

Abstract
Objective
To suggest an alternative strategy for deriving histocompatible stems cells without undertaking genetic manipulation.
Design
Prospective approach using an animal model.
Setting
Stem cell and bioevaluation laboratory, Seoul National University.
Animal(s)
F1 (C57BL6 × DBA2) and outbred (ICR) mice.
Intervention(s)
Ovarian stroma cells of less than 40 μm in diameter were subcultured with fibroblast monolayer, and colony-forming cells were characterized.
Main Outcome Measure(s)
Stemness, genotype, and imprinted gene methylation.
Result(s)
Two-lines of colony-forming cells were established, which expressed markers specific for embryonic stem cells (ESC) and formed embryoid bodies and teratomas. Complete matching of microsatellite markers with the cell donor strain confirmed their establishment from ovarian tissue, and identification of both homozygotic and heterozygotic chromosomes raised the possibility of their derivation from parthenogenetic oocytes. However, the use of cells smaller than mature oocytes for primary culture, the difference in imprinted gene methylation compared with parthenogenetic ESCs, and failure to establish the ESC-like cells by primary follicle culture collectively suggested the irrelevancy to gametes.
Conclusion(s)
Coculture of adult ovarian cells with somatic fibroblasts can yield colony-forming cells having ESC-like activity, which may provide an alternative for establishing autologous stem cells from adults that can be obtained without genetic manipulation.
Keywords: Cell transformation, meiosis, ovary, stem cell, stroma cell
Methods for the derivation of patient-specific stem cells that do not require somatic cell nuclear transfer are of tremendous clinical interest. Induction of induced pluripotent stem (iPS) cells from adult mouse and human somatic cells has been successfully achieved (1–3), but approaches that do not employ any type of gene manipulation would provide optimal clinical feasibility. In males, multipotent stem cell-like cells have been isolated from neonatal and adult testes (4–6), while germline stem cells have been identified in juvenile and adult mouse ovaries (7). These findings indicate that adult reproductive tissues may indeed harbor undiscovered endogenous pluripotent cells. We have been interested in establishing histocompatible stem cells from adult female tissues, and subsequently succeeded in establishing autologous embryonic stem cells (ESC) after in vitro folliculogenesis and parthenogenesis (8, 9). Based on these results, we have further attempted to derive histocompatible stem cells directly from culture of ovarian tissue in mice.
Our preliminary data show that there are cells expressing stemness-specific gene expression in mouse ovarian stromal tissue, from which mature oocytes and growing follicles of more than 40 μm in diameter are removed. In this study, we employed a coculture system and medium containing leukemia inhibitory factor (LIF) to create a favorable environment instead of using genetic manipulation for establishing stem cells. We addressed, first, whether ESC-like cells could be derived from ovarian tissue that does not contain mature oocytes. Second, cellular characteristics of the established lines were evaluated for elucidating cell origins and mechanisms related to stem cell derivation.
MATERIALS AND METHODS
Experimental Design
As a preliminary study, expression of stemness-related genes in individual ovaries was monitored by reverse transcriptase-polymerase chain reaction (RT-PCR) and quantitative PCR. A number of attempts were made to establish colony-forming cells by culturing of ovarian stroma cells under various culture conditions, with consequent characterization of established colony-forming cells using ESC and other cell-specific markers, differentiation analysis, telomerase assay, and karyotyping. To elucidate the origin of cells, strain-specific microsatellite analysis, single nucleotide polymorphism (SNP) genotyping, methylation analysis of imprinted genes, and culture of primordial follicles and blood monocytes were conducted.
Animals
All of the procedures for animal management, breeding, and surgery followed the standard operation protocols of Seoul National University; the review board of institutional animal care and use committee of Seoul National University approved the use of animals and the relevant experimental procedures (approval no. SNU-050331-2).
Cell Preparation
For the derivation of the murine embryonic fibroblast (MEF), 13.5-day postcoitus fetuses of the ICR strain were killed, and the visceral organs, heads, and extremities of the fetuses were removed under microscope. Embryonic fibroblasts were collected from the remaining tissue after dissociation. To prepare the ovarian cells, the ovaries of 10-week-old B6D2F1 (C57BL/6 × DBA2) female mice were collected and chopped using a surgical blade after the removal of adherent tissue. The specimens were incubated initially for 30 minutes in a dissociation medium that consisted of a 50:50 (v:v) mixture of 0.25% (v/w) trypsin-ethylenediaminetetraacetic acid (EDTA; GIBCO Invitrogen, Grand Island, NY) and Dulbecco’s modified eagle’s medium (DMEM; Gibco Invitrogen) that was supplemented with 750 units/mL collagenase type I (Sigma-Aldrich, St. Louis, MO) and 0.03% (v/v) fetal bovine serum (FBS; Hyclone, Logan, UT) at 37°C. The dissociated cells were filtered through a 40-μm cell strainer (BD Falcon, Franklin Lakes, NJ) and were subsequently centrifuged at 390 × g for 4 minutes. The prepared cells were precultured onto 60 × 15 mm culture dishes, and fibroblasts that attached quickly to the bottom of the dishes were discarded by collecting of buoyant cells 30 minutes after seeding. The buoyant cells collected were transferred onto a mitotically inactivated MEF monolayer. In some replications, the filtered cells were directly seeded onto the MEF monolayer without removing of fibroblasts.
Coculture for Establishing Colony-forming Cells
The ovarian cells seeded onto the MEF monolayer treated with 10 μg/mL mitomycin C (Sigma-Aldrich) were cultured in DMEM, which was supplemented with 0.1 mM β-mercaptoethanol (GIBCO Invitrogen), 1% (v/v) nonessential amino acids (GIBCO Invitrogen), 2 mM L-glutamine (Sigma-Aldrich), 1% (v/v) lyophilized mixture of penicillin and streptomycin (GIBCO Invitrogen), 5000 units/mL mouse LIF (Chemicon, Temecula, CA), and 15% (v/v) FBS at 37 °C under 5% CO2 in a humidified air atmosphere. At the end of the primary culture (on day 7 of culture), colony-forming cells were mechanically removed with a capillary pipette and placed on a newly prepared MEF monolayer; this was subsequently subpassaged at intervals of 3 days, during which period the medium was changed daily. The LIF concentration added to the culture medium was reduced to 1000 units/mL after the primary culture.
