 5b were obtained. For example, employing BINAP as ligand at 115 °C in neat bromoarene, only 17% of the desired diarylated product 5b was obtained, with monoarylated product 6b being the major product obtained in 47% yield (Table 1, entry 1). Similarly, a number of NHC ligands (IMes, SIMes, SIPr, and IPr*OMe) in combination with Pd(dba)2 failed to produce significant yields of the desired bis-arylated product 5 (see supporting information).11 The unsaturated bulky ligand IPr, however, provided some level of improvement and was thus selected for further optimization (Table 1).
Scheme 2.
NHC templates for Steric Control and Asymmetric Catalysis.
Table 1.
Optimization of Diamine Bis-arylation.
| |||||
|---|---|---|---|---|---|
| entry | ligand (mol %)a | solvent | time (h) | 5b yield (%)b | 6b yield (%)b |
| 1 | BINAP (24) | neat | 20 | 17 | 47 |
| 2 | IPr•HCl (20) | PhMe | 60 | 17 | 12 |
| 3 | IPrCl•HCl (20) | PhMe | 60 | 16 | 39 |
| 4 | IPrMe•HCl (20) | PhMe | 60 | 73 | --c |
| 5 | IPrMe•HCl (10) | PhMe | 60 | 12 | 56 |
| 6 | IPrMe•HCl (30) | PhMe | 60 | 93 | --c |
| 7 | IPrMe•HCl (30) | PhMe | 20 | 77 | --c |
| 8 | IPrMe•HCl (30) | dioxane | 20 | 49 | --c |
| 9 | IPrMe•HCl (30) | C6H5CF3 | 20 | 94 | --c |
| 10d | IPrMe•HCl (30) | C6H5CF3 | 20 | 93 | --c |
| 11d,e | IPrMe•HCl (30) | C6H5CF3 | 20 | 77 | --c |
Ligand scaffolds are shown in Scheme 2. IPr = ligand 2a, R1 = i-Pr, R2, R3 = H; IPrCl = ligand 2a, R1 = i-Pr, R2 = Cl, R3 = H; IPrMe = ligand 2a, R1 = i-Pr, R2 = Me, R3 = H)
NMR yields with di-bromomethane as the internal standard.
Not observed.
3 equiv of bromide was used.
Reaction was conducted at 90 °C.
Using IPr as ligand in toluene at 115 °C afforded the desired bis-arylation product 5b in 17% NMR yield together with 12% NMR yield of the mono-arylated product 6b (Table 1, entry 2). Other structurally related NHC ligands (IPrCl and IPrMe) were then evaluated (Table 1, entries 3–4). Promising results were obtained with the IPrMe ligand,12 with which the reaction afforded product 5b in 73% NMR yield with no observation of the mono-arylated product. It was then found that extra amounts of the NHC ligands were necessary. As illustrated (Table 1, entry 5), when a 1:1 ratio of IPrMe ligand to Pd(dba)2 was used, the yield of 5b decreased to 12% NMR yield and the major product of the reactions is the mono-arylated compound 6b. On the contrary, when the ratio of IPrMe/Pd was increased to 3:1, the NMR yield of 5b rose to 93% after stirring the reaction at designated temperature for 60 hours (Table 1, entry 6). Reducing the reaction time to 20 hours decreased the NMR yield of 5b to 77% yield (Table 1, entry 7). While dioxane as solvent lowered the chemical yield (Table 1, entry 8), further study showed that more polar solvent α,α,α-trifluorotoluene was the optimal solvent for the reaction. With this solvent, the NMR yield of the reaction rose to 94% (Table 1, entry 9). Although all previous study used large excess amount of the aryl bromide reactant (9 equiv), the same level of the reaction yield was observed when the amount of the bromide was reduced to 3 equiv (Table 1, entry 10). Reaction temperature is important for the reaction yield, and a lower yield of product 5b (77%) was obtained after decreasing the reaction temperature to 90 °C (Table 1, entry 11). Therefore, the optimized conditions from entry 10 were selected for the preparation of a range of new ligand motifs.
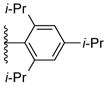
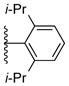
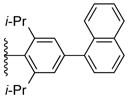
Using the optimized conditions, the synthesis of a range of bulky NHC ligands was carried out (Table 2). The reaction of 1,2-diphenylethane-1,2-diamine with 1-bromo-2,4,6-triisopropylbenzene was conducted on a 10 mmol scale. To simplify the purification procedure, the arylated diamine product 5b was purified by recrystallization, which furnished the product in 78% isolated yield. Diamine 5b was then successful cyclized to its corresponding NHC ligand 4b in 79% yield employing previously reported conditions (Table 2, entry 1). The initially explored ligand 4a, previously utilized in exploratory studies as material obtained by chiral chromatography, was also successfully synthesized under these conditions (Table 2, entry 2). Increasing the electron density by changing the para-substituent to 1-naphthyl and 9-anthracenyl decrease the yields for the dual arylation reaction and diamine 4c and 4d were obtained in 39% and 36% yield respectively. No mono-arylated product was observed in either case. Compared to the synthesis of ligands 4a and 4b, NHC ligands 4c and 4d were obtained in lower yields (Table 2, entries 3–4). The decrease in preparative efficiency may result from the extended conjugation of the N-aryl moiety in compounds 5c and 5d, which decreased the nucleophilicity of the nitrogen towards the cyclization reaction. Further increasing the electron density on the bromide reactant by introducing a para-methoxy group resulted in no formation of arylated products (Table 2, entry 5). We were also unable to apply these reaction conditions to the reaction of 1-bromo-2,4,6-tri(tert-butyl)benzene, suggesting that 2,6-diisopropyl substitution marks the limit of steric hindrance tolerated by this procedure. (Table 2, entry 6).
