

Abstract
The digestive tract of triatomines (DTT) is an ecological niche favored by microbiota whose enzymatic profile is adapted to the specific substrate availability in this medium. This report describes the molecular enzymatic properties that promote bacterial prominence in the DTT. The microbiota composition was assessed previously based on 16S ribosomal DNA, and whole sequenced genomes of bacteria from the same genera were used to calculate the GC level of rare and prominent bacterial species in the DTT. The enzymatic reactions encoded by coding sequences of both rare and common bacterial species were then compared and revealed key functions explaining why some genera outcompete others in the DTT. Representativeness of DTT microbiota was investigated by shotgun sequencing of DNA extracted from bacteria grown in liquid Luria-Bertani broth (LB) medium. Results showed that GC-rich bacteria outcompete GC-poor bacteria and are the dominant components of the DTT microbiota. In addition, oxidoreductases are the main enzymatic components of these bacteria. In particular, nitrate reductases (anaerobic respiration), oxygenases (catabolism of complex substrates), acetate-CoA ligase (tricarboxylic acid cycle and energy metabolism), and kinase (signaling pathway) were the major enzymatic determinants present together with a large group of minor enzymes including hydrogenases involved in energy and amino acid metabolism. In conclusion, despite their slower growth in liquid LB medium, bacteria from GC-rich genera outcompete the GC-poor bacteria because their specific enzymatic abilities impart a selective advantage in the DTT.
Keywords: GC content, genome size, gene number, EC number, ecological niche, midgut
Introduction
Chagas disease remains a serious health concern in South American countries, with approximately 8 million people in the chronic phase of this parasitosis. Trypanosoma cruzi, the causative agent, is mainly transmitted to humans by insects from the Triatominae subfamily distributed throughout the American continent.1
The host-parasite relationship between T cruzi and vertebrate hosts has been extensively studied and studies continue to develop new drugs and vaccines.2,3 In contrast, there are still few studies on the host-parasite relationship involving T cruzi and its interaction with the microbiota in the triatomine vector gut. Pioneer work by Azambuja et al4 showed that Serratia marcescens, belonging to the family Enterobacteriaceae, is a major component of the bacterial microbiota in the digestive tract of triatomines (DTT) that may kill T cruzi through mannose-sensitive fimbriae5,6 and could thus affect the epidemiology of Chagas disease. An investigation of the bacterial composition in the DTT was only recently undertaken at a molecular level through 16S ribosomal DNA (rDNA) characterization by da Mota et al7 and Gumiel et al.8 These 2 investigations found that the bacterial microbiota diversity is low (less than 10 major species) and varies in composition depending on the species of host triatomine. Apart from the intracellular endosymbiont genera, Arsenophonus, Wolbachia, and Candidatus Rohrkolberia, the major bacterial species found in the DTT were from Serratia genera and from the suborder Corynebacterineae (Mycobacterium, Rhodococcus, Gordonia, Corynebacterium, and Dietzia). Another microbiota with a low level of diversity has been described in female mosquitoes (Anopheles gambiae and Aedes aegypti), which are also hematophagous insects.9
This low number of major bacterial species found in the DTT provides an opportunity to investigate their molecular determinants. Aside from the major bacterial species mentioned above, da Mota et al7 and Gumiel et al8 also found several bacterial species that previously have not been reported to reproduce significantly in DTT. These species belong to the following genera: Acinetobacter, Actinomyces, Adhaeribacter, Bradyrhizobium, Chryseobacterium, Comamonas, Diaphorobacter, Enterococcus, Erwinia, Geobacillus, Haemophilus, Hydrogenophilus, Janthinobacterium, Marinomonas, Microvirga, Pectobacterium, Propionibacterium, Providencia, Pseudomonas, Shinella, Sphingomonas, Staphylococcus, Stenotrophomonas, Streptococcus, Streptophyta, Williamsia, and Xanthobacter. If some bacterial species reproduce optimally in the DTT, there must be a biochemical basis and it needs elucidation due to the possibility of controlling Chagas disease through paratransgenesis to reduce vector competence with genetically modified symbionts.10,11
The aim of this investigation was to identify enzymatic determinants resulting in the success of bacterial genera in the DTT, such as Serratia and the members of Corynebacterineae, in contrast to minor genera, including Staphylococcus, Streptococcus, Haemophilus, or Enterococcus. Thus, the biochemical differences were analyzed between the GC-rich (rich in guanine + cytosine) and GC-poor bacterial species found in the DTT, emphasizing specific enzymatic functions that could explain the success of the former compared with the latter bacteria.12–16
Materials and Methods
The successive methodological steps followed to analyze the genome properties and enzymatic determinants in the particular niche of DTT were as follows:
To set up a metagenomic model of wild microbiota in DTT from species of bacteria having their genome completely sequenced and being as close as possible (from the same genera) to those identified by 16S rDNA sequencing;
To characterize the gross genomic features (genome size, gene number, GC level, enzymatic annotation) from bacteria of the metagenomic model;
To set up a working hypothesis based on the metagenomic model to justify the enzymatic determinants that make certain bacteria outcompete others in the DTT niche;
To validate the inference drawn from the metagenomic model through a bench experiment using a quantitative marker. The bench experiment was a shotgun sequencing characterization of the bacterial population from DTTs after Luria-Bertani broth (LB) culture, whereas the quantitative marker was the ratio of the enzyme annotations that are overrepresented in GC-rich compared with GC-poor bacterial species relative to the DNA-encoded protein samples from these bacteria.
Ethics statement
The animals used for blood feeding the triatomines at FIOCRUZ were treated according to the Ethical Principles in Animal Experimentation approved by the Ethics Committee in Animal Experimentation (CEUA/FIOCRUZ) under the license numbers LW-24/2013 and following the protocol from Conselho Nacional de Experimentação Animal/Ministério de Ciência e Tecnologia. Triatomines were captured under the license L14323-7 given by the Sistema de Autorização e Informação em Biodiversidade (SISBIO) of the Instituto Chico Mendes de Conservação da Biodiversidade/Ministério do Meio Ambiente (MMA).
Triatomine colonies, gut dissection, and bacterial cultures
Triatoma infestans, Triatoma vitticeps, Panstrongylus megistus and Rhodnius neglectus are of epidemiologic importance.17 Dipetalogaster maximus has no epidemiologic importance, but presents good susceptibility to T cruzi, is used in xenodiagnosis, and is limited to Southern California and Mexico where it lives in a rocky habitat in association with lizards.18 The male and female triatomines used were in the fifth instar and maintained on chicken blood over approximately 20 generations in the Laboratório de Doenças Parasitárias (Instituto Oswaldo Cruz—IOC, Fundação Oswaldo Cruz—FIOCRUZ. Triatomines were dissected 7 to 10 days after feeding by opening the dorsal side from the posterior end of the abdomen to the last thoracic segment. Meticulous dissection of the midgut (stomach and intestine) and hindgut (rectum) was performed using a sterile ultrafine insulin syringe needle. Feces were obtained by abdominal compression or spontaneous ejections immediately after feeding. Guts and feces were collected together in sterile Eppendorf tubes and maintained at −20°C until use. All steps were performed under aseptic conditions.
Three guts and their feces of each T infestans, T vitticeps, D maximus, P megistus, and R neglectus were then incubated in 2-mL Eppendorf tubes filled with liquid LB (Sigma-Aldrich Brasil Ltda., Sao Paulo, Brazil) at 30°C without agitation for circa 48 hours until turbidity, due to bacterial growth, became evident.
DNA extraction from bacterial cultures
After incubation in LB medium for 48 hours at 30°C without agitation in 2-mL Eppendorf tubes, the triatomine digestive tracts were removed, and the DNA from the remaining bacterial suspensions was extracted with the Fast DNA Spin Kit for Soil (BIO 101 Systems; Qbiogene, Carlsbad, CA, USA) according to the manufacturer’s protocol. DNA concentrations were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). About 1 µg DNA for each sample of the 5 triatomine species was then amplified with a Nextera DNA Library Preparation Kit (Illumina, San Diego, CA, USA) and sequenced through 454 Titanium technology.
Microbial composition of triatomine digestive tract, sequence databases, and GC content
The predominant bacterial genera identified by denaturing gradient gel electrophoresis (DGGE) in the digestive tract of T infestans, T vitticeps, D maximus, P megistus, and R neglectus were Serratia, Erwinia, Candidatus Rohrkolberia, Providencia, Pectobacterium, and Arsenophonus.7 Of these 5 triatomine species, the genus Triatoma had a more diverse microbiota.
A previous more detailed study of the microbiota composition from digestive tracts of Triatoma brasiliensis collected in the field8 showed that the most abundant bacterial genera found by 454 sequencing of 16S rDNA were Gordonia sp. (36%), Serratia sp. (18%), Mycobacterium sp. (18%), Corynebacterium (6%), and Rhodococcus sp. (6%). Serratia was the most widely distributed genus among the triatomine species investigated here (Table 1). Complete genomes were sequenced for at least one species in most of the bacterial genera identified in this work (Table 1) and can be downloaded from ftp://ftp.ncbi.nih.gov/genomes/genbank/bacteria/. The coding sequences (CDS) were retrieved from the sequences of these genomes, available in *.fna files (see Table 1), by homologous comparison (tBLASTn) with their protein sequences, available in *.faa files (“*” stands for the name of the bacterial species under consideration). The average GC content was then calculated for (1) whole CDS or for (2) the first (GC1), second (GC2), and third (GC3) codon positions of each genome set, using a Perl script. When a complete genome sequence was not available for a given bacterial genus, as in the case of Arsenophonus sp. and Dietzia sp., a CDS sample was retrieved from GenBank (release 208—June 15, 2015) using the Infobiogen server (see http://www.infobiogen.fr) and the ACNUC/QUERY retrieval system19 with the options t = cds. The CDS samples used here for the GCx (x = 1, 2, 3, or the average of them) calculations are from bacterial species that, in most cases, were not the same as those diagnosed by da Mota et al7 and Gumiel et al,8 although the GC content obtained from these species was considered representative of the genus to which they belonged. Reference was made to Takahashi et al13 who stated that the construction of phylogenetic trees based on oligonucleotide frequency of bacterial species with similar GC contents led to topologies that were congruent at genus and family levels with those constructed from homologous genes. The bacterial genomes in GC-poor and GC-rich species were divided according to whether their GC3 was lower or higher than 50%.
Table 1.