Characterization of Colony-forming Cells
At the 20th subpassage, colony-forming cells were collected and immunostained with the antibodies for mouse ESC-specific markers: stage-specific embryonic antigen (SSEA)-1, Oct-4, integrin-α6, integrin-β1, and alkaline phosphatase (AP) as positive markers, and SSEA-3 and SSEA-4 as negative markers (10, 11). Telomerase activity was determined by use of the TRAPEZE Telomerase Detection Kit (Chemicon) according to the manufacturer’s instructions. Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA) and reverse-transcribed with the Reverse Transcription System (Promega, Madison, WI). Karyotypes of the cell lines were analyzed after they had been maintained in culture for 7 to 8 weeks, which corresponded to 16 to 18 passages. Colony-forming cells were immunostained with antibodies against the germline markers mouse Vasa homolog (MVH) and Fragilis and the follicular cell-specific marker antimüllerian hormone (AMH).
To confirm spontaneous differentiation in vitro, the colony-forming cells of two lines were dissociated and subsequently transferred in LIF-free DMEM containing 10% (v/v) FBS. The cultured embryoid bodies were stained with the antibodies for three germ layer–specific markers. To confirm in vivo differentiation, 1 × 107 colony-forming cells retrieved from each line at the 20th subpassage were injected subcutaneously into adult nonobese diabetic-severe combined immunodeficiency (NOD-SCID) mice. Teratomas that formed in the subcutaneous region were collected 6 weeks after transplantation. For in vitro differentiation into neuronal lineage cells, the colony-forming cells were cultured in modified N2B27 medium, and the differentiating cells were stained with the antibodies for neuronal lineage cell–specific markers.
Sex Determination by Genomic DNA-PCR Analysis
Total genomic DNA from each sample was extracted using the G-spin Genomic DNA Extraction Kit (iNtRON Biotechnology, Seoul, South Korea) according to the manufacturer’s instructions. The extracted genomic DNA was subjected to PCR amplification with primers for the Zfy1 (Y-chromosome specific) and Xist (X-chromosome specific) genes to determine the sex (12). The PCR products were size-fractionated by 1.2% agarose gel electrophoresis and visualized by ethidium bromide staining.
Elucidation of Origin of ESC-like, Colony-forming Cells
A number of analyses were conducted to elucidate the origin of colony-forming cells. To evaluate whether the colony-forming cells were derived from feeder or feeder-contaminated cells, DNA microsatellite analysis was performed with genomic DNA samples from B6D2F1 tail, ICR MEFs, and two lines of newly established colony-forming cells. The SNP genotyping that is polymorphic between C57BL/6 and DBA2 strains was performed using fibroblasts of DBA2 and C57BL/6, normally fertilized ESC, parthenogenetic ESC (pESC), colony-forming cells. Bisulfite DNA sequencing for determining methylation status of Nanog, Oct-4, H19, Gtl2, Peg3, and Snrpn genes was undertaken, and normally fertilized ESC, pESC, and colony-forming cells were subjected to this analysis. Culture of primary follicles, intrafollicular oocytes, a mixed population of stromal cells dissociated from the ovaries, follicular cells of primary follicles, and blood mononuclear cells were conducted using the same medium used for culturing of colony-forming cells.
RESULTS
Can ESC-like Cells Be Derived from the Culture of Ovarian Stromal Tissue?
We primarily surveyed the expression of three principal stem cell genes, Oct-4, Nanog, and Cripto, in the brain, heart, lung, liver, stomach, kidney, ovary, urinary bladder, small intestine, skin, and spleen of adult female mice. As shown in Supplementary Figure 1A (available online), the ovaries expressed all three genes. A total of five ovaries were individually provided for quantitative PCR, and every ovary consistently expressed Oct-4, Nanog, Cripto, Rex-1, Dnmt3b, Tert, and Lif Rc except for Nanog in one case (see Supplementary Fig. 1B, available online).
Consequently, the prefiltered, dissociated ovarian cells were cultured in DMEM containing β-mercaptoethanol, nonessential amino acids, L-glutamine, FBS, and antibiotics, to which increasing concentrations of LIF up to 1–5 × 103 units/mL were added. Thirty minutes after the initial seeding, buoyant cells were collected and then reseeded on a mitotically-inactivated fibroblast monolayer. Among the buoyant cell population, we detected the cells showing both Oct-4 and Nanog expressions (data not shown). In a total 30 trials, 18 (60%) yielded cell aggregates or colony-like cell clumps during primary culture, and of those two (11.1%) established primary colonies (see Supplementary Table 1, available online). Aggregation of several cells was initially detected, which led to the formation of cell clumps during primary culture. Subculturing of the clumps formed successfully established and maintained ESC-like cell colonies, which had similar morphology with ESCs and showed a well-delineated colony margin and large nucleus to cytoplasmic ratio (data not shown). These colony-forming cells, hereafter referred to as adult ovary-derived colony-forming cells (OCC), were morphologically similar to ESC (see Fig. 1A). An additional 28 trials were conducted with different LIF doses, use of gonadotropins or a calcium ionophore, or changing of the culture system and mouse strain for deriving OCC. Cell aggregation was observed in 20 cases (71%), but no colony-forming cell lines were established (see Supplementary Table 1, available online).
FIGURE 1.