Table 2.
Application of the bis-arylation reaction conditions in the synthesis of chiral bulky NHC ligands
| |||
|---|---|---|---|
| entry | R | yield of 5a–d | yield of 4a–d |
| 1 |
|
5b, 78%a | 4b, 79% |
| 2 |
|
5a, 73% | 4a, 97% |
| 3 |
|
5c, 39% | 4c, 55% |
| 4 |
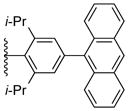
|
5d, 36% | 4d, 57% |
| 5 |
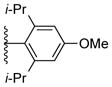
|
not observed | |
| 6 |
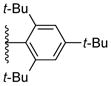
|
not observed | |
10 mmol scale and the product was isolated by recrystallization.
Regio- and Enantioselective Reductive Couplings
With the new classes of chiral ligands in hand, the asymmetric reductive coupling of aldehydes and alkynes was explored (Table 3). As highlighted above, the control of regio-chemistry in additions of internal alkynes that bear aliphatic substituents of similar size are exceptionally challenging in virtually all classes of catalytic reactions. For this reason, our initial explorations focused on alkyne 7, which possesses cyclohexyl and ethyl groups that must be differentiated during the coupling process. With this substrate, the catalyst must be able to distinguish a secondary from a tertiary propargylic substitution pattern in order to exert regiocontrol in the formation of product 8. While processes involving the influence of directing functionality are typically employed to obtain highly regioselective outcomes with this substitution pattern,13 non-directed processes involving internal alkynes of this type are exceedingly rare.
Table 3.
Optimization of the reductive coupling reaction with bulky NHC ligands
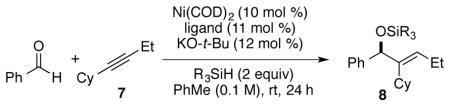
| ||||||
|---|---|---|---|---|---|---|
| entry | ligand | solvent | R3 | rra | yieldb (%) | eec (%) |
| 1 | 4b | THF | Et3 | >95:5 | 28 | 82 |
| 2 | 4b | PhMe | Et3 | >95:5 | 62 | 92 |
| 3 | 4b | PhMe | t-BuMe2 | >95:5 | 80 | 92 |
| 4 | 4b | PhMe | (i-Pr)3 | - | <5 | - |
| 5 | 4a | PhMe | t-BuMe2 | >95:5 | 65 | 86 |
| 6 | 4c | PhMe | t-BuMe2 | >95:5 | 53 | 81 |
| 7 | 4d | PhMe | t-BuMe2 | >95:5 | 85 | 84 |
| 8d | 4b | PhMe | t-BuMe2 | >95:5 | 47 | 91 |
| 9e | 4b | PhMe | t-BuMe2 | >95:5 | 46 | 87 |
Ratio of regioisomer.
isolated yield.
Determined by Supercritical Fluid Chromatography (SFC) analysis of the correspond alcohols generated by n-Bu4NF deprotection.
5 mol % catalyst loading; 72 hours.
2 mol % catalyst loading; 72 hours.
In initial explorations of regio- and enantioselective couplings with Et3SiH as the reducing reagent and tetrahydrofuran as the solvent, the nickel(0) catalyst of ligand 4b produced the desired regioisomer with high regioselectivity and excellent enantioselectivity (82% ee) but in low yield (Table 3, entry 1). It was then found that toluene was a superior solvent for this reaction, which improved the reaction yield to 62% and enantioselectivity to 92% ee (Table 3, entry 2). Further optimization showed that t-BuMe2SiH was the best reducing reagent for the reaction. With this silane, high reaction yield (80% yield) was obtained and the high enantioselectivity (92% ee) was maintained (Table 3, entry 3). The reaction mediated with bulky (i-Pr)3SiH afforded only trace amount of the reductive coupling product (Table 3, entry 4). This is likely due to inefficiencies of the bulky silane to participate in an effective σ–bond metathesis with the metallacycle intermediate.14 In addition to 4b, other ligands 4a, 4c, and 4d were also tested, and slightly decreased enantioselectivity was observed for all cases (Table 3, entries 5–7). Next, the possibility of decreasing the catalyst loading was examined. Although the level of enantioselective control was maintained by reducing the active catalyst loading to 5 mol %, the reaction yield decreased to 47% (Table 3, entry 8). A slight decrease in the enantioselectivity was observed after reducing the catalyst loading to 2 mol % (Table 3, entry 9). Therefore, the optimal conditions for the reductive coupling reaction (Table 3, entry 3) involves the use of 10 mol % active catalyst loading (10 mol % Ni(COD)2 and 11 mol % ligand 4b), toluene as the reaction solvent and t-BuMe2SiH as the reducing reagent. Slow addition of the substrates is not required. The absolute configuration of the product 8 was determined by Mosher ester analysis15 of the corresponding alcohol generated by TBAF deprotection, and S-configuration was concluded (see supporting information).