Sequence materials for GC and enzymatic characterization.
| Genera | Freq. | Whole genome sequences | GC |
|---|---|---|---|
| Acinetobacter a | 2 | NA | |
| Actinomyces a | 1 | NA | |
| Adhaeribacter a | 1 | NA | |
| Arsenophonus b | Endo | GenBank | Poor |
| Bradyrhizobium a | 1 | Bradyrhizobium_japonicum_USDA_6_uid158851/NC_017249.fna | Rich |
| Chryseobacterium a | 1 | NA | |
| Comamonas a | 1 | Comamonas_testosteroni_CNB_2_uid62961/NC_013446.fna | Rich |
| Corynebacterium a | 2015 | Corynebacterium_terpenotabidum_Y_11_uid210639/NC_021663.fna | Rich |
| Diaphorobacter a | 2 | NA | |
| Dietzia a | 5008 | GenBank | Rich |
| Enterococcus a | 6 | Enterococcus_faecalis_D32_uid171261/NC_018221.fna | Poor |
| Erwinia b | Low | Erwinia_amylovora_ATCC_49946_uid46943/NC_013971.fna | Rich |
| Geobacillus a | 2 | NA | |
| Gordonia a | 11825 | Gordonia_polyisoprenivorans_VH2_uid86651/NC_016906.fna | Rich |
| Haemophilus a | 1 | Haemophilus_somnus_2336_uid57979/NC_010519.fna | Poor |
| Hydrogenophilus a | 15 | NA | |
| Janthinobacterium a | 1 | NA | |
| Marinomonas a | 1 | Marinomonas_posidonica_IVIA_Po_181_uid67323/NC_015559.fna | Poor |
| Microvirga a | 2 | NA | |
| Mycobacterium a | 5737 | Mycobacterium_marinum_M_uid59423/NC_010612.fna | Rich |
| Pectobacterium b | Low | Pectobacterium_carotovorum_PC1_uid59295/NC_012917.fna | Rich |
| Propionibacterium a | 5 | Propionibacterium_propionicum_F0230a_uid170533/NC_018142.fna | Rich |
| Pseudomonas a | 4 | Pseudomonas_aeruginosa_RP73_uid209328/NC_021577.fna | Rich |
| Rhodococcus a | 1855 | Rhodococcus_opacus_B4_uid13791/NC_012522.fna | Rich |
| Serratia a | 4917 | Serratia_marcescens_FGI94_uid185180/NC_020064.fna | Rich |
| Shinella a | 2 | NA | |
| Sphingomonas a | 1 | Sphingomonas_wittichii_RW1_uid58691/NC_009511.fna | Rich |
| Staphylococcus a | 3 | Staphylococcus_saprophyticus_ATCC_15305_uid58411/NC_007350.fna | Poor |
| Stenotrophomonas a | 1 | Stenotrophomonas_maltophilia_D457_uid162199/NC_017671.fna | Rich |
| Streptococcus a | 1 | Streptococcus_oralis_Uo5_uid65449/NC_015291.fna | Poor |
| Streptophyta a | 2 | NA | |
| Williamsia a | 15 | NA | |
| Wolbachia b | Endo | Wolbachia_endosymbiont_of_Culex_quinquefasciatus_Pel_uid61645/NC_010981.fna | Poor |
| Xanthobacter a | 1 | Xanthobacter_autotrophicus_Py2_uid58453/NC_009720.fna | Rich |
Abbreviation: NA, not available.
Bacterial species detected by Gumiel et al.8
Bacterial species detected by da Mota et al.7
Freq. is for the numbers in Table 2 of Gumiel et al8 indicating the maximum absolute number of times a genus was detected over each of 4 samples. Low is for the low but uncharacterized level of detection of a genus by DGGE in da Mota et al.7 Endo is for the high but uncharacterized level of detection of an endosymbiont by DGGE in da Mota et al.7
Enzymatic profiling of bacterial genomes used as references
For this study, the classification Enzyme Commission (EC) numbers from the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) was used. If different enzymes catalyze the same reaction, then they receive the same EC number. Approximately 5500 enzyme reactions have already been classified according to a 4-digit hierarchy that is used to progressively refine classification descriptions. Briefly, the first digit reports on the type of reaction considered, which is divided into 6 main categories: Oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases. The second digit of the EC number describes the type of chemical object the reaction is acting on, whereas the third digit often describes the type of donor or acceptor group. Finally, the fourth digit associates the enzyme with its reaction name (see a complete description at http://www.chem.qmul.ac.uk/iubmb/enzyme/).
Protein sequences of files (1) NC_018221.faa (Enterococcus faecalis), NC_010519.faa (Haemophilus somnus), NC_007350.faa (Staphylococcus saprophyticus), NC_015291.faa (Streptococcus oralis) and (2) NC_010612.faa (Mycobacterium marinum), NC_012522.faa (Rhodococcus opacus), NC_016906.faa (Gordonia polyisoprenivorans), NC_020064.faa (Serratia marcescens), and NC_021663.faa (Corynebacterium terpenotabidum) were considered representative of GC-poor and GC-rich bacterial species found in the DTT, respectively.8 By taking only the best hits (E value < 0.0001) into account, the protein sequences (BLASTp) of each of the files outlined above were compared with the enzyme sequences from the database of the Kyoto Encyclopedia of Genes and Genomes (KEGG version from June 2015; http://www.genome.jp/kegg/) where the EC numbers are available. A homologous hit was considered significant when its identity rate was at least 60% for at least 33 amino acids. For each file of the homology comparison, the EC numbers (http://www.enzyme-database.org/class.php) were grouped according to their first (6 classes), second (67 subclasses), third (264 subclasses), and fourth digits (the whole EC number set of 5549 approved enzymes as available from BRENDA—online release as of June, 30, 2015; http://www.brenda-enzymes.org/all_enzymes.php; see the “Results” section). Finally, the relative frequency of EC numbers per functional category were compared between GC-poor and GC-rich genomes of bacterial species considered representative of the bacterial genera diagnosed in DTT.
Shotgun sequencing and analysis of DNA from bacterial cultures of triatomine guts
The shotgun sequencing of bacteria from DTT incubated in LB medium was performed to determine the validity of the model proposed above. At the time of the experiment, a representative amplification by polymerase chain reaction would have needed an amount of DNA that was not compatible with that obtained from direct extraction of triatomine feces. Thus, a culture step was introduced prior to shotgun library construction knowing that it would introduce a bias due to the different growth conditions in LB and DTT.
The 723 543 reads obtained by 454 sequencing were mounted into 16 435 contigs using Velvet20 according to http://ged.msu.edu/angus/tutorials-2011/short-read-assembly-velvet.html (k = 31, ie, 31mers were looking for overlaps between reads) and further assembled with CAP321 to finally obtain 14 269 nonredundant sequences (supplementary file S1). We then extracted the 738 297 open reading frames (ORF) from both positive and negative strands of these 14 269 sequences and filtered them out for CDS ORFs (cORFs) larger than 99 bp (base pairs) using the universal feature method (UFM),22,23 ending up with 35 105 cORFs compatible with the purine bias found in the CDSs (supplementary file S2). These sequences were then compared (BLASTx) with a data set composed by the protein sequences of GC-rich bacteria from the whole genomes of C terpenotabidum (NC_021663.fna), G polyisoprenivorans (NC_016906.fna), M marinum (NC_010612.fna), R opacus (NC_012522.fna), and S marcescens (NC_020064.fna), downloaded from ftp://ftp.ncbi.nih.gov/genomes/Bacteria/ and filtered out the homologies for identity rates ≥60% over ≥33 amino acids. We retrieved the sequence subset corresponding to these homologies with a Perl script and compared (BLASTx) them with the subset of protein sequences from KEGG corresponding to the list of EC numbers that are more frequent in GC-rich bacteria compared with GC-poor forms according to their first 3 digits. Finally, we did the same exercise with the protein sequences from the whole genomes of GC-poor bacteria, ie, S saprophyticus (NC_007350.fna), H somnus (NC_010519.fna), E faecalis (NC_018221.fna), and S oralis (NC_015291.fna), and compared the results.
Statistics
Due to the different environmental conditions, differential growth of the GC-poor and GC-rich bacteria in DTT and LB were to be expected. Thus, a marker is needed to verify that the model matches the bench experiment (shotgun sequences). Therefore, as a marker, the ratio (proportion) of (1) enzymatic functions that are overrepresented in GC-rich bacteria compared with GC-poor ones relative to (2) the whole DNA–encoded protein sample for the type of bacteria considered (GC-poor or GC-rich) were chosen. Because the relative ratio of these enzymatic functions is different in GC-rich and GC-poor bacteria, then rejection of the null hypothesis of equality of both proportions (1 for GC-rich and 1 for GC-poor bacteria) in the bench experiment is to be expected if it mirrors the model (where both proportions are different).
There are at least 4 different methods to test the equality of 2 proportions, but 1 based on the Z score (https://onlinecourses.science.psu.edu/stat414/node/268) is presented here. According to this method, the hypothesis of equality of 2 proportions H0: p1 = p2 can be rejected if a quantity, Z, is larger than a theoretical value (1.96) of reference for a probability risk α = 0.05. The quantity Z is calculated using formula (1):
| (1) |
where,
| (2) |
is the proportion of successes in the 2 samples combined (Y1 and Y2 are the absolute frequency of success in samples 1 and 2, respectively. The sample sizes of Y1 and Y2 are referred to as n1 and n2, respectively).
Results
In genera of bacteria isolated from Triatoma spp., those with GC-rich genomes surpass in relative number (75%) the genera of bacteria with GC-poor genomes (25%).8 In addition, among the genera described by Gumiel et al,8 the most widely represented include species with GC-rich genomes, whereas the genera only marginally represented include bacterial species with GC-poor genomes (Table 2). Obviously, being GC-rich is not sufficient for a bacterium to outperform others present in the gut of triatomines because 45% of the other minor bacterial species were also GC-rich.8 However, because all outperforming bacteria (6 belonging to 6 genera in 6 different families with 5 from Actinomycetales and 1 from Enterobacteriales) were GC-rich, the possibility of the GC level being a key factor for these bacteria in the DTT cannot be ignored (Table 2).
Table 2.
