

Abstract
The Curtius rearrangement is a classic, powerful method for converting carboxylic acids into protected amines, but its widespread use is impeded by safety issues (the need to handle azides). We have developed an alternative to the Curtius rearrangement that employs a copper catalyst in combination with blue-LED irradiation to achieve the decarboxylative coupling of aliphatic carboxylic acid derivatives (specifically, readily available N-hydroxyphthalimide esters) to afford protected amines under mild conditions. This C–N bond-forming process is compatible with a wide array of functional groups, including an alcohol, aldehyde, epoxide, indole, nitroalkane, and sulfide. Control reactions and mechanistic studies are consistent with the hypothesis that copper species are engaged in both the photochemistry and the key bond-forming step, which occurs through out-of-cage coupling of an alkyl radical.
TOC graphic
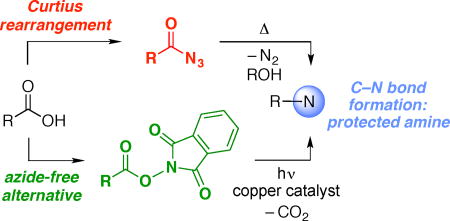
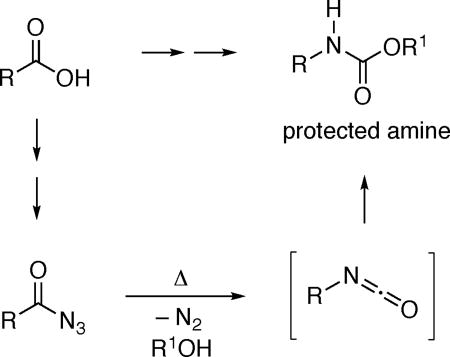
Due in part to the bioactivity of many amines,1 the construction of N–alkyl bonds is an important objective in organic synthesis; thus, processes such as reductive amination are among the most widely used reactions in the pharmaceutical industry.2 The Curtius rearrangement of acyl azides3 is a powerful method for the generation of amines from carboxylic acids (eq 1),4 which are a class of readily available compounds that have received a great deal of recent interest as feedstock starting materials for synthesis.5 However, safety concerns associated with azides are an impediment to the use of the Curtius rearrangement, leading to the pursuit of strategies such as flow chemistry in order to enable applications of this important transformation while mitigating potential hazards.6
 |
(1) |
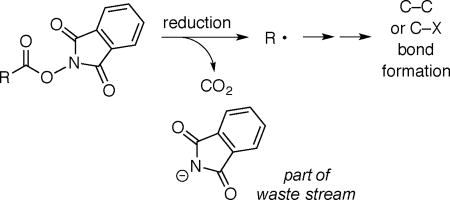
N-Hydroxyphthalimide (NHP) esters, which are readily prepared from carboxylic acids, have recently been intensively investigated as precursors to organic radicals, which then engage in intermolecular coupling reactions with a wide array of synthetically useful partners (eq 2).7,8,9 In these processes, the phthalimide unit of the original NHP ester becomes part of the waste stream.
 |
(2) |
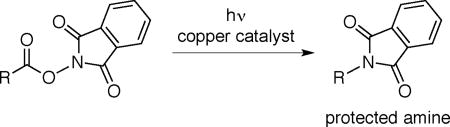
We have recently described a variety of photoinduced, copper-catalyzed coupling reactions that we believe are initiated by electron transfer to an electrophile to generate an organic radical, which then engages in copper-mediated bond formation with an added nucleophile.10,11,12 We became interested in expanding this approach to the development of an alternative to the Curtius rearrangement wherein NHP esters are employed as substrates (eq 3); this strategy has the attractive features of avoiding the use of azides and of efficiently utilizing the phthalimide of the NHP ester as the source of a protected amine.13,14 In this report, we describe the achievement of this objective.15,16
 |
(3) |
Thus, irradiation with blue LED lamps of a solution of the illustrated NHP ester in the presence of 10 mol% CuCN, 5 mol% 2,9-dimethyl-1,10-phenanthroline (dmp; neocuproine), and 15 mol% xantphos in 1,2-dichloroethane at 5–10 °C furnishes the desired protected amine in 77% yield (Table 1, entry 1). Control experiments establish the importance of light, CuCN, dmp, and xantphos for efficient C–N bond formation (entries 2–6). The coupling is sensitive to water (0.1 equiv; entry 7), but not to air (entry 8). If the NHP ester is heated, irradiated alone, irradiated in the presence of commonly employed ruthenium and iridium photoredox catalysts, or treated with a nickel catalyst,9a decarboxylative coupling is not observed (eq 4).17
Table 1.