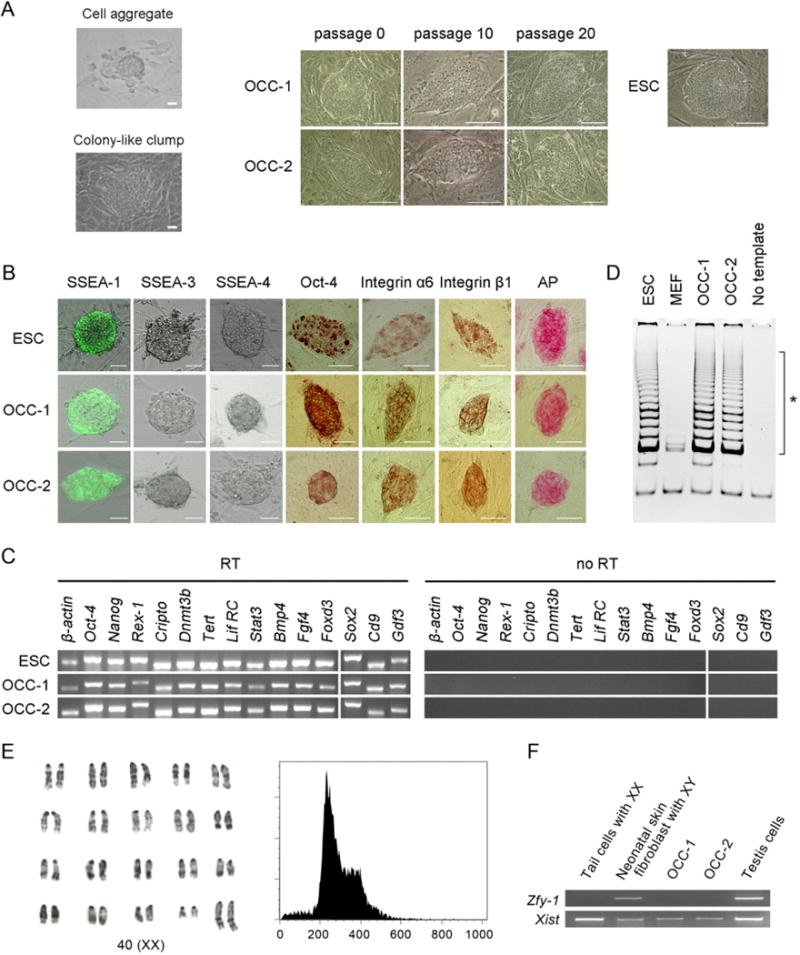
Initial characterization of ovary-derived colony-forming cells (OCC) derived from coculturing of adult ovarian cells and mouse embryonic fibroblast (MEF). (A) Morphology of cell aggregate, colony-like clump, and colony-forming cells on day 7 of primary culture, day 37 after 10 subpassages, and day 67 after 20 subpassages, with embryonic stem cells (ESC) as a reference. Scale bar = 50 μm. (B) Antibodies against stage-specific embryonic antigen (SSEA)-1, SSEA-3, and SSEA-4, Oct-4, integrin-α6, and integrin-β1 as well as alkaline phosphatase (AP) were used for characterization, and ESC were employed as the positive control. Similar to ESC, OCC are positive for SSEA-1, Oct-4, integrin-α6, integrin-β1, and AP. However, both ESC and OCC are negative for SSEA-3 and SSEA-4. Scale bar = 50 μm. (C) Pluripotent cell-specific gene expression by ESC and OCC was monitored by RT-PCR and similar gene expression profiles were detected. (D) Telomerase activities in OCC detected by the telomeric repeat amplification protocol assay. The ladder of telomerase products amplified by PCR is shown with six base increments starting at 50 nucleotides at the portion indicated by the asterisk. All lines express high levels of telomerase activity. NC, negative control without addition of template. (E, F) Karyotyping and sexing of OCC. (E) G-banding of air-dried chromosomes in OCC was undertaken for exact karyotyping, and the population of diploid cells was estimated with flow cytometry. The image from OCC-B6D2-SNU-1 was depicted on behalf of the established lines, and diploidy was detected in the established line. (F) PCR analysis was further conducted using the X-chromosome-specific Xist and Y-chromosome-specific Zfy primers. The OCC lines express the X-chromosome-specific Xist gene but not the Zfy gene.
Characterization of OCC
After more than 20 passages, both OCC lines remained immunopositive for AP, SSEA-1, integrin-α6, integrin-β1, and Oct-4, whereas no reactivity to anti-SSEA-3 or anti-SSEA-4 antibodies was detected (see Fig. 1B). The stem cell-associated Oct-4, Nanog, Rex-1, Cripto, Dnmt3b, Tert, Lif RC, Stat3, Bmp4, Fgf4, Foxd3, Sox2, CD9, and Gdf3 genes are all expressed in OCC (see Fig. 1C), as are high levels of telomerase activity (see Fig. 1D). Both OCC lines exhibited a normal diploid karyotype with XX sex chromosomes, as determined by G-banding of air-dried chromosomes, FACS, and PCR analysis using primers for Xist and Zfy1 (see Fig. 1E, 1F). Markers of the germline (Fragilis, MVH) or ovarian follicular somatic (granulosa) cells (AMH) were not detectable in OCC maintained in the presence of LIF (Supplementary Fig. 2A, available online). Further, OCC did not express tissue-specific stem cell markers, including Sca-1 and CD44 for mesenchymal stem cells or CD34 and CD45 for hematopoietic stem cells (see Supplementary Fig. 2B).
After culture in LIF-free medium, the OCC formed embryoid bodies that were positive for markers of cells derived from all three germ layers (see Fig. 2A). Subcutaneous transplantation of OCC into NOD-SCID mice generated teratomas consisting of cells derived from the three germ layers (see Fig. 2B, 2C), and the OCC differentiated into neuronal cells after treatment with N2B27 solution (see Fig. 2D).
FIGURE 2.