GC content of coding sequences associated with bacterial genera in digestive tract of triatomines.
| Phylum | Class | Order | Suborder | Family | Species | N | GC class | GC, % | σGC | GC1, % | σGC1 | GC2, % | σGC2 | GC3, % | σGC3 | Fra |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Firmicutes | Bacilli | Bacillales | NA | Staphylococcaceae | Staphylococcus saprophyticus | 1844 | Poor | 33.8 | 4.6 | 46.8 | 5.4 | 31.7 | 4.6 | 22.9 | 3.8 | Lb |
| Proteobacteria | α-Proteobacteria | Rickettsiales | NA | Anaplasmataceae | Wolbachia sp. | 1037 | Poor | 34.4 | 4.5 | 44.3 | 4.8 | 32.9 | 4.3 | 26.1 | 4.3 | Endoc |
| Proteobacteria | γ-Proteobacteria | Pasteurellales | NA | Pasteurellaceae | Haemophilus somnus | 1601 | Poor | 37.5 | 5.1 | 49.5 | 5.1 | 34.9 | 4.5 | 28.3 | 5.7 | L |
| Firmicutes | Bacilli | Lactobacillales | NA | Enterococcaceae | Enterococcus faecalis | 2249 | Poor | 37.8 | 4.7 | 49.4 | 5.0 | 34.3 | 4.7 | 29.8 | 4.5 | L |
| Firmicutes | Bacilli | Lactobacillales | NA | Streptococcaceae | Streptococcus oralis | 1451 | Poor | 41.3 | 5.9 | 52.2 | 5.8 | 34.0 | 4.6 | 37.7 | 7.2 | L |
| Proteobacteria | γ-Proteobacteria | Oceanospirillales | NA | Oceanospirillaceae | Marinomonas posidonica | 2795 | Poor | 44.6 | 4.6 | 54.0 | 4.4 | 37.9 | 3.9 | 41.7 | 5.3 | L |
| Proteobacteria | γ-Proteobacteria | Enterobacteriales | NA | Enterobacteriaceae | Arsenophonus nasoniae | 306 | Poor | 42.3 | 6.0 | 45.2 | 5.9 | 39.6 | 6.0 | 42.2 | 6.1 | Endo |
| Proteobacteria | γ-Proteobacteria | Enterobacteriales | NA | Enterobacteriaceae | Pectobacterium carotovorum | 3322 | Rich | 52.2 | 6.4 | 59.0 | 6.0 | 40.9 | 4.7 | 56.7 | 8.6 | L |
| Proteobacteria | γ-Proteobacteria | Enterobacteriales | NA | Enterobacteriaceae | Erwinia amylovora | 2478 | Rich | 53.9 | 6.7 | 60.2 | 6.4 | 41.7 | 4.9 | 59.6 | 9.0 | L |
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacterineae | Dietziaceae | Dietzia spp. | 157 | Rich | 68.0 | 4.7 | 69.1 | 5.3 | 66.3 | 4.3 | 68.6 | 4.4 | Hd |
| Proteobacteria | γ-Proteobacteria | Enterobacteriales | NA | Enterobacteriaceae | Serratia marcescens | 3620 | Rich | 59.4 | 7.5 | 63.2 | 6.6 | 42.7 | 5.1 | 72.5 | 10.8 | H |
| Proteobacteria | β-Proteobacteria | Burkholderiales | NA | Comamonadaceae | Comamonas testosteroni | 3941 | Rich | 62.0 | 6.2 | 65.2 | 5.3 | 45.9 | 4.9 | 74.8 | 8.3 | L |
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacterineae | Mycobacteriaceae | Mycobacterium marinum | 4464 | Rich | 65.6 | 5.4 | 68.0 | 4.9 | 50.6 | 5.5 | 78.0 | 5.8 | H |
| Proteobacteria | α-Proteobacteria | Rhizobiales | NA | Bradyrhizobiaceae | Bradyrhizobium japonicum | 7029 | Rich | 64.0 | 6.1 | 65.1 | 4.9 | 47.9 | 4.9 | 78.9 | 8.6 | L |
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacterineae | Gordoniaceae | Gordonia polyisoprenivorans | 3870 | Rich | 67.1 | 5.4 | 69.0 | 5.0 | 51.1 | 5.2 | 81.3 | 6.0 | H |
| Actinobacteria | Actinobacteria | Actinomycetales | NA | Propionibacteriaceae | Propionibacterium propionicum | 2292 | Rich | 66.4 | 6.3 | 68.4 | 5.5 | 49.2 | 5.4 | 81.6 | 8.0 | L |
| Proteobacteria | γ-Proteobacteria | Xanthomonadales | NA | Xanthomonadaceae | Stenotrophomonas maltophilia | 3275 | Rich | 67.2 | 5.7 | 69.8 | 5.6 | 48.8 | 5.4 | 83.0 | 6.2 | L |
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacterineae | Corynebacteriaceae | Corynebacterium terpenotabidum | 1861 | Rich | 67.3 | 5.4 | 68.8 | 5.1 | 49.7 | 5.3 | 83.3 | 5.7 | H |
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacterineae | Nocardiaceae | Rhodococcus opacus | 5908 | Rich | 67.9 | 5.7 | 69.7 | 4.9 | 50.6 | 5.3 | 83.5 | 6.7 | H |
| Proteobacteria | α-Proteobacteria | Rhizobiales | NA | Xanthobacteraceae | Xanthobacter autotrophicus | 3783 | Rich | 67.6 | 6.0 | 69.1 | 5.6 | 49.7 | 5.3 | 84.0 | 7.3 | L |
| Proteobacteria | γ-Proteobacteria | Pseudomonadales | NA | Pseudomonadaceae | Pseudomonas aeruginosa | 4627 | Rich | 66.6 | 6.4 | 68.7 | 5.8 | 46.8 | 5.8 | 84.3 | 7.5 | L |
| Proteobacteria | α-Proteobacteria | Sphingomonadales | NA | Sphingomonadaceae | Sphingomonas wittichii | 4069 | Rich | 68.7 | 5.9 | 69.3 | 5.6 | 50.0 | 5.2 | 86.7 | 6.9 | L |
Abbreviation: NA, not available.
Fr is for the frequency of bacterial species reported in Gumiel et al.8
L is for low frequency.
Endo is for endosymbiont.
H is for high frequency and is highlighted in gray background.
The shading regions in Table 2 is to improve the contrast between GC-rich (gray) and GC-poor (white) genomes.
On comparing GC3 with genome size and gene number for the species of the genera identified by Gumiel et al8 for which a complete genome sequence was available, a significant positive correlations was found for GC3 vs genome size (r = .66, P < .01), GC3 vs gene number (r = .61, P < .01), and genome size vs gene number (r = .99, P < .01) (Table 3).
Table 3.
Relationships between GC3, genome size, and gene number in the representative bacterial species with complete genome sequence of bacterial genera found in the intestinal tract of triatomines.
| Species | GC3 | Genome, bp | Gene, nb |
|---|---|---|---|
| Staphylococcus saprophyticus (NC_007350) | 22.9 | 2 516 573 | 2445 |
| Haemophilus somnus (NC_010519) | 28.3 | 2 263 855 | 1980 |
| Enterococcus faecalis (NC_018221) | 29.8 | 2 987 449 | 2876 |
| Streptococcus oralis (NC_015291) | 37.7 | 1 958 688 | 1905 |
| Marinomonas posidonica (NC_015559) | 41.7 | 3 899 938 | 3491 |
| Pectobacterium carotovorum (NC_012917) | 56.7 | 4 862 911 | 4246 |
| Erwinia amylovora (NC_013971) | 59.6 | 3 805 872 | 3437 |
| Serratia marcescens (NC_020064) | 72.5 | 4 858 215 | 4361 |
| Comamonas testosteroni (NC_013446) | 74.8 | 5 373 642 | 4802 |
| Mycobacterium marinum (NC_010612) | 78.0 | 6 636 826 | 5423 |
| Bradyrhizobium japonicum (NC_017249) | 78.9 | 9 207 382 | 8826 |
| Gordonia polyisoprenivorans (NC_013441) | 81.3 | 5 669 804 | 4945 |
| Propionibacterium propionicum (NC_018142) | 81.6 | 3 449 358 | 2938 |
| Stenotrophomonas maltophilia (NC_017671) | 83.0 | 4 769 154 | 4101 |
| Corynebacterium terpenotabidum (NC_021663) | 83.3 | 2 751 232 | 2369 |
| Rhodococcus opacus (NC_012522) | 83.5 | 7 913 449 | 7246 |
| Xanthobacter autotrophicus (NC_009720) | 84.0 | 5 308 932 | 4746 |
| Pseudomonas aeruginosa (NC_021577) | 84.3 | 6 342 033 | 5762 |
| Sphingomonas wittichii (NC_009511) | 86.7 | 5 382 259 | 4850 |
| Correlations | |||
| rGC3 × GenomSza | 0.66 | — | — |
| r GC3 × GeneNb b | — | 0.61 | — |
| r GenomSz × GeneNb c | — | — | 0.99 |
Correlation for GC3 vs genome size.
Correlation for GC3 vs gene number.
Correlation for genome size vs gene number.
The shading regions in Table 3 is to improve the contrast between GC-rich (gray) and GC-poor (white) genomes.
If the last correlation may seem trivial in bacteria, the first one is not (the second is a consequence of the first given the third). As a consequence of the positive correlation between GC3 and genome size, GC-rich genomes of DTT bacterial microbiota have a potentially more complex metabolism than that of GC-poor genomes, which seems to be an advantage in this system. A more careful analysis of Table 3 shows that Corynebacterium has a small genome (at least in the species considered here), but this fact is not necessarily a contradiction because several other Corynebacterineae in DTT have large genomes. It simply suggests that Corynebacterium (at least the species considered here) may be in a process of genome reduction on the basis of the enzymatic apparatus of the family.
When comparing enzymatic activities in GC-poor and GC-rich bacteria through the evaluation of their relative frequency according to the first digit of the EC numbers (Table 4), oxidoreductases might explain the success of GC-rich bacteria because they were 2 times more frequent, on average, than in GC-poor ones.
Table 4.