Photoinduced, Copper-Catalyzed Decarboxylative C–N Coupling: Effect of Reaction Parameters.
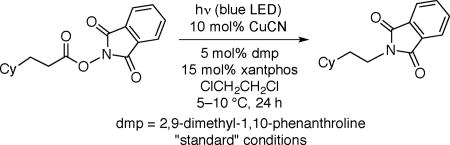
| ||
|---|---|---|
|
| ||
| entry | variation from the "standard" conditions | yield (%)a |
| 1 | none | 77 |
| 2 | no light | <1b |
| 3 | no light, 80 °C | <1b |
| 4 | no CuCN | <1b |
| 5 | no dmp | <1b |
| 6 | no xantphos | <1b |
| 7 | H2O (0.1 equiv) | 7 |
| 8 | capped vial under air | 76 |
The yield was determined through GC analysis with the aid of a calibrated internal standard (average of two experiments).
Recovered starting material: >90%.
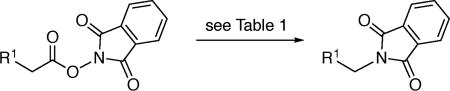
This photoinduced, copper-catalyzed decarboxylative C–N coupling can be applied to a range of NHP esters. Table 2 provides examples wherein the R group is a primary alkyl substituent. Although the efficiency of the process is impacted by the size of the substituent (entries 1–4), a reasonable yield is obtained even when R = neopentyl (entry 4). A wide variety of functional groups, including an olefin, ether, aryl bromide, aryl iodide, alkyl chloride, alkyl bromide, ester, ketone, carbamate, and thiophene, are compatible with the reaction conditions (entries 5–14). The method can be applied to a more complex structure (entry 15).
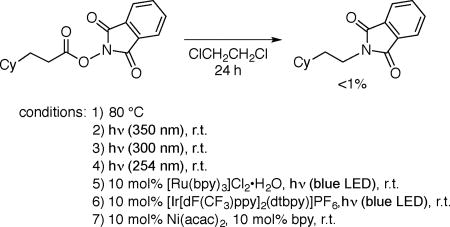 |
(4) |
Table 2.
Scope of the Decarboxylative C–N Coupling: Primary Alkyl Groups.
| |||||
|---|---|---|---|---|---|
|
|
|
||||
| entry | R1 | yield (%)a | entry | R1 | yield (%)a |
|
|
|
||||
| 1 | n-octyl | 74 | 9 |
|
71 |
| 2 |
|
71 | 10 |
|
50 |
| 3 |
|
65b | 11 |
|
66 |
| 4 |
|
50b | 12 |
|
64 |
| 5 |
|
70 | 13 |
|
68 |
| 6 |
|
72 | 14 |
|
57 |
| 7 |
|
65 | 15 |
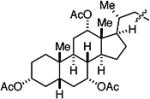
|
52 |
| 8 |
|
67 | |||
Yield of purified product (average of two experiments).
Catalyst loading: 20 mol% CuCN, 10 mol% dmp, 30 mol% xantphos.
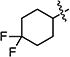
Decarboxylative amination can be achieved under the same conditions when the R group of the NHP ester is a secondary alkyl substituent (Table 3). Although C–N bond formation proceeds in modest yield if the group is acyclic (entry 1), improved efficiency is observed if it is cyclic (entries 2–7). The substrate can bear functional groups such as an olefin, alkyl fluoride, and ether (entries 5–7). The product of the coupling depicted in entry 3 can be isolated in comparable yield (60%) in a gram-scale reaction.18
Table 3.