Compared to the excellent enantioselectivity obtained with the alkyne having an ethyl group as the small substituent, the reaction of methyl cyclohexylacetylene provided the TBS protected allylic alcohol in 79% yield with >95:5 regioselectivity but only 28% ee (Table 4, entry 1). Similar results were obtained from the reaction of 2-hexyne (Table 4, entry 2). The role of silane structure and temperature was explored, but these changes failed to improve the reaction enantioselectivity (Table 4, entries 3–6). Comparing the data in Tables 3 and 4, this outcome clearly demonstrates that the small substituent of the alkyne strongly influences enantioselectivity. The origin of this effect is discussed in a computational analysis below.
Table 4.
Couplings involving methyl-substituted alkynes
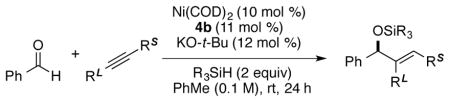
| |||||||
|---|---|---|---|---|---|---|---|
| entry | RL | RS | R | temp (°C) | rr | yield (%) | ee (%) |
| 1 | Cy | Me | t-BuMe2 | rt | >95:5 | 79 | 28 |
| 2 | n-Pr | Me | t-BuMe2 | rt3 | >95:5 | 83 | 38 |
| 3 | n-Pr | Me | i-Pr | rt | >95:5 | 74 | 11 |
| 4 | n-Pr | Me | t-BuMe2 | 50 | >95:5 | 78 | 38 |
| 5 | n-Pr | Me | t-BuMe2 | −20 | >95:5 | 74 | 33 |
| 6 | n-Pr | Me | t-BuMe2 | −78 | >95:5 | 70 | 32 |
With the understanding that substituents larger than methyl are essential as the small, distal substituent, the scope of enantioselective and regioselective couplings was explored with a range of aldehyde-alkyne combinations (Table 5). To first focus on enantioselectivity, the reactions of symmetrical alkynes were examined to probe the influence of the length of the alkyl chain on the enantioselectivity. Compared to the reaction of ethyl cyclohexylacetylene, the removal of the pro-pargylic branched substituent of the alkyne produced the silyl protected allylic alcohols in high yield with slightly decreased enantioselectivity (compounds 9a–9c). Compared to the results of the reaction with unbranched alkynes, improved enantioselectivity was observed for the reaction of the alkyne having homopropargylic branched substituent (compound 9d). Good regioselectivity (88:12) was also obtained. Alkynes with phenyl, silyloxy, and phthalimido groups were tolerated under the reaction conditions. Compounds 9e–9g were all obtained with high enantioselectivity. For compounds 9e–9f, the enantioselectivity of the minor regioisomers was also determined, which is the same level as that of the major regioisomers. The absolute configurations of the silyl-protected allylic alcohols were assumed to be the same as compound 8 by analogy.
Table 5.
Exploration of alkyne scopea
|
Enantioselectivity was determined by SFC analysis of the corresponding alcohols generated by Bu4NF deprotection.
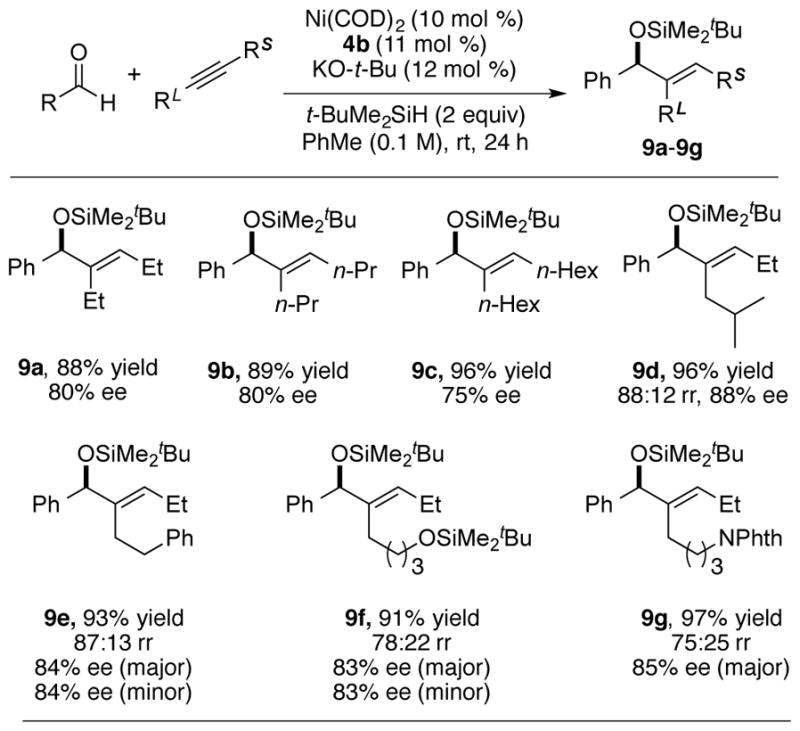
The scope of the aldehydes was found to be quite broad (Table 6). Both electron-rich and electron-poor aldehydes were tolerated under the reaction conditions, and compounds 10a–10c, possessing para-methoxy, fluoro, and methyl ester substituents were all formed with >90% ee. For the synthesis of compounds 10a and 10c, 20 mol % catalyst loading was required to ensure full conversion. In addition to benzaldehyde derivatives, naphthalenyl and heteroaryl aldehydes were also tolerated, and compounds 10d, 10e, 10f and 10g were obtained with 90%, 84%, 90% and 90% ee respectively. The reactivity of aliphatic aldehydes was also evaluated. To facilitate the analysis of enantioselectivity, the alkyne having a phenethyl substituent was employed. Both linear and α-branched aldehydes, such as octanal and cyclohexyl carboxaldehyde, reacted with the alkyne under the standard conditions to furnish products 10h and 10i with moderate regioselectivity and good enantioselectivity. The decrease of the regioselectivity of 10i compared to 10h illustrates that increasing steric interactions between the aldehyde substituent and the large group on the alkyne compromises the normal regiochemistry preference for the more hindered regioisomer.