Relative frequency (%) of enzymatic functions in GC-poor and GC-rich bacterial species found in triatomine digestive tract according to the first EC number digit.
| ECNO. |
Ss |
Hs |
Ef |
So |
Average | SD | Sm |
Mm |
Gp |
Ct |
Ro |
Average | SD | AvGCr/AvGCpa | Class of enzyme function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC3,% | 22.9 | 28.3 | 29.8 | 37.7 | 72.5 | 78.0 | 81.3 | 83.3 | 83.5 | ||||||
| 1. -.-.- | 17.9 | 14.4 | 12.7 | 12.8 | 14.4 | 2.4 | 21.2 | 31.6 | 30.5 | 20.3 | 34.2 | 27.5 | 6.4 | 1.9 | Oxidoreductases |
| 2. -.-.- | 31.9 | 33.8 | 31.6 | 33.7 | 32.8 | 1.2 | 29.5 | 28.8 | 27.6 | 32.4 | 25.3 | 28.7 | 2.6 | 0.9 | Transferases |
| 3. -.-.- | 29.3 | 27.2 | 35.5 | 33.9 | 31.5 | 3.9 | 28.3 | 21.2 | 19.6 | 25.3 | 19.8 | 22.8 | 3.8 | 0.7 | Hydrolases |
| 4. -.-.- | 8.2 | 10.0 | 6.9 | 7.2 | 8.1 | 1.4 | 10.1 | 8.5 | 9.6 | 9.3 | 8.9 | 9.3 | 0.6 | 1.1 | Lyases |
| 5. -.-.- | 5.3 | 7.5 | 6.7 | 5.1 | 6.1 | 1.1 | 6.2 | 3.3 | 4.5 | 4.3 | 4.3 | 4.5 | 1.0 | 0.7 | Isomerases |
| 6. -.-.- | 7.5 | 7.1 | 6.6 | 7.3 | 7.1 | 0.4 | 4.7 | 6.6 | 8.3 | 8.4 | 7.6 | 7.1 | 1.5 | 1.0 | Ligases |
Abbreviations: Ct, Corynebacterium terpenotabidum; EC no., Enzyme Commission number; Ef, Enterococcus faecalis; Gp, Gordonia polyisoprenivorans; Hs, Haemophilus somnus; Mm, Mycobacterium marinum; Ro, Rhodococcus opacus; Sm, Stenotrophomonas maltophilia; So, Streptococcus oralis; Ss, Staphylococcus saprophyticus.
Factor difference where AvGCr is for average of GC-rich and AvGCp is for average of GC-poor.
The shading regions in Table 4 is to improve the contrast between GC-rich (gray) and GC-poor (white) genomes.
According to this observation, the enzymatic comparison of the second digit (Table 5) revealed a larger number of subcategories (6, ie, acting on the CH-CH group of donors—EC:1.3.-.-, acting on the CH-NH2 group of donors—EC:1.4.-.-, acting on single donors with incorporation of molecular oxygen—EC:1.13.-.-, acting on paired donors with incorporation or reduction of molecular oxygen—EC:1.14.-.-, acting on iron-sulfur proteins as donors—EC:1.18.-.-) with a larger EC number frequency (≥2 times more frequent) in GC-rich compared with GC-poor bacteria in oxidoreductases than in the other 4 categories of the first digit level, ie, 2 (acting on ether bonds—EC:3.3.-.-, acting on carbon-carbon bonds—EC:3.7.-.-) in hydrolases, 1 (intramolecular lyases—EC:5.5.-.-) in isomerases, and 1 (forming carbon-sulfur bonds—EC:6.2.-.-) in ligases. The largest differences of EC relative frequency, according to the second digit within those of the first digit category, between GC-poor and GC-rich bacteria were due to acting on single donors with incorporation of molecular oxygen—EC:1.13.-.- (difference of ~24 times) and acting on paired donors with incorporation or reduction of molecular oxygen—EC:1.14.-.- (difference of ~8 times). The differences due to acting on iron-sulfur proteins as donors—EC:1.18.-.- (difference of ~5 times), acting on ether bonds—EC:3.3.-.- (difference of ~6 times), acting on carbon-carbon bonds—EC:3.7.-.- (difference of ~4 times), and forming carbon-sulfur bonds—EC:6.2.-.- (difference of ~4 times) were also relatively large (the other differences being around 2 times). Thus, the functional variability of the enzymatic apparatus seems to be important for a bacterium to be able to outperform the others in the intestinal environment of triatomines. In addition, we found that even if the function acting on diphenols and related substances as donors (EC:1.10.-.-) exists only at a low rate in all GC-rich bacteria of Table 5, it is simply absent from the GC-poor bacteria found in DTT. This kind of function is an example of a larger metabolic complexity in GC-rich bacteria in the DTT environment.
Table 5.
Relative frequency (%) of enzymatic functions in GC-poor and GC-rich bacterial species found in triatomine digestive tract according to the 2 first EC number digits.
| ECNO. |
Ss |
Hs |
Ef |
So |
Average | SD | Sm |
Mm |
Gp |
Ct |
Ro |
Average | SD | AvGCr/AvGCpa | Class of enzyme function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC3,% | 22.9 | 28.3 | 29.8 | 37.7 | 72.5 | 78.0 | 81.3 | 83.3 | 83.5 | ||||||
| 1.3.-.- | 1.3 | 1.4 | 1.2 | 1.3 | 1.3 | 0.1 | 1.6 | 5.1 | 4.6 | 2.8 | 5.2 | 3.8 | 1.6 | 2.9 | Acting on the CH-CH group of donors |
| 1.4.-.- | 0.9 | 0.2 | 0.3 | 0.3 | 0.4 | 0.3 | 0.9 | 0.9 | 0.9 | 1.3 | 1.6 | 1.1 | 0.3 | 2.7 | Acting on the CH-NH2 group of donors (amino acid oxidoreductase) |
| 1.10.-.- | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.1 | 0.2 | 0.3 | 0.1 | 0.2 | 0.1 | ∞ | Acting on diphenols and related substances as donors (diphenol oxidoreductases) |
| 1.13.-.- | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.6 | 0.6 | 0.8 | 0.1 | 1.5 | 0.7 | 0.5 | 24.5 | Acting on single donors with incorporation of molecular oxygen (monooxygenases) |
| 1.14.-.- | 0.8 | 0.6 | 0.1 | 0.1 | 0.4 | 0.4 | 1.4 | 4.3 | 4.9 | 2.1 | 3.8 | 3.3 | 1.5 | 8.2 | Acting on paired donors, with incorporation or reduction of molecular oxygen (dioxygenases) |
| 1.18.-.- | 0.1 | 0.0 | 0.2 | 0.0 | 0.1 | 0.1 | 0.3 | 0.2 | 0.4 | 0.4 | 0.5 | 0.4 | 0.1 | 5.0 | Acting on iron-sulfur proteins as donors |
| 3.3.-.- | 0.1 | 0.0 | 0.1 | 0.0 | 0.0 | 0.1 | 0.1 | 0.4 | 0.2 | 0.3 | 0.4 | 0.3 | 0.1 | 6.1 | Acting on ether bonds |
| 3.7.-.- | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.2 | 0.0 | 0.2 | 0.1 | 0.1 | 4.4 | Acting on carbon-carbon bonds |
| 5.5.-.- | 0.0 | 0.1 | 0.0 | 0.1 | 0.1 | 0.1 | 0.2 | 0.1 | 0.2 | 0.1 | 0.1 | 0.1 | 0.0 | 2.2 | Intramolecular lyases |
| 6.2.-.- | 0.6 | 0.5 | 0.5 | 0.1 | 0.4 | 0.2 | 0.5 | 2.3 | 1.7 | 0.6 | 3.3 | 1.7 | 1.2 | 4.0 | Forming carbon-sulfur bonds |
Abbreviations: Ct, Corynebacterium terpenotabidum; EC no., Enzyme Commission number; Ef, Enterococcus faecalis; Gp, Gordonia polyisoprenivorans; Hs, Haemophilus somnus; Mm, Mycobacterium marinum; Ro, Rhodococcus opacus; Sm, Stenotrophomonas maltophilia; So, Streptococcus oralis; Ss, Staphylococcus saprophyticus.
Factor difference where AvGCr is for average of GC-rich and AvGCp is for average of GC-poor.
The shading regions in Table 5 is to improve the contrast between GC-rich (gray) and GC-poor (white) genomes and is expected to improve table readability.
The comparison of the third digit place of EC numbers (Table 6) for difference of enzymatic activity between GC-poor and GC-rich bacteria of the DTT showed a consistently larger number of enzymes involved in oxygen and nitrogen processing suggesting a much larger ability to cope with the degradation of complex substrates of higher chemical stability such as those containing aromatic rings (aryls). These enzymes can be mainly grouped under EC numbers 1.13.11.-, 1.4.3.-, 1.3.99.-, and 1.14.99.- but also to a lesser extent in 1.13.12.-, 1.14.11.-, 1.14.12.-, 1.14.13.-, 1.1.99.-, and 1.7.99.-.
Table 6.
Relative frequency (%) of enzymatic functions in GC-poor and GC-rich bacterial species found in the triatomine digestive tract according to the 3 first EC number digits.