Scope of the Decarboxylative C–N Coupling: Secondary Alkyl Groups.
Yield of purified product (average of two experiments); gram-scale reaction in parentheses.
We have further examined the functional-group tolerance of this new method by conducting a reaction in the presence of various additives (Table 4). We have determined that the presence of an alkene, alkyne, alkylboronate ester, indole, nitrile, aldehyde, benzofuran, epoxide, nitroalkane, secondary alcohol, tertiary amide, aryl tosylate, aryl triflate, or sulfide has essentially no impact on C–N bond formation, and that the additive can be recovered virtually quantitatively at the end of the reaction (≥90% yield). On the other hand, C–N bond formation is impeded by the presence of a primary amine or an alkylthiol.
Table 4.
Photoinduced, Copper-Catalyzed Decarboxylative C–N Coupling: Functional-Group Tolerance.

|
An outline of a possible pathway for this photoinduced, copper-catalyzed decarboxylative C–N coupling is outlined in Figure 1, involving a sequence of: photoexcitation of a copper(I) complex;19,20 electron transfer by the excited copper(I) complex to the NHP ester to form a copper(II) complex and the radical anion of the NHP ester, which successively fragments to produce the phthalimide anion (which binds to copper), CO2, and R•; recombination of R• and a copper(II)–phthalimide complex to afford the coupling product and regenerate a copper(I) complex.11,21,22 According to this mechanism, copper species are engaged in both the photochemistry and in the key bond-forming step.23 Although a number of photoinduced coupling reactions of NHP esters have been described,7,8,13 to the best of our knowledge this would be the first wherein a copper complex likely serves as the light absorber.24
Figure 1.

Outline of a possible pathway for photoinduced, copper-catalyzed decarboxylative C–N coupling (phth = phthalimidate).21
Various observations (Table 1 and eq 4) are consistent with the mechanistic hypothesis outlined in Figure 1; for example, in the absence of CuCN, the NHP ester does not react to a significant extent (>90% recovery; entry 4 of Table 1). The blue LED lamps used in this study do not emit at wavelengths less than 380 nm, whereas CuCN, dmp, the NHP ester, and xantphos do not individually absorb appreciably at wavelengths greater than 380 nm (Figure 2). In contrast, the combination of CuCN, dmp, and xantphos does absorb in the range from 380–460 nm (Figure 2),19 and the resulting emission can be quenched by the addition of the NHP ester.
Figure 2.

Absorption spectra in ClCH2CH2Cl of: (A) CuCN; (B) dmp; (C) NHP ester (R = CH2CH2Cy); (D) xantphos; (E) CuCN:dmp:xantphos (2:1:3) (reaction mixture without the NHP ester); (F) reaction mixture; (G) [Cu(dmp)2]PF6 (not observed in the reaction mixture).
The loss of CO2 from a carboxyl radical occurs with a rate constant of ~2–7 × 109 s−1 (R = ethyl or isopropyl in MeOH at 20 °C),25 which is similar to typical rate constants for diffusion (generally >108 s−1).26 In order to gain insight into whether escape of the reactants from the solvent cage occurs prior to C–N bond formation, we have conducted crossover experiments (Figures 3a and 3b) and a trapping experiment with TEMPO (Figure 3c); our observations of crossover and of trapping by TEMPO are consistent with cage escape. Furthermore, we have carried out radical-clock studies that indicate that, once R• is formed, it undergoes coupling more slowly than the illustrated ring-opening and ring-forming reactions (Figures 3d and 3e).27,28,29,30
Figure 3.

Photoinduced, copper-catalyzed decarboxylative C–N coupling: Mechanistic studies.