Table 6.
Exploration of aldehyde scopea
|
Enantioselectivity was determined by SFC analysis.
20 mol % Ni(COD)2 and 4b were used.
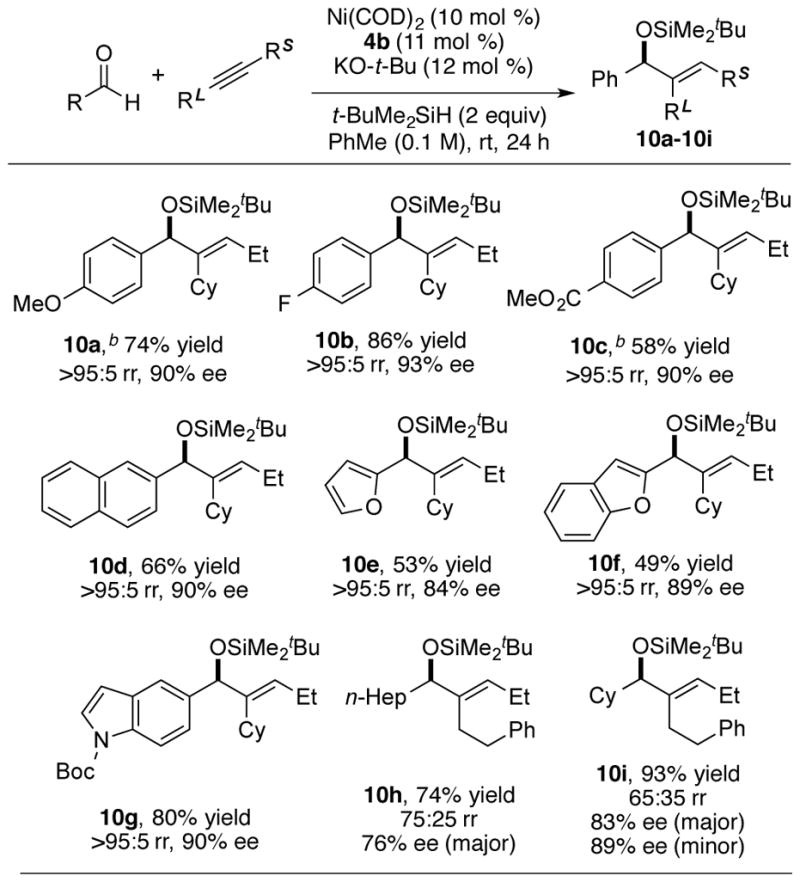
As expected, based on the above observations that groups larger than methyl are required as the small alkyne substituent for high enantioselectivity, the reaction of a terminal alkyne afforded the terminal methylene product 11a with low enantioselectivity (Scheme 3). Compared to reaction of internal alkyne, the slow addition of the solution of the reactants benzaldehyde, cyclohexylacetylene and tert-butyldimethylsilane in toluene is necessary for the reaction of terminal alkynes to achieve good yield with minimal alkyne trimerization. As seen in the reactions of ethyl cyclohexylacetylene and methyl cyclohexylacetylene, (i-Pr)3SiH was not an ideal reducing reagent for this branched terminal alkyne, affording a very low yield of the product 11b.
Scheme 3.
Comparison of terminal alkynes and allenes
As an alternative, the terminal methylene product can be synthesized by the reaction of aldehydes with allenes (Scheme 3). The nickel-catalyzed reductive coupling reaction of aldehydes and allenes were previously developed by Jamison.16 While excellent chirality transfer utilizing enantiopure allenes was exploited in that study, highly enantioselective variants using achiral allenes have not been developed. Employing the reaction conditions developed above for the reaction of aldehydes and alkynes, the reaction of benzaldehyde and cyclohexylallene using t-BuMe2SiH as reductant provided the product 12a in promising yield and enantioselectivity (79% ee). The absolute stereochemistry was determined to be of the S-configuration through Mosher ester analysis of the corresponding alcohol generated by TBAF deprotection of 12a. In this instance, the use of the bulkier silane i-Pr3SiH afforded considerably improved results, with product 12b being obtained with enantioselectivity (94% ee), albeit with lower regioselectivity (79:21).