| ECNO. |
Ss |
Hs |
Ef |
So |
Average | SD | Sm |
Mm |
Gp |
Ct |
Ro |
Average | SD | AvGCr/AvGCpa | Class of enzyme function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC3,% | 22.9 | 28.3 | 29.8 | 37.7 | 72.5 | 78.0 | 81.3 | 83.3 | 83.5 | ||||||
| 1.1.2.- | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.2 | 0.1 | 0.1 | 0.0 | 0.1 | 0.1 | ∞ | With a cytochrome as acceptor |
| 1.1.3.- | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.0 | 0.2 | 0.3 | 0.0 | 0.4 | 0.2 | 0.2 | 2.8 | With oxygen as acceptor |
| 1.1.99.- | 0.1 | 0.0 | 0.1 | 0.0 | 0.1 | 0.1 | 0.5 | 0.1 | 0.3 | 0.1 | 0.2 | 0.2 | 0.2 | 4.8 | With unknown physiological acceptors |
| 1.2.1.- | 1.5 | 0.9 | 0.9 | 0.3 | 0.9 | 0.5 | 1.5 | 2.1 | 2.6 | 1.8 | 3.5 | 2.3 | 0.8 | 2.6 | With NAD+ or NADP+ as acceptor |
| 1.3.99.- | 0.3 | 0.6 | 0.2 | 0.1 | 0.3 | 0.2 | 0.6 | 4.5 | 3.4 | 1.8 | 4.7 | 3.0 | 1.8 | 9.6 | With unknown physiological acceptors |
| 1.4.1.- | 0.6 | 0.1 | 0.3 | 0.1 | 0.3 | 0.2 | 0.5 | 0.3 | 0.5 | 0.7 | 0.9 | 0.6 | 0.2 | 2.0 | With NAD+ or NADP+ as acceptor |
| 1.4.3.- | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.3 | 0.5 | 0.4 | 0.3 | 0.6 | 0.4 | 0.1 | 14.2 | With oxygen as acceptor |
| 1.4.4.- | 0.2 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.2 | 0.0 | 0.3 | 0.1 | 0.1 | 0.1 | 2.8 | With a disulfide as acceptor |
| 1.5.3.- | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.2 | 0.1 | 0.5 | 0.1 | 0.2 | 0.2 | 0.2 | ∞ | With oxygen as acceptor |
| 1.6.1.- | 0.0 | 0.2 | 0.0 | 0.0 | 0.1 | 0.1 | 0.2 | 0.3 | 0.3 | 0.1 | 0.1 | 0.2 | 0.1 | 3.5 | With NAD+ or NADP+ as acceptor |
| 1.7.99.- | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.7 | 0.1 | 0.0 | 0.0 | 0.3 | 0.2 | 0.3 | 6.7 | With unknown physiological acceptors |
| 1.9.3.- | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.2 | 0.3 | 0.1 | 0.1 | 0.1 | 5.3 | With oxygen as acceptor |
| 1.13.11.- | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.5 | 0.4 | 0.6 | 0.0 | 1.2 | 0.5 | 0.4 | 17.5 | With incorporation of 2 atoms of oxygen |
| 1.13.12.- | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.3 | 0.3 | 0.1 | 0.5 | 0.2 | 0.2 | ∞ | With incorporation of 1 atom of oxygen (internal monooxygenases or internal mixed function oxidases) |
| 1.14.11.- | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.2 | 0.1 | 0.3 | 0.0 | 0.2 | 0.1 | 0.1 | 4.3 | With 2-oxoglutarate as 1 donor, and incorporation of 1 atom each of oxygen into both donors |
| 1.14.12.- | 0.2 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.2 | 0.0 | 0.2 | 0.4 | 0.4 | 0.2 | 0.2 | 4.5 | With NADH or NADPH as 1 donor, and incorporation of 2 atoms of oxygen into 1 donor |
| 1.14.13.- | 0.5 | 0.5 | 0.1 | 0.0 | 0.3 | 0.3 | 0.6 | 1.0 | 3.0 | 0.6 | 1.7 | 1.4 | 1.0 | 4.9 | With NAD or NADH as 1 donor, and incorporation of 1 atom of oxygen |
| 1.14.99.- | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.3 | 0.3 | 0.4 | 0.2 | 0.2 | 9.2 | Miscellaneous |
| 1.17.7.- | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.0 | 0.0 | 0.1 | 0.0 | 2.1 | With an iron-sulfur protein as acceptor |
| 1.18.1.- | 0.1 | 0.0 | 0.2 | 0.0 | 0.1 | 0.1 | 0.2 | 0.2 | 0.4 | 0.4 | 0.6 | 0.4 | 0.2 | 5.1 | With NAD+ or NADP+ as acceptor |
| 2.7.11.- | 0.4 | 0.0 | 0.2 | 0.3 | 0.2 | 0.2 | 0.2 | 0.8 | 1.1 | 0.6 | 1.3 | 0.8 | 0.4 | 3.6 | Protein-serine/threonine kinases |
| 2.8.3.- | 0.1 | 0.2 | 0.0 | 0.0 | 0.1 | 0.1 | 0.2 | 0.7 | 0.5 | 0.6 | 1.4 | 0.7 | 0.4 | 7.7 | CoA-transferases |
| 2.10.1.- | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.0 | 0.0 | 0.1 | 0.0 | 2.1 | Molybdenumtransferases or tungstentransferases with sulfide groups as acceptors |
| 3.1.6.- | 0.0 | 0.2 | 0.0 | 0.1 | 0.1 | 0.1 | 0.2 | 0.3 | 0.1 | 0.1 | 0.4 | 0.2 | 0.1 | 2.4 | Sulfuric ester hydrolases |
| 3.3.2.- | 0.1 | 0.0 | 0.1 | 0.0 | 0.1 | 0.1 | 0.1 | 0.4 | 0.2 | 0.1 | 0.4 | 0.3 | 0.2 | 5.1 | Ether hydrolases |
| 4.3.3.- | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.3 | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.1 | 4.1 | Amine-lyases |
| 5.1.99.- | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.1 | 0.3 | 0.1 | 0.3 | 0.2 | 0.1 | ∞ | Acting on other compounds |
| 5.3.3.- | 0.1 | 0.0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.3 | 0.4 | 0.3 | 0.1 | 0.2 | 0.3 | 0.1 | 3.2 | Transposing C=C bonds |
| 5.5.1.- | 0.0 | 0.1 | 0.0 | 0.1 | 0.1 | 0.1 | 0.2 | 0.1 | 0.2 | 0.1 | 0.1 | 0.2 | 0.0 | 2.4 | Miscellaneous |
| 6.2.1.- | 0.6 | 0.5 | 0.5 | 0.1 | 0.4 | 0.2 | 0.6 | 2.6 | 1.9 | 0.6 | 3.6 | 1.9 | 1.3 | 4.3 | Miscellaneous |
Abbreviations: Ct, Corynebacterium terpenotabidum; EC no., Enzyme Commission number; Ef, Enterococcus faecalis; Gp, Gordonia polyisoprenivorans; Hs, Haemophilus somnus; Mm, Mycobacterium marinum; Ro, Rhodococcus opacus; Sm, Stenotrophomonas maltophilia; So, Streptococcus oralis; Ss, Staphylococcus saprophyticus.
Factor difference where AvGCr is for average of GC-rich and AvGCp is for average of GC-poor.
The shading regions in Table 6 is to improve the contrast between GC-rich (gray) and GC-poor (white) genomes.
With the list of EC numbers (n = 30) that are overrepresented in GC-rich bacteria according to the first 3 digits (Table 6), the protein sequences were retrieved corresponding to the EC numbers fully described on the 4 digits (n = 778) in (1) S saprophyticus (n = 39), H somnus (n = 25), E faecalis (n = 29), and S oralis (n = 11), ie, 26 EC numbers on average (σ = 11.6) for GC-poor bacteria and (2) C terpenotabidum (n = 55), G polyisoprenivorans (n = 96), M marinum (n = 73), R opacus (n = 116), and S marcescens (n = 95), ie, 87 EC numbers on average (σ = 23.0) for GC-rich bacteria. This statistic means that GC-rich bacteria have enzymes with 3.3 times more enzymatic functionalities than GC-poor ones, on average, according to the list of Table 6, which sustains the hypothesis that GC-rich bacteria outperform GC-poor bacteria because of their more complex metabolism, which seems to be an advantage in the DTT. Most enzymatic activities were found in dehydrogenases (aldehyde and amino acid), oxygenases (mono and di), and ligase (acetate-CoA), which are enzymatic activities involved in the very first steps of molecular degradation and synthesis.
Table 7 shows that among 35 enzymatic reactions that are overrepresented in GC-rich bacteria, the large majority are from oxidoreductases (74%) followed by transferases and ligases (9% each) with hydrolases and lyases in last position accounting for only 6% and 3%, respectively. A closer look at Table 7 enables understanding that the enzyme groups, which most explain the differences between GC-rich and GC-poor bacteria, are ranked by decreasing level of factor difference (AvGCr/AvGCp) and that the data of Table 7 can be reorganized as shown in Table 8.
Table 7.
Relative frequency (%) of enzymatic functions in GC-poor and GC-rich bacterial species found in the triatomine digestive tract according to the 4 EC number digits.
| ECNO. |
Ss |
Hs |
Ef |
So |
Average | SD | Sm |
Mm |
Gp |
Ct |
Ro |
Average | SD | AvGCr/AvGCpa | Class of enzyme function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC3,% | 22.9 | 28.3 | 29.8 | 37.7 | 72.5 | 78.0 | 81.3 | 83.3 | 83.5 | ||||||
| 1.1.99.1 | 1.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.5 | 2.0 | 1.0 | 1.0 | 1.0 | 2.0 | 1.4 | 0.5 | 5.6 | Choline dehydrogenase |
| 1.2.1.2 | 4.0 | 1.0 | 1.0 | 0.0 | 1.5 | 1.7 | 7.0 | 8.0 | 7.0 | 2.0 | 9.0 | 6.6 | 2.7 | 4.4 | Formate dehydrogenase |