In conclusion, we have developed a photoinduced, copper- catalyzed decarboxylative C–N coupling that, by achieving the overall conversion of an aliphatic carboxylic acid to a protected amine, provides an alternative to the Curtius rearrangement that avoids the use of hazardous azides. The reaction proceeds under mild conditions (5–10 °C; blue LED) with a catalyst derived from commercially available components (CuCN, dmp, and xantphos) and is compatible with a very wide array of functional groups (e.g., an alcohol, aldehyde, indole, nitroalkane, and sulfide); in contrast, the Curtius rearrangement typically involves heating to ≥80 °C and generates a reactive isocyanate intermediate. We suggest a possible mechanism wherein copper plays a key role both in the photochemical step and in C–N coupling, and we present experiments that are consistent with the formation of radical intermediates that escape from the solvent cage prior to C–N bond formation. Our current efforts are directed at continuing to expand the scope of photoinduced, copper-catalyzed coupling reactions, including developing enantioconvergent processes of racemic substrates.
Supplementary Material
Acknowledgments
Support has been provided by Amgen and by the National Institutes of Health (NIGMS: R01–109194). We thank Jun Myun Ahn, Dr. Victor J. Cee, Bradley J. Gorsline, Carson D. Matier, Dr. Paul H. Oyala, and Dr. Haolin Yin for assistance and helpful discussions.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Procedures and characterization data (PDF)
The authors declare no competing financial interests.
References
- 1.For example, see: Funayama S, Cordell GA, editors. Alkaloids: A Treasury of Poisons and Medicines. Academic Press; Waltham, MA: 2014.
- 2.For example, see Figure 1 in: Brown DG, Boström J. J. Med. Chem. 2016;59:4443–4458. doi: 10.1021/acs.jmedchem.5b01409.
- 3.For leading references, see: Ahmad NM. In: Name Reactions for Functional Group Transformations. Li JJ, Corey EJ, editors. John Wiley & Sons: Hoboken, NJ; 2007. pp. 438–450.
- 4.For leading references to related processes, including the Lossen rearrangement, see: Aube J, Fehl C, Liu R, McLeod MC, Motiwala HF. In: Comprehensive Organic Synthesis. Knochel P, Molander GA, editors. Vol. 6. Elsevier; Amsterdam: 2015. pp. 598–635.
- 5.For recent illustrative examples, see: Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525.Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;352:801–805. doi: 10.1126/science.aaf6123.
- 6.For a recent example and discussion, see: Marsini MA, Buono FG, Lorenz JC, Yang B-S, Reeves JT, Sidhu K, Sarvestani M, Tan Z, Zhang Y, Li N, Lee H, Brazzillo J, Nummy LJ, Chung JC, Luvaga IK, Narayanan BA, Wei X, Song JJ, Roschangar F, Yee NK, Senanayake CH. Green Chem. 2017;19:1454–1461.
- 7.For pioneering studies of photoinduced activation of NHP esters see: Okada K, Okamoto K, Oda M. J. Am. Chem. Soc. 1988;110:8736–8738.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J. Am. Chem. Soc. 1991;113:9401–9402.
- 8.For an early application of photoinduced activation of NHP esters to achieve C–C bond formation in total synthesis, see: Schnermann MJ, Overman LE. Angew. Chem. Int. Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977.
- 9.For representative examples of recent studies, see: Edwards JT, Merchant RR, McClymont KS, Knouse KW, Qin T, Malins LR, Vokits B, Shaw SA, Bao D-H, Wei F-L, Zhou T, Eastgate MD, Baran PS. Nature. 2017;545:213–218. doi: 10.1038/nature22307.Fawcett A, Pradeilles J, Wang Y, Mutsuga T, Myers EL, Aggarwal VK. Science. 2017;357:283–286. doi: 10.1126/science.aan3679.Tlahuext-Aca A, Garza-Sanchez RA, Glorius F. Angew. Chem. Int. Ed. 2017;56:3708–3711. doi: 10.1002/anie.201700049.Suzuki N, Hofstra JL, Poremba KE, Reisman SE. Org. Lett. 2017;19:2150–2153. doi: 10.1021/acs.orglett.7b00793.Huihui KMM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, Ackerman LKG, Weix DJ. J. Am. Chem. Soc. 2016;138:5016–5019. doi: 10.1021/jacs.6b01533.