Computational Evaluation
A number of key mechanistic questions emerge from the above empirical data. First, ligands 4a and 4b demonstrated unique regiochemical control in the enantioselective couplings examined in this study as well in the racemic reactions in previous studies.2a While the steric bulk of the 2,6-disubstitution pattern of the ligand N-aryl group is well documented as a key factor in ligand sterics of the widely used ligands IPr and SIPr,17 the identical 2,6-diisopropylphenyl substituent of ligands 4a and 4b exerts a regiochemical influence consistent with a much greater degree of steric demand. To reveal the effects of backbone substitution on the steric properties of the NHC ligands, we calculated the percent buried volumes (%Vbur, Table 7) of a series of ligands using structures optimized with density functional theory (DFT).18 Percent buried volume describes the percentage of space occupied by the NHC ligand within the first coordination sphere that is 3.5 Å from the metal center.19 Both 4a and 4b have greater %Vbur than SIPr and IPr, indicating an increased steric demand of the C2 symmetric ligands. Since the backbone phenyl substituents of 4a and 4b are outside of the first coordination sphere, they indirectly influence the steric properties of these NHC ligands by controlling the conformation of the N-diisopropylphenyl groups and placing the ortho-isopropyl groups closer to the metal center. This reveals an effective approach to increasing the steric demand of NHC ligands, in addition to altering the size of the N-aryl group.17b
Table 7.
Percent Buried volumes (%Vbur) of NHC ligands.
| |
|---|---|
| ligand | %Vbur |
| 4b | 37.5 |
| 4a | 37.5 |
| SIPr | 36.1 |
| 3a | 31.6 |
| IPr | 31.6 |
Furthermore, the above experimental study demonstrates that the small alkyne substituents, which are positioned as the distal substituent with respect to the bond-forming process in aldehyde alkyne couplings, play a significant role in the reaction enantioselectivity. This feature is illustrated by the simple change of an ethyl to a methyl substituent (compare Tables 3 and 4), which results in enantioselectivities dropping from 92% to 28% ee under otherwise identical conditions in couplings of benzaldehyde with cyclohexyl acetylene derivatives. Coupling with cyclohexylacetylene further reduced the enantioselectivity to 13% ee (Scheme 3). We performed DFT calculations of the oxidative cyclization transition states to investigate the origin of enantioselectivity and the effects of the alkyne substituents.20 We first investigated the reaction of alkyne 7 and benzaldehyde using ligand 4b (Figure 1a). The transition state that eventually leads to the (S)-product (TS1) is 2.6 kcal/mol more stable than the transition state that gives the (R)-product (TS2), in agreement with the high level of enantioselectivity for the (S)-product in experiment. Considering the chiral center on the NHC backbone is more than 6 Å away from the pro-chiral aldehyde, the level of enantiocontrol is remarkable. This remote chiral induction operates through the conformational change of the N-diisopropylphenyl groups on the C2 symmetric ligand. In the disfavored transition state TS2, the highlighted N-diisopropylphenyl group is significantly tilted towards the benzaldehyde and causes unfavorable steric clashes between the o-iPr on the NHC ligand and the phenyl group on the benzaldehyde. This tilted N-aryl conformation is induced by the phenyl substituent on the NHC backbone as well as the distal ethyl group on the alkyne. In the favored transition state TS1, the N-aryl group is almost perfectly horizontal. Thus, the steric clashes between the NHC ligand and the aldehyde are diminished.21 The cooperative effects of the NHC backbone and the distal alkyne substituent on enantioselectivity are confirmed by examining the TS structures with a terminal alkyne, cyclohexylacetylene, and the same NHC ligand 4b (Figure 1b). With the distal ethyl group replaced with a hydrogen, the N-aryl group is not tilted in either transition state. Thus, the level of chiral induction is diminished.
Figure 1. Remote chiral induction by the C2 symmetric ligand 4b in the oxidative cyclization transition states.

Energies are computed at the M06/SDD–6-311+G(d,p)/SMD (toluene) level of theory with geometries optimized at the B3LYP/LANL2DZ–6-31G(d) level.
To obtain a better understanding of the steric environment of the C2-symmetric ligand, we created the 2D steric contour plot of ligand 4b in the oxidative cyclization transition states TS1 and TS2 (Figure 2). Following previously reported procedure,22–23 the ligand steric contour was derived from the van der Waals surface of the NHC ligand and color-coded based on the distance from the substrate – red and yellow indicate regions on the ligand surface that are closer to the half-space containing the substrate, while blue and green indicate regions that are more distant from the substrate. In TS1, the alkyne and the nickel catalyst attack the (Si)-face of the benzaldehyde, placing the phenyl group in the less sterically encumbered quadrant (II) and the hydrogen in the occupied quadrant (III). In contrast, in the disfavored (Re)-face attack (TS2), the phenyl group is positioned in the occupied quadrant (III), leading to much more significant steric repulsions with the NHC ligand. The ligand contour plots highlight the conformational charge of the N-diisopropylphenyl groups induced by the backbone phenyl substituents that are positioned in quadrants I and III. The distal ethyl substituent on the alkyne is positioned in quadrant IV and accentuates the rotation of the N-diisopropylphenyl group towards the substrate in the occupied quadrant III. As a result, quadrant III becomes highly encumbered, leading to the unfavorable steric repulsion with benzaldehyde in TS2. In accordance with the analysis in Figure 1, these results again indicate that the enantioselectivity is influenced by the cooperative effects of the chiral NHC backbone and the distal alkyne substituent.
Figure 2. van der Waals surface and steric contour plot of ligand 4b in the oxidative cyclization transition states TS1 and TS2.

The geometries of the benzaldehyde and the alkyne are overlaid onto the contour plots. Red and yellow in the contour plots indicate regions that are occupied by the NHC ligand.