| 1.2.1.3 | 3.0 | 0.0 | 1.0 | 0.0 | 1.0 | 1.4 | 3.0 | 4.0 | 5.0 | 2.0 | 14.0 | 5.6 | 4.8 | 5.6 | Aldehyde dehydrogenase (NAD+) |
| 1.2.1.7 | 1.0 | 1.0 | 0.0 | 0.0 | 0.5 | 0.6 | 1.0 | 3.0 | 2.0 | 2.0 | 1.0 | 1.8 | 0.8 | 3.6 | Benzaldehyde dehydrogenase (NADP+) |
| 1.2.1.8 | 1.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.5 | 5.0 | 0.0 | 1.0 | 1.0 | 2.0 | 1.8 | 1.9 | 7.2 | Betaine-aldehyde dehydrogenase |
| 1.2.1.10 | 0.0 | 0.0 | 3.0 | 0.0 | 0.8 | 1.5 | 1.0 | 1.0 | 1.0 | 0.0 | 5.0 | 1.6 | 1.9 | 2.1 | Acetaldehyde dehydrogenase (acetylating) |
| 1.2.1.16 | 1.0 | 1.0 | 0.0 | 0.0 | 0.5 | 0.6 | 1.0 | 1.0 | 2.0 | 1.0 | 2.0 | 1.4 | 0.5 | 2.8 | Glyceraldehyde-3-phosphate dehydrogenase (NADP+) (phosphorylating) |
| 1.2.1.27 | 0.0 | 1.0 | 0.0 | 0.0 | 0.3 | 0.5 | 1.0 | 1.0 | 3.0 | 0.0 | 5.0 | 2.0 | 2.0 | 8.0 | Methylmalonate-semialdehyde dehydrogenase (acylating) |
| 1.2.1.38 | 1.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.5 | 1.0 | 0.0 | 1.0 | 1.0 | 1.0 | 0.8 | 0.4 | 3.2 | N-acetyl-g-glutamyl-phosphate reductase |
| 1.2.1.70 | 1.0 | 1.0 | 0.0 | 0.0 | 0.5 | 0.6 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 0.0 | 2.0 | Glutamyl-tRNA reductase |
| 1.3.99.1 | 2.0 | 3.0 | 1.0 | 0.0 | 1.5 | 1.3 | 8.0 | 6.0 | 9.0 | 2.0 | 7.0 | 6.4 | 2.7 | 4.3 | Succinate dehydrogenase |
| 1.4.1.1 | 3.0 | 0.0 | 1.0 | 0.0 | 1.0 | 1.4 | 3.0 | 4.0 | 6.0 | 4.0 | 8.0 | 5.0 | 2.0 | 5.0 | Alanine dehydrogenase |
| 1.4.1.2 | 2.0 | 0.0 | 0.0 | 0.0 | 0.5 | 1.0 | 2.0 | 1.0 | 0.0 | 0.0 | 6.0 | 1.8 | 2.5 | 3.6 | Glutamate dehydrogenase |
| 1.4.1.13 | 2.0 | 0.0 | 1.0 | 0.0 | 0.8 | 1.0 | 3.0 | 3.0 | 5.0 | 2.0 | 6.0 | 3.8 | 1.6 | 5.1 | Glutamate synthase (NADPH) |
| 1.4.3.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 | 1.0 | 1.0 | 2.0 | 3.0 | 1.6 | 0.9 | ∞ | d-aspartate oxidase |
| 1.4.4.2 | 2.0 | 0.0 | 0.0 | 0.0 | 0.5 | 1.0 | 2.0 | 3.0 | 0.0 | 2.0 | 3.0 | 2.0 | 1.2 | 4.0 | Glycine dehydrogenase (decarboxylating) |
| 1.5.3.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 4.0 | 1.0 | 5.0 | 1.0 | 5.0 | 3.2 | 2.0 | ∞ | Sarcosine oxidase |
| 1.6.1.2 | 0.0 | 2.0 | 0.0 | 0.0 | 0.5 | 1.0 | 2.0 | 2.0 | 3.0 | 1.0 | 3.0 | 2.2 | 0.8 | 4.4 | NAD(P)+ transhydrogenase (Re/Si-specific) |
| 1.7.99.4 | 0.0 | 1.0 | 0.0 | 0.0 | 0.3 | 0.5 | 12.0 | 1.0 | 0.0 | 0.0 | 6.0 | 3.8 | 5.2 | 15.2 | Nitrate reductase |
| 1.13.11.2 | 0.0 | 1.0 | 0.0 | 0.0 | 0.3 | 0.5 | 3.0 | 4.0 | 2.0 | 0.0 | 11.0 | 4.0 | 4.2 | 16.0 | Catechol 2,3-dioxygenase |
| 1.13.11.24 | 0.0 | 1.0 | 0.0 | 0.0 | 0.3 | 0.5 | 2.0 | 2.0 | 0.0 | 0.0 | 3.0 | 1.4 | 1.3 | 5.6 | Quercetin 2,3-dioxygenase |
| 1.14.12.1 | 2.0 | 0.0 | 0.0 | 0.0 | 0.5 | 1.0 | 3.0 | 0.0 | 1.0 | 1.0 | 3.0 | 1.6 | 1.3 | 3.2 | Anthranilate 1,2-dioxygenase (deaminating, decarboxylating) |
| 1.14.13.1 | 1.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.5 | 5.0 | 1.0 | 4.0 | 1.0 | 6.0 | 3.4 | 2.3 | 13.6 | Salicylate 1-monooxygenase |
| 1.14.13.8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 | 1.0 | 4.0 | 1.0 | 3.0 | 2.0 | 1.4 | ∞ | Dimethyl aniline monooxygenase (N-oxide-forming) |
| 1.14.99.3 | 1.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.5 | 0.0 | 1.0 | 2.0 | 2.0 | 2.0 | 1.4 | 0.9 | 5.6 | Heme oxygenase (decyclizing) |
| 1.17.7.1 | 0.0 | 1.0 | 0.0 | 0.0 | 0.3 | 0.5 | 1.0 | 2.0 | 1.0 | 0.0 | 1.0 | 1.0 | 0.7 | 4.0 | 4-Hydroxy-3-methylbut-2-en-1-yldiphosphate synthase |
| 1.18.1.2 | 1.0 | 0.0 | 2.0 | 0.0 | 0.8 | 1.0 | 2.0 | 1.0 | 3.0 | 2.0 | 5.0 | 2.6 | 1.5 | 3.5 | Ferredoxin—NADP+ reductase |
| 1.18.1.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 | 1.0 | 1.0 | 1.0 | 6.0 | 2.0 | 2.2 | ∞ | Ferredoxin—NAD+ reductase |
| 2.7.11.1 | 1.0 | 0.0 | 1.0 | 2.0 | 1.0 | 0.8 | 2.0 | 12.0 | 12.0 | 4.0 | 29.0 | 11.8 | 10.6 | 11.8 | Nonspecific serine/threonine protein kinase |
| 2.8.3.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 | 9.0 | 4.0 | 1.0 | 17.0 | 6.4 | 6.8 | ∞ | Propionate CoA-transferase |
| 2.10.1.1 | 0.0 | 1.0 | 0.0 | 0.0 | 0.3 | 0.5 | 1.0 | 2.0 | 1.0 | 0.0 | 1.0 | 1.0 | 0.7 | 4.0 | Molybdopterin molybdotransferase |
| 3.1.6.1 | 0.0 | 1.0 | 0.0 | 1.0 | 0.5 | 0.6 | 2.0 | 5.0 | 0.0 | 1.0 | 7.0 | 3.0 | 2.9 | 6.0 | Arylsulfatase |
| 3.3.2.1 | 1.0 | 0.0 | 1.0 | 0.0 | 0.5 | 0.6 | 1.0 | 4.0 | 1.0 | 0.0 | 1.0 | 1.4 | 1.5 | 2.8 | Isochorismatase |
| 4.3.3.7 | 0.0 | 0.0 | 1.0 | 0.0 | 0.3 | 0.5 | 5.0 | 0.0 | 0.0 | 1.0 | 0.0 | 1.2 | 2.2 | 4.8 | 4-Hydroxy-tetrahydrodipicolinate synthase |
| 6.2.1.1 | 2.0 | 0.0 | 0.0 | 0.0 | 0.5 | 1.0 | 2.0 | 2.0 | 6.0 | 1.0 | 15.0 | 5.2 | 5.8 | 10.4 | Acetate-CoA ligase |
| 6.2.1.3 | 1.0 | 1.0 | 1.0 | 0.0 | 0.8 | 0.5 | 4.0 | 5.0 | 6.0 | 2.0 | 9.0 | 5.2 | 2.6 | 6.9 | Long-chain-fatty-acid-CoA ligase |
| 6.2.1.26 | 0.0 | 1.0 | 1.0 | 0.0 | 0.5 | 0.6 | 1.0 | 1.0 | 1.0 | 0.0 | 3.0 | 1.2 | 1.1 | 2.4 | o-Succinylbenzoate-CoA ligase |
Abbreviations: Ct, Corynebacterium terpenotabidum; EC no., Enzyme Commission number; Ef, Enterococcus faecalis; Gp, Gordonia polyisoprenivorans; Hs, Haemophilus somnus; Mm, Mycobacterium marinum; Ro, Rhodococcus opacus; Sm, Stenotrophomonas maltophilia; So, Streptococcus oralis; Ss, Staphylococcus saprophyticus.
Factor difference where AvGCr is for average of GC-rich and AvGCp is for average of GC-poor.
The shading regions in Table 7 is to improve the contrast between GC-rich (gray) and GC-poor (white) genomes.
Table 8.
Enzyme reactions of Table 7 classified by decreasing AvGCr/AvGCp.
| AvGCr/AvGCpa | S. no. | EC no. | Enzymatic function |
|---|---|---|---|
| 10 to 16 | 1 | 1.13.11.2 | Catechol 2,3-dioxygenase |
| 2 | 1.7.99.4 | Nitrate reductase | |
| 3 | 1.14.13.1 | Salicylate 1-monooxygenase | |
| 4 | 2.7.11.1 | Nonspecific serine/threonine protein kinase | |
| 5 | 6.2.1.1 | Acetate—CoA ligase | |
| 5 to <10 | 1 | 1.2.1.27 | Methylmalonate-semialdehyde dehydrogenase |
| 2 | 1.2.1.8 | Betaine-aldehyde dehydrogenase | |
| 3 | 6.2.1.3 | Long-chain-fatty-acid-CoA ligase | |
| 4 | 3.1.6.1 | Arylsulfatase | |
| 5 | 1.1.99.1 | Choline dehydrogenase | |
| 6 | 1.2.1.3 | Aldehyde dehydrogenases | |
| 7 | 1.13.11.24 | Quercetin 2,3-dioxygenase | |
| 8 | 1.14.99.3 | Heme oxygenase—biliverdin-producing | |
| 9 | 1.4.1.13 | Glutamate synthase—NADPH | |
| 10 | 1.4.1.1 | Alanine dehydrogenase | |
| 4 to <5 | 1 | 4.3.3.7 | 4-Hydroxy-tetrahydrodipicolinate synthase |
| 2 | 1.2.1.2 | Formate dehydrogenase | |
| 3 | 1.6.1.2 | NAD(P)+ transhydrogenase—Re/Si-specific | |
| 4 | 1.3.99.1 | Succinate dehydrogenase | |
| 5 | 1.4.4.2 | Glycine dehydrogenase—aminomethyl-transferring | |
| 6 | 1.17.7.1 | Cytidine diphosphate-4-dehydro-6-deoxyglucose reductase | |
| 2 to <4 | 1 | 2.10.1.1 | Molybdopterin molybdotransferase |
| 2 | 1.2.1.7 | Benzaldehyde dehydrogenase | |
| 3 | 1.4.1.2 | Glutamate dehydrogenase | |
| 4 | 1.18.1.2 | Ferredoxin—NADP+ reductase | |
| 5 | 1.2.1.38 | N-acetyl-g-glutamyl-phosphate reductase | |
| 6 | 3.3.2.1 | Isochorismatase | |
| 7 | 6.2.1.26 | o-Succinylbenzoate—CoA ligase | |
| 8 | 1.2.1.10 | Acetaldehyde dehydrogenase | |
| 9 | 1.2.1.70 | Glutamyl-tRNA reductase | |
| <2 | 1 | 1.4.3.1 | d-Aspartate oxidase |
| 2 | 1.5.3.1 | Sarcosine oxidase | |
| 3 | 1.14.13.8 | Flavin-containing monooxygenase | |
| 4 | 1.18.1.3 | Ferredoxin—NAD+ reductase | |
| 5 | 2.8.3.1 | Propionate CoA-transferase |
Abbreviation: EC no., Enzyme Commission number.
Factor difference where AvGCr is for average of GC-rich and AvGCp is for average of GC-poor.