- 10.(a) arylation of nitrogen nucleophiles: Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647–651. doi: 10.1126/science.1226458.(b) alkylation of nitrogen nucleophiles: Bissember AC, Lundgren RJ, Creutz SE, Peters JC, Fu GC. Angew. Chem. Int. Ed. 2013;52:5129–5133. doi: 10.1002/anie.201301202.(c) arylation of sulfur nucleophiles: Uyeda C, Tan Y, Fu GC, Peters JC. J. Am. Chem. Soc. 2013;135:9548–9552. doi: 10.1021/ja404050f.(d) arylation, alkenylation, and alkynylation of nitrogen nucleophiles: Ziegler DT, Choi J, Munoz-Molina JM, Bissember AC, Peters JC, Fu GC. J. Am. Chem. Soc. 2013;135:13107–13112. doi: 10.1021/ja4060806.(e) alkylation of nitrogen nucleophiles: Do H-Q, Bachman S, Bissember AC, Peters JC, Fu GC. J. Am. Chem. Soc. 2014;136:2162–2167. doi: 10.1021/ja4126609.(f) arylation of oxygen nucleophiles: Tan Y, Munoz-Molina JM, Fu GC, Peters JC. Chem. Sci. 2014;5:2831–2835.(g) alkylation of carbon nucleophiles: Ratani TS, Bachman S, Fu GC, Peters JC. J. Am. Chem. Soc. 2015;137:13902–13907. doi: 10.1021/jacs.5b08452.(h) enantioconvergent alkylation of nitrogen nucleophiles: Kainz QM, Matier CM, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313.
- 11.For mechanistic studies, see: Johnson MW, Hannoun KI, Tan Y, Fu GC, Peters JC. Chem. Sci. 2016;7:4091–4100. doi: 10.1039/c5sc04709a.Ahn JM, Ratani TS, Hannoun KI, Fu GC, Peters JC. submitted for publication.
- 12.For leading references to the use of copper complexes in photocatalysis, see: Paria S, Reiser O. ChemCatChem. 2014;6:2477–2483.
- 13.For reviews of decarboxylative reactions, see: (a) C–C bond formation: Patra T, Maiti D. Chem. Eur. J. 2017;23:7382–7401. doi: 10.1002/chem.201604496.Rodriguez N, Goossen LJ. Chem. Soc. Rev. 2011;40:5030–5048. doi: 10.1039/c1cs15093f.(b) photoredox processes: Jin Y, Fu H. Asian J. Org. Chem. 2017;6:368–385.
- 14.Decarboxylative reactions that involve the net extrusion of CO2 from a substrate are well-known (e.g. transition metal-catalyzed allylation and benzylation: Weaver JD, Recio A, Grenning AJ, Tunge JA. Chem. Rev. 2011;111:1846–1913. doi: 10.1021/cr1002744.) and have been demonstrated in photoredox chemistry (e.g., Le C, MacMillan DWC. J. Am. Chem. Soc. 2015;137:11938–11941. doi: 10.1021/jacs.5b08304.
- 15.For examples of metal-catalyzed intermolecular decarboxylative C–N couplings, see: Lang SB, Cartwright KC, Welter RS, Locascio TM, Tunge JA. Eur. J. Org. Chem. 2016:3331–3334. doi: 10.1002/ejoc.201600620.See also: Zhang Y, Patel S, Mainolfi N. Chem. Sci. 2012;3:3196–3199.propiolic acids: Jia W, Jiao N. Org. Lett. 2010;12:2000–2003. doi: 10.1021/ol1004615.