Conclusions
In conclusion, an effective synthesis of a novel class of enantiopure NHC ligands with exceptional steric demand has been developed. The catalytic reactivity of these ligands was evaluated in the Ni-catalyzed asymmetric reductive coupling reactions of aldehydes and alkynes. This study has identified ligands that are able to distinguish electronically and sterically similar alkyne substituents in a highly regioselective manner while also inducing high levels of enantioselectivity in the process. Computational studies have elucidated the origin of the substantial steric influence exerted by the ligand class explored. Additionally, the origin of enantioselectivity is explained, including the significant role that alkyne sterics play in influencing the ligand-aldehyde interactions Future work will focus on employing these ligands in other organic transformations and developing other NHC ligands designed to simultaneously control enantioselectivity and regioselectivity.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (R35GM118133; JM) and the National Science Foundation (CHE-1654122; PL) for support of this work. We greatly appreciate assistance from Christina Kraml from Lotus Separations, LLC for the preparative separation of the enantiomers of chiral ligand 4a for preliminary study. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF.
Footnotes
ASSOCIATED CONTENT
Experimental details and copies of spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of regiodivergent reactions: Jackson EP, Malik HA, Sormunen GJ, Baxter RD, Liu P, Wang H, Shareef AR, Montgomery J. Acc Chem Res. 2015;48:1736. doi: 10.1021/acs.accounts.5b00096.Montgomery J. Organonickel Chemistry. In: Lipshutz BH, editor. Organometallics in Synthesis. John Wiley & Sons, Inc; Hoboken: 2013. pp. 319–428.Mahatthananchai J, Dumas AM, Bode JW. Angew Chem, Int Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787.
- 2.For regiodivergent reactions from our laboratory: Malik HA, Sormunen GJ, Montgomery J. J Am Chem Soc. 2010;132:6304–6305. doi: 10.1021/ja102262v.Jackson EP, Montgomery J. J Am Chem Soc. 2015;137:958–963. doi: 10.1021/ja511778a.Wang H, Negretti S, Knauff AR, Montgomery J. Org Lett. 2015;17:1493–1496. doi: 10.1021/acs.orglett.5b00381.Miller ZD, Dorel R, Montgomery J. Angew Chem, Int Ed. 2015;54:9088–9091. doi: 10.1002/anie.201503521.Miller ZD, Li W, Belderrain TR, Montgomery J. J Am Chem Soc. 2013;135:15282–15285. doi: 10.1021/ja407749w.Shareef AR, Sherman DH, Montgomery J. Chem Sci. 2012;3:892–895. doi: 10.1039/C2SC00866A.Kerchner HA, Montgomery J. Org Lett. 2016;18:5760–5763. doi: 10.1021/acs.orglett.6b03090.
- 3.For other recent reports of regiodivergent reactions: Kennemur JL, Kortman GD, Hull KL. J Am Chem Soc. 2016;138:11914–11919. doi: 10.1021/jacs.6b07142.Ding D, Mou T, Feng M, Jiang X. J Am Chem Soc. 2016;138:5218–5221. doi: 10.1021/jacs.6b01707.Xu B, Tambar UK. J Am Chem Soc. 2016;138:12073–12076. doi: 10.1021/jacs.6b08624.Du X, Zhang Y, Peng D, Huang Z. Angew Chem, Int Ed. 2016;55:6671. doi: 10.1002/anie.201601197.Sakae R, Hirano K, Miura M. J Am Chem Soc. 2015;137:6460–6463. doi: 10.1021/jacs.5b02775.Donets PA, Cramer N. Angew Chem, Int Ed. 2015;54:633–637. doi: 10.1002/anie.201409669.Cahard E, Male HPJ, Tissot M, Gaunt MJ. J Am Chem Soc. 2015;137:7986–7989. doi: 10.1021/jacs.5b03937.Tani Y, Fujihara T, Terao J, Tsuji Y. J Am Chem Soc. 2014;136:17706–17709. doi: 10.1021/ja512040c.Moragas T, Cornella J, Martin R. J Am Chem Soc. 2014;136:17702–17705. doi: 10.1021/ja509077a.Shiroodi RK, Koleda O, Gevorgyan V. J Am Chem Soc. 2014;136:13146–13149. doi: 10.1021/ja507054j.Köpfer A, Sam B, Breit B, Krische MJ. Chem Sci. 2013;4:1876–1880.Zhao Y, Weix DJ. J Am Chem Soc. 2013;136:48–51. doi: 10.1021/ja410704d.Yang Y, Buchwald SL. J Am Chem Soc. 2013;135:10642–10645. doi: 10.1021/ja405950c.Worthy AD, Sun X, Tan KL. J Am Chem Soc. 2012;134:7321–7324. doi: 10.1021/ja3027086.Bausch CC, Patman RL, Breit B, Krische MJ. Angew Chem, Int Ed. 2011;50:5687–5690. doi: 10.1002/anie.201101496.