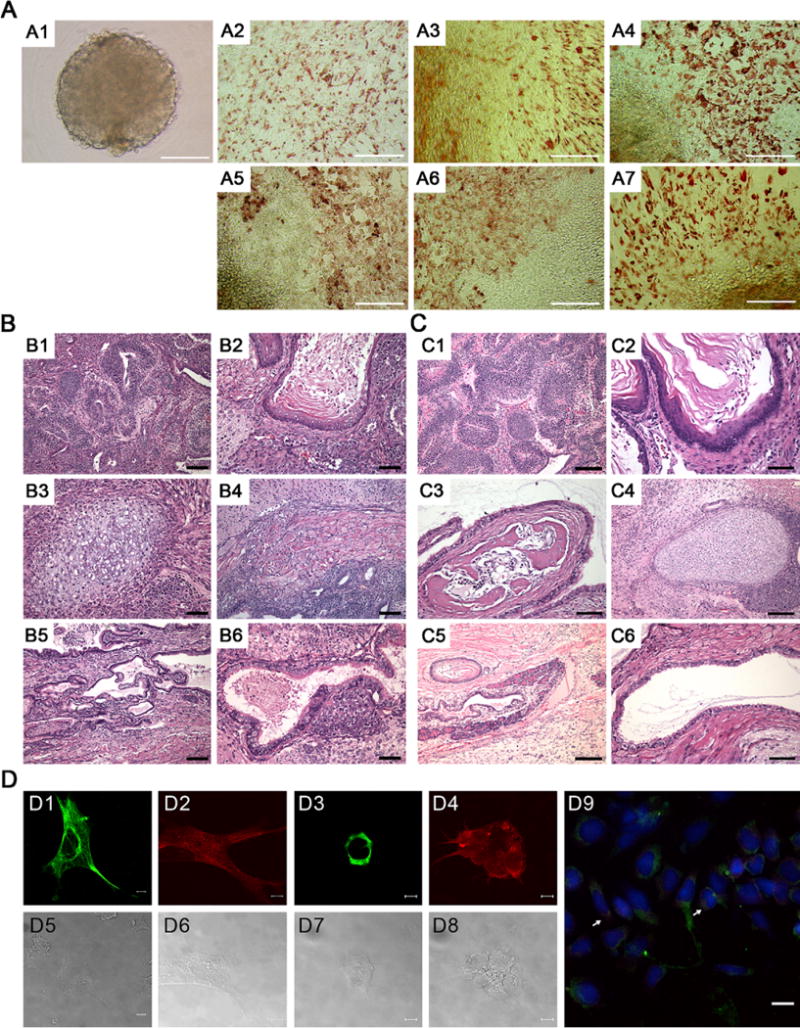
Spontaneous and inducible differentiation of ovary-derived colony-forming cells (OCC) in vitro and in vivo. (A) In vitro differentiation of OCC into embryoid bodies by culturing in leukemia inhibitory factor (LIF)-free medium. (A1) Embryoid body observed on day 4 of culture. Immunocytochemistry of embryoid bodies was undertaken to detect differentiation along all three germ layer using the specific markers of S-100 (A2, ectodermal), nestin (A3, ectodermal), smooth muscle actin (A4, mesodermal), Desmin (A5, mesodermal), α-fetoprotein (A6, endodermal), and Troma-1 (A7, endodermal). Results from analysis of OCC-1 are depicted on behalf of both established cell lines. Scale bar = 100 μm. (B–C) In vivo differentiation of OCC by subcutaneous transplantation of OCC-1 and OCC-2 into NOD-SCID mice. (B) A representative OCC-1-derived teratoma, stained with hematoxylin and eosin, contained (B1) neuroepithelial rosettes, (B2) keratinized stratified squamous epithelial cells, (B3) cartilage, (B4) muscle, (B5) glandular cuboidal to columnar epithelium, and (B6) ciliated columnar epithelial cells. Scale bar = 50 μm for B1, B4, and B5, and 25 μm for B2 and B3. (C) A representative OCC-2-derived teratoma contained (C1) neuroepithelial rosettes, (C2) keratinized stratified squamous epithelial cells, (C3) osteoid island showing bony differentiation, (C4) cartilage, (C5) pancreatic tissue, and (C6) ciliated cuboidal epithelial cells. Scale bar = 100 μm for C1 and C5, and 50 μm for C2, C3, C4, and C6. (D) Neuronal cell differentiation of OCC. (D1) Nestin-positive and (D2) Tuj1-positive neurons generated 14 days after replating on fibronectin. (D3) O4-positive oligodendrocyte generated 8 days and (D4) GFAP-positive astrocytes generated 15 days after replating. (D5–D8) Phase contrast images of D1–D4. (D9) Merged image of TH-positive and Tuj1-positive (arrow) neurons generated 19 days after replating on fibronectin. OCC were differentiated in modified N2B27 medium. DAPI (4,6-diamidino-2-phenylindole), blue; TH, green; Tuj1, red. Both established lines exhibited neuronal cell differentiation, and results from OCC-1 are shown on behalf of both established cells. Scale bar = 10 μm for D1–D8 and 20 μm for D9.
Elucidation of Cell Origin and Mechanism Related to OCC Establishment
Four markers of short tandem repeat microsatellite analysis, to distinguish cell donor and feeder cell lines, were selected from a public database. As shown in Supplementary Table 2 (available online), the two OCC lines were an identical match to those of the ovary donor (B6D2F1; C57BL/6 × DBA2), and thus we could exclude the feeder fibroblasts from the list of candidates.
Next, SNP genotyping and methylation analysis were executed for discerning the origination. Heterozygosity and homozygosity of SNP loci in OCC were compared with those of somatic fibroblasts of DBA2 and C57BL/6 strains, normally fertilized ESC, and pESC. Both homozygotic and heterozygotic chromosome recombination were detected in OCC and pESC lines (Fig. 3A). The ESC of the F1 strain possessed only heterozygotic loci, but homozygotic recombination was detected in fibroblasts of both inbred strains. The methylation status of the imprinted genes (H19, Snrpn, and Gtl2) except for Peg3 was different between OCC and pESC (see Fig. 3B). Different methylation of Peg3 and Snrpn genes between OCC and the control ESC was also detected, but stemness-related Nanog and Oct-4 were completely demethylated in all lines examined.
FIGURE 3.