- 16.For examples of intramolecular decarboxylative C–N couplings, see: Jin Y, Yang H, Fu H. Org. Lett. 2016;18:6400–6403. doi: 10.1021/acs.orglett.6b03300.Liu Z-J, Lu X, Wang G, Li L, Jiang W-T, Wang Y-D, Xiao B, Fu Y. J. Am. Chem. Soc. 2016;138:9714–9719. doi: 10.1021/jacs.6b05788.Dai Q, Li P, Ma N, Hu C. Org. Lett. 2016;18:5560–5563. doi: 10.1021/acs.orglett.6b02724.
- 17.While our study was underway, H. Fu reported that thiophenols can catalyze photoinduced decarboxylative C–N couplings of the type illustrated in eq 3, wherein the NHP ester is derived from an α-amino acid, via an N-acylimine intermediate (Reference 16a); however, application of these conditions (CFL irradiation, 10 mol% 4-trifluoromethylthiophenol, 0.5 equiv Cs2CO3, DMF, r.t.) to the NHP ester illustrated in eq 4 furnishes none of the desired product (<1%).
- 18.Deprotection of the product under conventional conditions affords cyclohexylamine in 83% isolated yield.
- 19.For reports of the use of [Cu(dmp)(xantphos)]BF4 as a photocatalyst in organic chemistry, see: Hernandez-Perez AC, Collins SK. Angew. Chem. Int. Ed. 2013;52:12696–12700. doi: 10.1002/anie.201306920.Xiao P, Dumur F, Zhang J, Fouassier JP, Gigmes D, Lalevee J. Macromolecules. 2014;47:3837–3844.
- 20.When the mixture of CuCN, dmp, and xantphos shown in Table 1 is replaced with 10 mol% [Cu(dmp)(xantphos)]BF4, essentially none of the desired product is formed (<1% yield). We hypothesize that [Cu(dmp)(xantphos)]+, which we have identified by UV–vis spectroscopy and ESI–MS under the reaction conditions, is serving as the photoreductant (e.g. see Reference 19), but that an independent copper complex is involved in the C–N bond-forming step.
- 21.Notes: (a) A number of variations in this mechanistic outline can be envisioned, including fragmentation of the NHP ester through copper-bound intermediates, as well as C–N bond formation via reductive elimination from a copper(III) complex that bears R and phth as ligands. (b) In Figure 1, the copper(II)–phthalimide complex that engages in C–N bond formation is not formed from the same copper(I) complex that undergoes excitation. Diffusion out of the solvent cage, ligand exchange, etc. occur in between (vide infra).
- 22.For a non-photoinduced, copper-catalyzed intermolecular C– H phthalimidation of unactivated alkanes, see: Tran BL, Li B, Driess M, Hartwig JF. J. Am. Chem. Soc. 2014;136:2555–2563. doi: 10.1021/ja411912p.
- 23.For leading references on photoredox catalysis, see: Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r.Skubi KL, Blum TR, Yoon TP. Chem. Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018.
- 24.For a method that employs [Ru(bpy)3]Cl2 for photoactivation and copper for C–C bond formation, see: Zhang H, Zhang P, Jiang M, Yang H, Fu H. Org. Lett. 2017;19:1016–1019. doi: 10.1021/acs.orglett.6b03888.
- 25.Hilborn JW, Pincock JA. J. Am. Chem. Soc. 1991;113:2683–2686. [Google Scholar]
- 26.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. p. 156. [Google Scholar]
- 27.For leading references on radical clocks, see: Newcomb M. In: Encyclopedia of Radicals in Chemistry, Biology and Materials. Chatgilialoglu C, Studer A, editors. Vol. 1. John Wiley & Sons; Chichester: 2012. pp. 107–124.
- 28.This discussion is based on the assumption that ring opening and ring formation are proceeding through radical rather than organometallic, pathways.
- 29.An EPR study of a C–N coupling after 30 minutes at 10 °C showed a weak signal that is consistent with a copper-containing radical.
- 30.For the C–N coupling illustrated in Table 1, ethylcyclohexane and vinylcyclohexane, which may form via the disproportionation of R•, comprise the majority of the mass balance (15%).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.