- 4.For nickel-catalyzed asymmetric reactions involving alkynes: Kumar R, Tokura H, Nishimura A, Mori T, Hoshimoto Y, Ohashi M, Ogoshi S. Org Lett. 2015;17:6018–6021. doi: 10.1021/acs.orglett.5b02983.Fu W, Nie M, Wang A, Cao Z, Tang W. Angew Chem, Int Ed. 2015;54:2520–2524. doi: 10.1002/anie.201410700.Nie M, Fu W, Cao Z, Tang W. Org Chem Front. 2015;2:1322–1325.Check CT, Jang KP, Schwamb CB, Wong AS, Wang MH, Scheidt KA. Angew Chem, Int Ed. 2015;54:4264–4268. doi: 10.1002/anie.201410118.Ahlin JSE, Donets PA, Cramer N. Angew Chem, Int Ed. 2014;53:13229–13233. doi: 10.1002/anie.201408364.Zhou CY, Zhu SF, Wang LX, Zhou QL. J Am Chem Soc. 2010;132:10955–10957. doi: 10.1021/ja104505t.Yang Y, Zhu SF, Zhou CY, Zhou QL. J Am Chem Soc. 2008;130:14052–14053. doi: 10.1021/ja805296k.Chaulagain MR, Sormunen GJ, Montgomery J. J Am Chem Soc. 2007;129:9568–9569. doi: 10.1021/ja072992f.Patel SJ, Jamison TF. Angew Chem, Int Ed. 2004;43:3941–3944. doi: 10.1002/anie.200460044.Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y.
- 5.For a rare exception, see: Wipf P, Janjic J, Stephenson CRJ. Org Biomol Chem. 2004;2:443–445. doi: 10.1039/b316741k. (b)
- 6.Liu P, Montgomery J, Houk KN. J Am Chem Soc. 2011;133:6956–6959. doi: 10.1021/ja202007s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For examples of chiral NHC ligands: Ahlin JSE, Cramer N. Org Lett. 2016;18:3242–3245. doi: 10.1021/acs.orglett.6b01492.Ho CY, Chan CW, He L. Angew Chem, Int Ed. 2015;54:4512–4516. doi: 10.1002/anie.201411882.Jang H, Jung B, Hoveyda AH. Org Lett. 2014;16:4658–4661. doi: 10.1021/ol5022417.Grassi D, Dolka C, Jackowski O, Alexakis A. Chem Eur J. 2013;19:1466–1675. doi: 10.1002/chem.201202318.Jung B, Hoveyda AH. J Am Chem Soc. 2012;134:1490–1493. doi: 10.1021/ja211269w.Liu L, Ishida N, Ashida S, Murakami M. Org Lett. 2011;13:1666–1669. doi: 10.1021/ol200149s.Luan X, Mariz R, Robert C, Gatti M, Blumentritt S, Linden A, Dorta R. Org Lett. 2008;10:5569–5572. doi: 10.1021/ol8021808.Brown MK, May TL, Baxter CA, Hoveyda AH. Angew Chem, Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511.Funk TW, Berlin JM, Grubbs RH. J Am Chem Soc. 2006;128:1840–1846. doi: 10.1021/ja055994d.Clavier H, Coutable L, Toupet L, Guillemin JC, Mauduit M. J Organomet Chem. 2005;690:5237–5254.
- 8.Anderson DR, O’Leary DJ, Grubbs RH. Chem Eur J. 2008;14:7536–7544. doi: 10.1002/chem.200701362. [DOI] [PubMed] [Google Scholar]
- 9.(a) Mercer GJ, Sturdy M, Jensen DR, Sigman MS. Tetrahedron. 2005;61:6418–6424. [Google Scholar]; (b) Zinner SC, Herrmann WA, Kühn FE. Tetrahedron Asymm. 2008;19:1532–1535. [Google Scholar]; (c) Liu L, Ishida N, Ashida S, Murakami M. Org Lett. 2011;13:1666–1669. doi: 10.1021/ol200149s. [DOI] [PubMed] [Google Scholar]
- 10.Lotus Separations, LLC. conducted preparative separation of the enantiomers of chiral ligand 4a for preliminary study.
- 11.For reviews on N-arylation reactions: Ruiz-Castillo P, Blackmond DG, Buchwald SL. J Am Chem Soc. 2015;137:3085–3092. doi: 10.1021/ja512903g.Surry DS, Buchwald SL. Chem Sci. 2011;2:27–50. doi: 10.1039/C0SC00331J.Klinkenberg JL, Hartwig JF. Angew Chem, Int Ed. 2011;50:86–95. doi: 10.1002/anie.201002354.Surry DS, Buchwald SL. Angew Chem, Int Ed. 2008;47:6338–6361. doi: 10.1002/anie.200800497.Hartwig JF. Acc Chem Res. 2008;41:1534–1544. doi: 10.1021/ar800098p.Hartwig JF. Nature. 2008;455:314. doi: 10.1038/nature07369.Valente C, Pompeo M, Sayah M, Organ MG. Org Process Res Dev. 2014;18:180–190.Valente C, Çalimsiz S, Hoi KH, Mallik D, Sayah M, Organ MG. Angew Chem, Int Ed. 2012;51:3314–3332. doi: 10.1002/anie.201106131.Fortman GC, Nolan SP. Chem Soc Rev. 2011;40:5151–5169. doi: 10.1039/c1cs15088j.Marion N, Nolan SP. Acc Chem Res. 2008;41:1440–1449. doi: 10.1021/ar800020y.For N-arylation reactions using IPr*OMe ligand: Martin AR, Nelson DJ, Meiries S, Slawin AMZ, Nolan SP. Eur J Org Chem. 2014;2014:3127–3131.Meiries S, Speck K, Cordes DB, Slawin AMZ, Nolan SP. Organometallics. 2013;32:330–339.