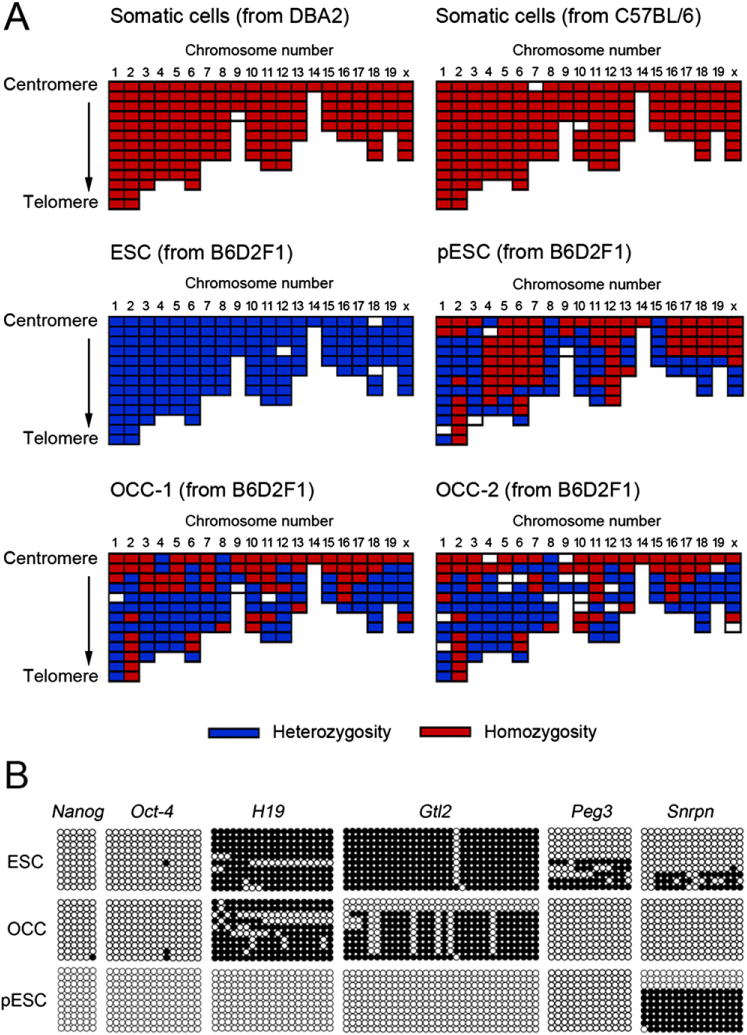
Analyses on elucidating the origins of ovary-derived colony-forming cells (OCC). (A) Single nucleotide polymorphism (SNP) genotyping of OCC and control cell lines. Heterozygosity or homozygosity of SNP loci was analyzed, and the SNP pattern of OCC-1 and OCC-2 of B6D2F1 was compared with that of somatic fibroblasts of DBA2 and C57BL6, B6D2F1 embryonic stem cells (ESC), and parthenogenetic ESC (pESC). Both homozygosity and heterozygosity were concomitantly detected in the OCC line. The ESC of the F1 strain showed heterozygosity alone, and only homozygotic SNP loci were detected in the fibroblasts of the inbred strain. The pESC line possessed both homozygotic and heterozygotic chromosomes. (B) Methylation status of OCC, ESC, and pESC. Genomic DNA isolated from these cell lines was subjected to bisulfite genomic sequencing analysis. We compared the methylation levels of the promoter regions of stemness-related genes (Oct-4 and Nanog) and the imprinted genes that expressed differentially after parthenogenetic activation (H19, Peg3, Snrpn, and Gtl2). The PCR products were cloned, and 10 plasmid clones were sequenced for each sample. Open and closed circles indicate unmethylated and methylated CpG dinucleotides, respectively. Stemness-related genes were demethylated in all cell lines while the methylation status of other genes was prominently different among the cell lines. Methylation of OCC was different from that of ESC or pESC: more methylated H19 and Gtl2 than pESC, and less methylated Peg3 and Snrpn than ESC.
Primary follicles and their components were cultured under the same conditions of OCC establishment, but only a monolayer formation of follicular cells without transformation of the oocyte into OCC was observed (Fig. 4). The monolayer did not react with any stem cell marker antibodies. Furthermore, to identify the possibility that OCC are derived from the circulating stem or progenitor cells in circulatory blood, mononuclear cells derived from whole blood were cultured, but they did not transform into colony-forming cells during primary culture over a total 13 days (see Supplementary Fig. 3, available online).
FIGURE 4.