In conclusion, we can say from the divisions in Table 8 that oxygenases (incorporation of oxygen in organic substrates) and CoA ligases (a central function in energy storage) make up the main significant differences, in comparison with the more basic metabolic functions, between the GC-poor and GC-rich bacteria. In addition, this difference emphasizes the existence of a larger metabolic variety of enzymatic systems in GC-rich bacteria than in GC-poor ones in the DTT. Also, the relative frequency of the enzymes ranked AvGCr/AvGCp < 2 is low in GC-rich bacteria, but because they are absent in GC-poor bacteria, they are probably of significance too.
The shotgun sequencing of bacteria from DTT grown in LB medium produced a total number of 723 543 readings whose size followed a bimodal distribution with the significant mode at 350 bp (Figure 1A). The average size significantly increased after contig assembling as can be seen in Figure 1B; however, most of the ORFs remained below 100 bp (Figure 1C). Filtering of cORFs with UFM resulted in a final sample of 35 105 cORFs mostly in the range of 100 to 300 bp (Figure 1D), which is in the acceptable limit to perform homology comparison.
Figure 1.
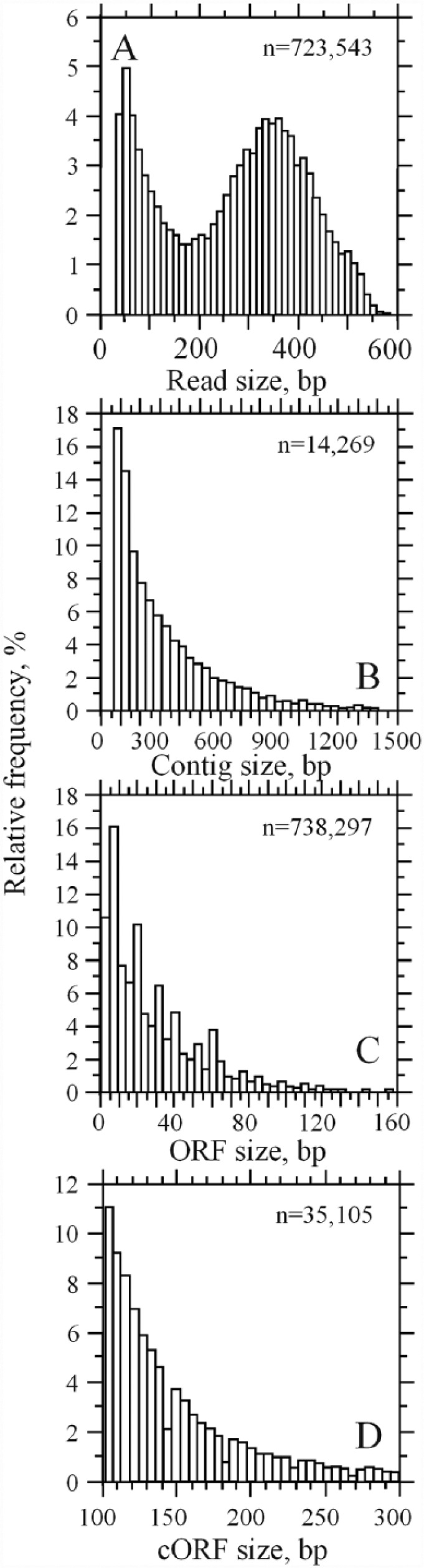
Relative frequency of shotgun sequences associated with bacteria found in the digestive tract of triatomines. (A) Size of reads obtained by 454 Titanium technology, (B) contig size after successive read assembling with Velvet and CAP3, (C) size of ORFs extracted from read contigs, and (D) size of coding ORFs after UFM filtering. cORFs indicate coding open reading frames; ORFs, open reading frames.
From the shotgun sequence samples, 2233 significant homologies (best hit) were found among the putative 35 105 cORFs with the representative species of the genera of GC-rich bacteria reported by Gumiel et al.8 Among the 2233 sequences, 425 (19.0%) had significant homologies with KEGG for the EC number list of Table 6. In contrast, 19 715 significant homologies (best hit) were found among the putative 35 105 cORFs with the representative species of the genera of GC-poor bacteria reported by Gumiel et al.8 Among the 19 715 sequences, 1424 (7.2%) had significant homologies with KEGG for the EC number list of Table 6. Because the sample sizes are very different, one must be concerned with a statistical consistency of the factor ~2.7 (close to the theoretical value of 3.3 found with the model) of overrepresented enzymes (Tables 6 and 7) observed in GC-rich compared with GC-poor bacteria. The Z test applied to the comparison of 2 proportions allows the formal conclusion that the null hypothesis of proportion equality must be rejected because Zobs (19) > Zth (1.96). Thus, despite a bias introduced by LB fermentation is favorable for the growth of GC-poor bacteria, the conclusion from the shotgun DNA sequencing is that overrepresented enzymatic activities are more frequent, in relative terms, in a medium fermented by GC-rich than in a medium fermented by GC-poor bacteria, as suggested by the model analysis based on complete genome sequences available from National Center for Biotechnology Information (NCBI).
Discussion
A recent investigation compared the microbiota of DTT in the presence or absence of T cruzi.24 Globally, it showed a predominance of GC-rich bacterial species (without considering the intracellular endosymbiont Arsenophonus)25 as previously described7,8 except for Staphylococcus which predominated in some individuals of P megistus and T infestans. However, the results of Díaz et al24 may be equivocal because the V3-V4 hypervariable region of 16S rDNA produces a less accurate quantitative description than with the V1-V3 region used by Gumiel et al,8 particularly for Staphylococcus. Also, with the V3-V4 region, differences between expected and observed frequencies of 10 to 300 times were reported by Zheng et al26 for this latter genus, whereas the measure obtained using V1-V3 region was close to the expected value.
The GC level of a genome is an interesting variable to consider because it is robust in the sense that it is expected to be globally conserved at the level of the family rank.13 Thus, if one GC-poor bacterial species is present in one family, there is a major likelihood that another species of that family will also be GC-poor. The same reasoning also applies to GC-rich organisms. However, in special situations, such as in endosymbiosis where the selective constraints are not those normally encountered by the members of the family, the above tendency is violated because endosymbionts are generally GC-poor, independent of the family they belong to. The GC-poor trait of endosymbionts may be due to an evolutionary convergence induced by the peculiar constraints imposed by the intracellular environment although this is debateable.27 The fact that the luminal environment of DTT in which T cruzi thrives is very different compared with that of intestinal epithelial cells was sufficient to eliminate the endosymbionts, Arsenophonus, Wolbachia, Candidatus, and Rohrkolberia from the present analysis.
Predominant bacterial species are GC-rich,8 which raises the question of whether a cause and effect relationship exists between a bacterial species being GC-rich and its growth success28,29 in the DTT environment. In fact, the positive correlation between GC3 and genome size suggests that, in the DTT, bacterial species with GC-rich genomes have a potentially more complex metabolism than those with GC-poor genomes, which would be an advantage for the bacteria in this niche.
The most significant differences that we found between the bacterial groups were due to oxidoreductase enzymes that are much more numerous in GC-rich than GC-poor bacteria and seem to confer a metabolic advantage to GC-rich bacteria in an environment such as blood, in particular, nitrate reductases and oxygenases that are common in GC-rich bacteria.
In the enzymatic reaction involving nitrate reductases (EC 1.7.99.4, KEGG map 910),30 the electron transport system is similar to that of aerobic respiration.31,32 It can be complemented by vitamin K to generate the energy required to survive in anaerobic conditions.33
Oxygenases are enzymes that oxidize a substrate by the transference of gaseous oxygen. Dioxygenases transfer both oxygen atoms of O2 into the substrate,34 whereas monooxygenases, such as phenolases (cytochrome P450 oxidases), incorporate only one atom of molecular oxygen into a substrate, such as phenols, and the other atom is reduced to H2O.35 Oxygenases are usual in soil bacteria because oxygen reactivity plays important roles in the degradation of complex substrates. In particular, ring-cleaving dioxygenases catalyze key reactions in the aerobic microbial degradation of aromatic compounds. Many pathways converge to catecholic intermediates. An example of the degradadation of a complex substrate is catechol 2,3-dioxygenases (EC 1.13.11.2) that catalyzes the opening of the benzene ring (KEGG maps 361, 362, 622, 643) and converts catechol into semialdehyde (OHC-R-COOH).36,37 Ring-cleaving dioxygenases that are active toward ring compounds belong to the cupin superfamily. Cupin-type dioxygenases also involve quercetinases (flavonol 2,4-dioxygenases), which open up 2 C-C bonds of the heterocyclic ring of quercetin, a widespread plant flavonol.38 In GC-rich bacteria, several other enzymes involved in ring modification or heteroatom oxidation are also available such as (1) arylsulfatases (EC 3.1.6.1), (2) benzaldehyde dehydrogenases—NADP+ (EC 1.2.1.7),39 and (3) flavin-containing monooxygenases (EC 1.14.13.8), which can oxidize a wide array of heteroatoms, particularly soft nucleophiles, such as amines, sulfides, and phosphites from xeno-substrates, with no common structural features, to facilitate their excretion.40,41
In the DTT, oxygenases have been shown to allow the access to iron of bacteria that encode that enzymatic system via hemoglobin degradation with heme oxygenases—biliverdin-producing (EC 1.14.99.3).42 Heme oxygenase is an enzyme that catalyzes the degradation of heme and produces biliverdin, iron, and carbon monoxide.43–45 Biliverdin is subsequently converted to bilirubin by biliverdin reductases. Iron is an essential nutrient required for the survival of most bacteria.46 Bioavailability of iron in many environments such as soil or sea is limited by the very low solubility of the Fe3+ ion. Microbes release siderophores to scavenge iron from these mineral phases by formation of soluble Fe3+ complexes that can be taken up by active transport mechanisms. Many siderophores are nonribosomal peptides,47,48 although several are biosynthesized independently. Some pathogenic bacteria, such as S marcescens, can use heme and hemoproteins as iron sources, independently of siderophore production, by mechanisms involving outer membrane heme-binding proteins and heme transport systems.49,50 The iron-binding protein, transferrin, produces a marked increase in S marcescens hemolytic activity.51
The levels of extracellular iron available within a host are limited, with most of the free iron being complexed to high-affinity binding proteins such as transferrin. To circumvent this low iron availability, pathogens have developed sophisticated mechanisms to use the host’s iron-containing and heme-containing proteins. The mechanism by which gram-positive bacteria, such as Corynebacterium diphtheriae, acquire heme is similar to the heme transport with siderophore52 and involves iron-chelating molecules excreted in the bacterial environment. Once the heme has been transported across the outer membrane and is localized within the cytoplasm, it is degraded by heme oxygenase.53,54
In contrast to oxygenases, oxidases (that reduce molecular oxygen to hydrogen peroxide or to water) and dehydrogenases (by transferring hydrogen from one substance to another) are mainly, if not exclusively, involved in energy metabolism. Many of the hydrogenases predominating in GC-rich bacteria are involved in many different central pathways such as glycolysis, the tricarboxylic acid cycle and oxidative phosphorylation that are essential to cell success in their environment.