- 12.For the synthesis and application of IPrMe in transition metal catalysis: Gaillard S, Bantreil X, Slawin AMZ, Nolan SP. Dalton Trans. 2009:6967–6971. doi: 10.1039/b907109a.Harkal S, Jackstell R, Nierlich F, Ortmann D, Beller M. Org Lett. 2005;7:541–544. doi: 10.1021/ol048025g.
- 13.(a) Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Eur J Org Chem. 2010;2010:391–409. doi: 10.1002/ejoc.200901094. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]; (c) Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307–1370. [Google Scholar]
- 14.For mechanistic studies on the nickel-catalyzed redcutive coupling reactions: Baxter RD, Montgomery J. J Am Chem Soc. 2011;133:5728–5731. doi: 10.1021/ja200867d.Liu P, McCarren P, Cheong PHY, Jamison TF, Houk KN. J Am Chem Soc. 2010;132:2050–2057. doi: 10.1021/ja909562y.Ogoshi S, Arai T, Ohashi M, Kurosawa H. Chem Commun. 2008:1347–1349. doi: 10.1039/b717261c.Hoshimoto Y, Ohashi M, Ogoshi S. Acc Chem Res. 2015;48:1746–1755. doi: 10.1021/acs.accounts.5b00061.Liu T, Bi S. Organometallics. 2016;35:1114–1124.
- 15.(a) Hoye TR, Jeffrey CS, Shao F. Nat Protocols. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]; (b) Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- 16.(a) Ng SS, Jamison TF. Tetrahedron. 2006;62:11350–11359. [Google Scholar]; (b) Ng SS, Jamison TF. J Am Chem Soc. 2005;127:7320–7321. doi: 10.1021/ja0521831. [DOI] [PubMed] [Google Scholar]
- 17.(a) Ragone Francesco, Poater Albert, Cavallo Luigi. J Am Chem Soc. 2010;132:4249–4258. doi: 10.1021/ja909441x. [DOI] [PubMed] [Google Scholar]; (b) Nelson DJ, Collado A, Manzini S, Meiries S, Slawin AMZ, Cordes DB, Nolan SP. Organometallics. 2014;33:2048–2058. [Google Scholar]
- 18.The %Vbur for ligands 4a, 4b, 3a, IPr and SIPr were calculated using the web-based SambVca 2.0 program (https://www.molnac.unisa.it/OMtools/sambvca2.0/index.html); Falivene L, Credendino R, Poater A, Petta A, Serra L, Oliva R, Scarano V, Cavallo L. Organometallics. 2016;35:2286–2293.) based on the geometries of (NHC)Ir(CO)2Cl, which were optimized using the BP86 functional and a mixed basis set of SDD for Ir and 6-31G(d) for other atoms. The default parameters in SambVca 2.0 were used as suggested by Cavallo: sphere radius: 3.5 Å, distance from the center of the sphere: 2.1 Å, mesh spacing: 0.1 Å, H atoms omitted, atom radii: Bondi radii scaled by 1.17.
- 19.Poater A, Cosenza B, Correa A, Giudice S, Ragone F, Scarano V, Cavallo L. Eur J Inorg Chem. 2009;2009:1759–1766. [Google Scholar]; Clavier H, Nolan SP. Chem Commun. 2010;46:841–861. doi: 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]
- 20.Previous mechanistic studies indicated the oxidative cyclization of the alkyne and aldehyde is the rate-determining step in reactions with sterically less hindered silanes, while this step may be reversible in reactions with more sterically demanding silanes (see Ref. 2b; for a computational study on the effects of silane on the rate-determining step, see Ref. 14e). The identical ee obtained with two different silanes in Table 3, entries 2 and 3 is consistent with the oxidative cyclization being enantioselectivity-determining. However, the decrease of ee with TIPSH in Table 4 (entries 2 versus 3) suggests that metallacycle formation becomes reversible and is no longer enantioselectivity-determining under these conditions. In the DFT calculations, we considered the enantioselectivity in the oxidative cyclization step, which is expected to determine the ee in reactions with sterically less hindered silanes. See reference 14e for a detailed study of the question of the influence of silane sterics on the rate-determining step.
- 21.It should be noted that the steric interactions between the terminal Et group and the NHC ligand are comparable in TS1 and TS2. In both transition states, one of the N-diisopropylphenyl groups is tilted to avoid the steric repulsion with the terminal Et group. In TS2, the tilted N-aryl group (highlighted and pointing out-of-the-paper in Figure 1a) is on the same side with the phenyl group on the aldehyde. In TS1, the tilted N-aryl group (not highlighted and pointing into the paper in Figure 1a) is on the opposite side, and thus does not cause steric repulsions with the aldehyde.
- 22.(a) Lu G, Fang C, Xu T, Dong G, Liu P. J Am Chem Soc. 2015;137:8274–8283. doi: 10.1021/jacs.5b04691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang G, Liu P. ACS Catal. 2016;6:809–820. [Google Scholar]
- 23.Falivene L, Credendino R, Poater A, Petta A, Serra L, Oliva R, Scarano V, Cavallo L. Organometallics. 2016;35:2286–2293. [Google Scholar]; Wang T, Yu Z, Hoon DL, Huang KW, Lan Y, Lu Y. Chem Sci. 2015;6:4912–4922. doi: 10.1039/c5sc01614b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.