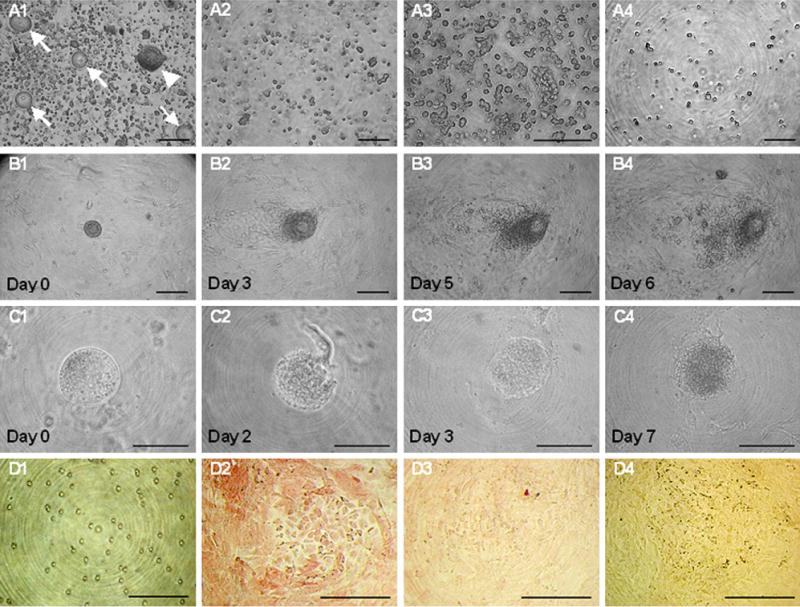
Culture of primary follicles, intrafollicular oocytes, and follicular cells in the primary follicle. (A) The cells dissociated from ovarian tissue by the standard protocol of our laboratory were passed through a 40-μm cell strainer (A2). Numerous primary follicles (arrowhead) and primary oocytes (arrow) were observed before filtering (A1), but only stromal cells were visible after filtering. (A3) Enlarged image of A2 further confirms there were no preantral follicles or oocytes after the filtration. (A4) Buoyant cells were collected from culturing of stromal cells on tissue culture plate 30 minutes after the filtration, and a decrease in the cell number was noticed. (B) (B1–B4) Primary follicles were cultured on a MEF monolayer for up to 6 days. Proliferation of follicular cells was detected from day 3 (B2), but degeneration of intrafollicular oocytes was observed on day 5 (B3) and day 6 (B4) of culture. (C) (C1–C4) Culturing of intrafollicular oocytes collected from the enzymatic digestion of primary follicles. The zona pellucida of the oocyte was removed before culture, and cell lysis and degeneration of the ooplasm was detected as early as from day 2 (C2) of culture. To observe darkened ooplasm on day 3 (C3) and day 7 (C4), the zona-free oocyte was exposed to an excessive lightening field; breakdown of the ooplasmic membrane was prominent on day 7. (D) Follicle granulosa cells dissociated from primary follicles (D1) were cultured on a MEF monolayer for 7 days and were subsequently stained for cells with antimüllerian hormone (AMH) (D2) and SSEA-1 antibodies (D3) and AP (D4). The granulosa cells proliferated vigorously but did not form colonies during coculture with the MEF monolayer. Strong reactivity of the cultured cells with AMH was detected, but no reactivity with SSEA-1 or AP was observed. Scale bars = 100 μm (A, B, and D) and 30 μm (C).
DISCUSSION
The results of this study clearly demonstrate that there are cells expressing stemness-related genes in ovarian stromal tissue. Two lines of ESC-like, colony-forming cells were subsequently established from coculturing of the ovarian cells with a fibroblast monolayer. There are several possible origins of the OCC explored herein: meiotic germ cells, parthenogenetic oocytes, follicular cells, medullary stromal cells, circulating blood cells that migrate to the ovaries, or even the feeder cells. Apparently, parthenogenesis of mature oocytes was not the origin of established ESC-like cells. The methylation status of imprinted genes (H19, Snrpn, and Gtl2) in OCC was different from that of pESC, a difference reported previously elsewhere (13, 14). On the other hand, the results of microsatellite analysis, immunostaining, and in vitro culture excluded maturing oocytes, follicular cells, feeder fibroblasts, and blood-derived mononuclear cells as candidates for the cells’ origins.
The colony-forming cells may be derived directly from ovarian stromal cells expressing stemness-related genes or from their progenitor cells. From this initial interpretation, we presume two possible originations relating to the OCC establishment: isolation and repopulation of ESC-like cells from ovarian stromal tissue, and reprogramming of nongermline ovarian stromal cells under a certain environment. In both cases, additional analysis of genetic instability and cell reprogramming is necessary to confirm the clinical feasibility of the suggested system, because chromosomal configuration is changed during stem cell transformation. Ovarian stroma cells may be transformed into ESC-like cells by in vitro oncogenesis under a certain environment. Either isolation of normal diploid cells having stemness from cancer cell or acquisition of differentiated cells without genetic instability would be important for acquiring clinical feasibility in this case. From both theoretical and practical viewpoints, reprogramming of germ cells during early meiosis shortly after chromosome recombination is barely possible.
Recent findings on the role of cell cycle regulators in iPS cell establishment (15) demonstrated that not all iPS cells have pluripotency. The established OCC can differentiate both in vivo and in vitro, but that cannot guarantee pluripotency of established cell lines. In fact, we found discrepancies in the methylation status of imprinted genes between OCC and the control ESC. Probably, incomplete reprogramming occurs under the specific environment reported in this study. Nevertheless, the results of our study may provide the information to establish induced stem cells by reprogramming without genetic manipulation. Further study is necessary to confirm whether the established cells have pluripotency or how these cells may be applied for clinical practice in human regenerative medicine.
MATERIALS AND METHODS
RT-PCR and Realtime PCR for Stemness-related Gene Expression in Various Organ and Characterization of Colony-forming Cells
Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA) and reverse-transcribed with Reverse Transcription System (Promega, Madison, WI). Synthesized complementary deoxyribonucleic acid (cDNA) was subjected to PCR amplification with specific primers (Supplementary Table 3). Primary3 software (Whitehead Institute/MIT Center for Genome Research, Cambridge, MA) was used to design all the primers in these experiments. For identifying specific genes and exclusion of pseudo genes, the PCR primers were designed based on mouse cDNA and genomic DNA sequences obtained from the U.S. National Institutes of Health GenBank database. The specificities of the designed primers were tested by conducting 40 PCR cycles of 95°C for 30 seconds, the annealing temperature for 45 seconds, and 72°C for 30 seconds.
To compare relative messenger ribonucleic acid (mRNA) level of samples, the cDNA synthesized from total RNA was quantified by real-time PCR using the DyNAmo SYBRGreen qPCR Kit (Finnzymes, Espoo, Finland). The mRNA level of each gene in the samples was normalized to that of β-actin and defined as 2−ΔΔCt, where Ct = threshold cycle for target amplification, ΔCt = Cttarget gene − Ctinternal reference (β–actin), and ΔΔCt = ΔCtsample − ΔCtcalibrator.
Karyotyping and DNA Content Analysis by Fluorescence-Activated Cell Sorter (FACS)
The cells were treated in culture medium supplemented with 0.05 μg/mL of Colcemid (Wako Pure Chemical, Osaka, Japan) for 1 to 2 hours and were subsequently harvested for trypsinization. The cells were treated with 0.56% KCl solution for 15 minutes and fixed in a cold methanoacetic acid (3:1) mixture for 30 minutes on ice. The fixative solution was changed twice by centrifuging the cells at a 30-minute interval. Chromosomes were spread onto heat-treated slides. The modified method of Seabright (1) was used for G-banding of air-dried chromosomes. The chromosome spread on glass slides was aged for approximately a week at room temperature, dipped in a 0.025% trypsin solution for 10 seconds, rinsed in distilled water, and stained with Giemsa’s solution in phosphate buffer (pH 6.8) for 10 minutes. After being washed in distilled water and air-dried, at least 50 spreads were counted for chromosome number, and 10 banding patterns were analyzed at 300 to 500 band resolution.
Immunoassay of Colony-forming Cells
Both lines of colony-forming cells were analyzed along with crude-dispersed ovarian cells before seeding, and embryonic stem cells (ESC) were used as control cells. By contrast, the colony-forming cells dissociated by 1 mM EDTA (Bioneer, Seoul, South Korea) were reacted for 1 hour at room temperature with primary antibodies to PE-conjugated Sca-1 (BD Biosciences, San Jose, CA), FITC-conjugated CD44 (BD Biosciences), biotin-conjugated CD34 (BD Biosciences), and biotin-conjugated CD45 (BD Biosciences). The Sca-1 and CD44 were specific markers for mesenchymal stem cells, and CD34 and CD45 were used for hematopoietic stem cell–specific markers. After washing twice, CD34 and CD45 were additionally reacted with streptavidin-phycoerythrin (SAv-PE; BD Biosciences). The antigen-antibody complexes were analyzed by flow cytometry (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ).
Test of Differentiation Potency
After formation of embryoid bodies, the embryoid bodies were seeded separately into four-well culture plates and cultured for 10 to 14 days. The cultured embryoid bodies were stained with the antibodies for S-100 (ectodermal), nestin (ectodermal), smooth muscle actin (mesodermal), Desmin (mesodermal), α-fetoprotein (endodermal), and Troma-1 (endodermal) of three germ layer–specific markers. To confirm in vivo differentiation, teratomas that formed in the subcutaneous region were fixed with 4% (v/v) paraformaldehyde (Sigma-Aldrich, St. Louis, MO). After they were embedded in paraffin blocks, the tissues were stained with hematoxylin and eosin for examination under a phase-contrast microscope (BX51TF; Olympus, Kogaku, Japan). For in vitro differentiation into neuronal lineage cells, the colony-forming cells were cultured in modified N2B27 medium which consisted of DMEM/F12 (GIBCO Invitrogen, Grand Island, NY) containing N2 (GIBCO Invitrogen) and B27 (GIBCO Invitrogen). Morphologic evaluation was conducted throughout the culture period, and the culture medium was changed at intervals of 2 days. Differentiating cells were maintained by replating into fibronectin-coated tissue culture dishes, and they were subsequently stained with the antibodies for Nestin and Tuj-1 for neuron, O4 for oligodendrocyte, glial fibrillary acidic protein (GFAP) for astrocyte, and tyrosine hydroxylase (TH) for dopaminergic neuron. Analyses of neuronal differentiation were replicated three times.
DNA Microsatellite Analysis
Four specific mouse microsatellite primers (D3Mit200, D11Mit4, D19Mit33, and D4Mit251) were collected from a public database (2). The genomic DNA from each sample was amplified by PCR for the four microsatellite loci. Forward primers were synthesized with a fluorescent tag (TET or HEX) at the 5′-end, and fluorescent PCR amplification was performed with the PC808 program TEMP control system (ASTEC, Fukuoka, Japan). The PCR products were subsequently analyzed in the ABI Prism 310 DNA automated sequencer (Applied Biosystems, Foster City, CA). Digital images were obtained using the Genescan Data Collection version 2.5 software (Applied Biosystems). Each fluorescent peak was quantified for base-pair size, peak height, and peak area.
Preparation of ESC and Parthenogenetic ESC as Control
For establishing of XX ESC, zona-free B6D2F1 3.5-day-old blastocysts were cultured on a MEF monolayer; after 4 to 5 days of culture, colonies derived from the inner cell mass cells were subcultured in knockout Dulbecco’s minimal essential medium supplemented with β-mercaptoethanol, nonessential amino acids, L-glutamine, mouse LIF, a 3:1 mixture of FBS and knockout serum replacement, and antibiotics. For deriving parthenogenetic ESC (pESC), mature oocytes were freed from the cumulus cells and subsequently activated by culturing in Ca2+-free potassium simplex optimized medium (KSOM) supplemented with 10 mM SrCl2 and 5 μg/mL of cytochalasin B for 4 hours. Parthenogenotes were cultured in Chatot, Ziomek, and Bavister (CZB) medium for deriving blastocysts, and inner cell mass cells of blastocysts were provided for pESC establishment.
SNP Genotyping
The SNP genotyping to distinguish genetic polymorphism between C57BL/6 and DBA2 was performed by iPLEXTM for use with the MassARRAY platform (Sequenom, San Diego, CA) (3). Genomic DNA was extracted from each sample using AccuPrep Genomic DNA extraction kit (Bioneer, Seoul, South Korea). All primers specific to each SNP and assay were designed by MassARRAY assay design software (Sequenom).
Bisulfite DNA Sequencing
We incubated 1 μg of genomic DNA with 3 M sodium bisulfite (pH 5.0), then 50 ng of bisulfite-modified DNA was subjected to PCR amplification of the Nanog, Oct-4, H19, Gtl2, Peg3, and Snrpn genes by the use of specific primer sets (Supplementary Table 3). The PCR products were cloned into pCRII vectors (Invitogen, Carlsbad, CA), and 10 clones of each specimen were sequenced by automated fluorescence-based DNA sequencing to determine the methylation status.
Culture of Cells Consisting of Primary Follicles
After we isolated primary follicles from the adult mouse ovaries, some were dissociated into oocytes and granulosa cells by treating 0.25% trypsin (GIBCO) for 30 minutes. Primary follicles themselves and the primary oocytes and granulosa cells derived from the follicles were cultured on mitomycin C–treated MEF monolayer containing DMEM, which was supplemented with 0.1 mM β-mercaptoethanol (GIBCO Invitrogen), 1% (v/v) nonessential amino acids (GIBCO Invitrogen), 2 mM L-glutamine (Sigma-Aldrich), 1% (v/v) lyophilized mixture of penicillin and streptomycin (GIBCO Invitrogen), 5000 units/mL of LIF (Chemicon), and 15% (v/v) fetal bovine serum (FBS; HyClone, Logan, UT). In several trials, zona-free, primary oocytes were also cultured for colony formation. The granulosa cells were cultured for 7 days, fixed by 4% paraformaldehyde (Sigma-Aldrich), and subsequently assessed for alkaline phosphatase (AP) activity and expression of stage-specific embryonic antigen 1 (SSEA-1) and antimüllerian hormone (AMH). The AP activity was assessed with Fast Red TR/naphthol AS-MX phosphate (Sigma-Aldrich) and localization of SSEA-1 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) and AMH (Abcam, Cambridge, United Kingdom) antibodies was performed using the DakoCytomation kit (Dako, Glostrup Denmark).
Culture of Mononuclear Cells Derived from Whole Blood
To collect whole blood, adult female mice (B6D2F1) were anesthetized by ether, and blood was drawn via cardiac puncture. For purifying the mononuclear cells, the blood was divided by density gradient centrifugation using Ficoll-Paque PLUS (Amersham Biosciences, Uppsala, Sweden) according to the manufacturer’s instructions. Buffy coat fraction containing the mononuclear cells was isolated and washed in DPBS. These cells were subsequently cultured under the same conditions as used for establishing OCC.
Supplementary Material
Acknowledgments
Supported by a grant (SC-5160) from Stem Cell Research Center of the 21st Century Frontier Research Program funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea the National Institute on Aging (NIH R37-AG012279), the National Institute of Child Health and Human Development (NIH R01-HD057873), the Henry and Vivian Rosenberg Philanthropic Fund, and Vincent Memorial Research Funds. This study was also supported by educational grants of Brain Korea 21 and WCU (World Class University) programs (R31-10056) through the National Research Foundation of Korea funded by MEST.
Footnotes
S.P.G. has nothing to disclose. S.T.L. has nothing to disclose. E.J.L. has nothing to disclose. D.Y.K. has nothing to disclose. G.L. has nothing to disclose. S.G.C. has nothing to disclose. B-K.R. has nothing to disclose. C.H.L. has nothing to disclose. K.E.Y. has nothing to disclose. H-J.L. has nothing to disclose. J.Y.H. has nothing to disclose. J.L.T. has nothing to disclose. J.M.L. has nothing to disclose.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Conrad S, Renninger M, Hennenlotter J, Wiesner T, Just L, Bonin M, et al. Generation of pluripotent stem cells from adult human testis. Nature. 2008;456:344–9. doi: 10.1038/nature07404. [DOI] [PubMed] [Google Scholar]
- 5.Guan K, Nayernia K, Maier LS, Wagner S, Dressel R, Lee JH, et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature. 2006;440:1199–203. doi: 10.1038/nature04697. [DOI] [PubMed] [Google Scholar]
- 6.Kanatsu-Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, Miki H, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119:1001–12. doi: 10.1016/j.cell.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 7.Johnson J, Canning J, Kaneko T, Pru JK, Tilly JL. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature. 2004;428:145–50. doi: 10.1038/nature02316. [DOI] [PubMed] [Google Scholar]
- 8.Gong SP, Lee EJ, Lee ST, Kim H, Lee SH, Han HJ, et al. Improved establishment of autologous stem cells derived from preantral follicle culture and oocyte parthenogenesis. Stem Cells Dev. 2008;17:695–712. doi: 10.1089/scd.2007.0168. [DOI] [PubMed] [Google Scholar]
- 9.Lee ST, Choi MH, Lee EJ, Gong SP, Jang M, Park SH, et al. Establishment of autologous embryonic stem cells derived from preantral follicle culture and oocyte parthenogenesis. Fertil Steril. 2008;90:1910–20. doi: 10.1016/j.fertnstert.2007.01.099. [DOI] [PubMed] [Google Scholar]
- 10.Cooper HM, Tamura RN, Quaranta V. The major laminin receptor of mouse embryonic stem cells is a novel isoform of the alpha 6 beta 1 integrin. J Cell Biol. 1991;115:843–50. doi: 10.1083/jcb.115.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonde S, Zavazava N. Immunogenicity and engraftment of mouse embryonic stem cells in allogeneic recipients. Stem Cells. 2006;24:2192–201. doi: 10.1634/stemcells.2006-0022. [DOI] [PubMed] [Google Scholar]
- 12.Lee JT. Disruption of imprinted X inactivation by parent-of-origin effects at Tsix. Cell. 2000;103:17–27. doi: 10.1016/s0092-8674(00)00101-x. [DOI] [PubMed] [Google Scholar]
- 13.Horii T, Kimura M, Morita S, Nagao Y, Hatada I. Loss of genomic imprinting in mouse parthenogenetic embryonic stem cells. Stem Cells. 2008;26:79–88. doi: 10.1634/stemcells.2006-0635. [DOI] [PubMed] [Google Scholar]
- 14.Li C, Chen Z, Liu Z, Huang J, Zhang W, Zhou L, et al. Correlation of expression and methylation of imprinted genes with pluripotency of parthenogenetic embryonic stem cells. Hum Mol Genet. 2009;18:2177–87. doi: 10.1093/hmg/ddp150. [DOI] [PubMed] [Google Scholar]
- 15.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–5. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971;2:971–2. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- 2.Center for Inherited Disease Research (CIDR) Mouse Marker Set. Available at http://www.cidr.jhmi.edu/mouse/mmset.html.
- 3.Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Koster H. Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci USA. 1999;96:10016–20. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.