As shown by Unrean and Srienc,55 a “cell system has a natural tendency to evolve with time towards an asymptotic state with maximum rate of entropy production.” In addition, De Martino et al56 showed that “growth rate can be explained in terms of a trade-off between the higher fitness of fast-growing phenotypes and the higher entropy of slow-growing ones.” From the results of this article and those of Unrean and Srienc55 and De Martino et al,56 it can be deduced that the success of GC-rich compared with GC-poor bacteria in the DTT is due to their enhanced ability to metabolize chemically complex substrates. The higher entropy of their metabolic networks may at least result from the predominance of hydrogenase functions in central metabolic pathways such as those for amino acid and nucleotide metabolism. In parallel with these increases in enzymatic functions, a conserved set of CoA enzymes was also found to be predominant in GC-rich bacteria and involved in different pathways such as the synthesis of chemical bond between large molecules (EC 6.2.1.1),57 toxic compound degradation (EC 2.8.3.1, EC 1.2.1.10),58 and fatty acid (EC 6.2.1.3) and amino acid (EC 4.3.3.7)59 metabolism.
The higher metabolic activity found in GC-rich bacteria suggests that signaling proteins should also be significantly increased. Indeed, a large difference was found for the nonspecific serine/threonine protein kinases (EC 2.7.11.1), which belong to the family of transferases, specifically protein-serine/threonine kinases. These enzymes transfer phosphates to the oxygen atom of a serine or threonine side chain in proteins. This process is called phosphorylation and is known to regulate most of the cellular pathways, especially those involved in signal transduction.60
In agreement with De Martino et al,56 the size inversion of GC-rich and GC-poor bacterial population found in the shotgun sequencing analysis is not surprising because the rich LB medium is more favorable for fast-growing bacteria with small genomes and less enzymatic abilities (lower metabolic network entropy). During experiments in this study, to have sufficient DNA, it was necessary to amplify it for sequencing and bacterial culturing were necessary steps, but, of course, at a cost of a bias. The population bias favored GC-poor bacteria and demonstrates the importance of using culture-independent techniques for in situ microbiota investigation. In this respect, the strategy of describing the microbiota composition by 16S rDNA sequencing prior to any further metagenomic description is surely the best, provided that the complete genome sequences for the metagenomes investigated are available. Thus, complete genome sequences allow the construction of a model suitable to determine what can be reasonably expected from the present experiments. Despite its bias, the shotgun analysis undertaken shows that the inferences proposed through the present model are still relevant. Therefore, the species believed to be representative of their respective genera are indeed representative in the context of this work because the proportion of predominant enzymes in the experiments is similar to that of the model.
Shotgun sequencing of microbiota is expensive, and the large amount of data provided can be difficult to analyze, especially when a eukaryote vector and blood meal source are involved as most of the sequences come from the host gut and not from the rare microbiota it contains.61 For instance, the genome of Rhodnius prolixus RproC1 was predicted to be about 733 Mb, whereas the average size of each bacterial genome sequence of its digestive tract is only about 4 to 5 Mb.62 Another limitation of shotgun sequencing is that the information it provides on the composition of a microbiota depends on a reference set of microbial genomes which is still only small, typically in the range of few thousand genomes.63 In contrast, large numbers of 16S rDNA gene sequences are available for comparative analyses. For example, the RDP Release 11.5 of September 30, 2016 consisted of 3 356 809 aligned and annotated 16S rDNA sequences (http://rdp.cme.msu.edu/).
The 1550 bp of the 16S gene consist of 8 highly conserved regions (U1-U8) and 9 variable regions across the bacterial domain.64 Identifying the organisms populating a microbial community and their relative abundances is the typical primary objective of investigations based on 16S rDNA amplicon characterization. A similarity comparison of 16S gene sequences is usually used as the gold standard for taxonomic identification at least at the genus level.65 The characterization of 16S amplicons by DGGE has been a useful technique for rapid assessment of the composition of DTT microbiota and is particularly suitable for a first-pass comparison of multiple samples.61 However, sequencing the 16S gene is currently the most common approach used in microbial classification.66 The application of next-generation sequencing to microbial ecology has shown that the diversity in microbial populations is significantly higher than previously estimated by traditional culture-based and conventional molecular methods.67 New technologies of DNA microarray (PhyloChip, Second Genome Inc, South San Francisco, USA) are now supporting microbiota investigations by 16S rDNA classification and offer the benefit of simultaneous detection of thousands of genes in a single shot.68,69 Core genes enriched for housekeeping functions are also used to enrich classification based on 16S rDNA and to improve the resolution of microbial community structure.70
Conclusions
The qualitative and quantitative description of a microbiota, as adapted from Gumiel et al,8 is more precise than a blind metagenomic analysis by DNA shotgun as long as complete genome sequences exist for the bacterial genera diagnosed by 16S rDNA. This is precisely the case in the present investigation as most of the genomes in the list of bacterial species identified by 16S rDNA sequencing by Gumiel et al8 had companion species effectively sequenced that can be downloaded from the NCBI server. The most striking differences in overrepresented enzymatic functions that are found in GC-rich bacteria (prominent in the colonization of the triatomine digestive tract compared with GC-poor ones) are for the most part due to oxidoreductases. We conclude that this group of enzymatic functions allows GC-rich bacteria to outcompete GC-poor ones in an environment where the fermentation of a medium such as fresh blood may need some specific metabolic activities such as iron recycling and oxygen management. In such a context, GC-rich bacteria would have a comparative advantage in the colonization of their environment, thanks to their more complex enzymatic apparatus, however, at the cost of a larger genome that is slower to replicate. In consequence, invertebrate vectors are valuable systems in which to study the properties that may favor one particular microbial community as opposed to another.71
Supplementary Material
Acknowledgments
The authors thank Norman A Ratcliffe for reading and editing the manuscript.
Footnotes
Peer review:Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 878 words, excluding any confidential comments to the academic editor.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: N.C. received a grant (PAPES-V) from Fiocruz/CNPq (http://portal.fiocruz.br/pt-br; http://cnpq.br/) and M.G. a PhD scholarship from CAPES (http://www.capes.gov.br/). The publication fees were paid by INCT-IDN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: NC, MG, FFdM, CJdCM, and PA conceived and designed the experiments. NC analyzed the data and wrote the first draft of the manuscript. PA contributed to the writing of the manuscript. All authors reviewed and approved the final manuscript.
Disclosures and Ethics: As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
- 1. World Health Organization. WHO media centre: Chagas disease (American trypanosomiasis). http://www.who.int/mediacentre/factsheets/fs340/en/. Published 2014. Accessed January 9, 2015.
- 2. Moraes CB, Giardini MA, Kim H, et al. Nitroheterocyclic compounds are more efficacious than CYP51 inhibitors against Trypanosoma cruzi: implications for Chagas disease drug discovery and development. Sci Rep. 2014;4:4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quijano-Hernandez I, Dumonteil E. Advances and challenges towards a vaccine against Chagas disease. Hum Vaccin. 2011;7:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Azambuja P, Feder D, Garcia ES. Isolation of Serratia marcescens in the midgut of Rhodnius prolixus: impact on the establishment of the parasite Trypanosoma cruzi in the vector. Exp Parasitol. 2004;107:89–96. [DOI] [PubMed] [Google Scholar]
- 5. Castro D, Morales C, Garcia E, Azambuja P. Inhibitory effects of d-mannose on trypanosomatid lysis induced by Serratia marcescens. Exp Parasitol. 2007;115:200–204. [DOI] [PubMed] [Google Scholar]
- 6. Garcia ES, Genta F, Azambuja P, Schaub GA. Interactions between intestinal compounds of triatomines and Trypanosoma cruzi. Trends Parasitol. 2010;26:499–505. [DOI] [PubMed] [Google Scholar]
- 7. da Mota FF, Marinho LP, Moreira CJC, et al. Cultivation-independent methods reveal differences among bacterial gut microbiota in triatomine vectors of Chagas disease. PLoS Negl Trop Dis. 2012;6:e1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gumiel M, da Mota FF, Rizzo VS, et al. Characterization of the microbiota in the guts of Triatoma brasiliensis and Triatoma pseudomaculata infected by Trypanosoma cruzi in natural conditions using culture independent methods. Parasite Vector. 2015;8:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dennison NJ, Jupatanakul N, Dimopoulos G. The mosquito microbiota influences vector competence for human pathogens. Curr Opin Insect Sci. 2014;3:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beard CB, Cordon-Rosales C, Durvasula RV. Bacterial symbionts of the triatominae and their potential use in control of Chagas disease transmission. Annu Rev Entomol. 2002;47:123–141. [DOI] [PubMed] [Google Scholar]
- 11. Durvasula RV, Sundaram RK, Kirsch P, et al. Genetic transformation of a corynebacterial symbiont from the Chagas disease vector Triatoma infestans. Exp Parasitol. 2008;119:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bentley SD, Parkhill J. Comparative genomic structure of prokaryotes. Annu Rev Genet. 2004;38:771–792. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi M, Kryukov K, Saitou N. Estimation of bacterial species phylogeny through oligonucleotide frequency distances. Genomics. 2009;93:525–533. [DOI] [PubMed] [Google Scholar]
- 14. Bohlin J, Snipen L, Hardy SP, et al. Analysis of intra-genomic GC content homogeneity within prokaryotes. BMC Genomics. 2010;11:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lightfield J, Fram NR, Ely B. Across bacterial phyla, distantly-related genomes with similar genomic GC content have similar patterns of amino acid usage. PLoS One. 2011;6:e17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu H, Zhang Z, Hu S, Yu J. On the molecular mechanism of GC content variation among eubacterial genomes. Biol Direct. 2012;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dias JCP, Ramos AN, Jr, Gontijo ED, et al. II Consenso Brasileiro em doença de Chagas, 2015. Epidemiol Serv Saúde. 2016;25:7–86. [DOI] [PubMed] [Google Scholar]
- 18. Cuba CAC, Alvarenga NJ, Barretto AC, Marsden PD, Gama MP. Dipetalogaster maximus (Hemiptera, Triatominae) for xenodiagnosis of patients with serologically detectable Trypanosoma cruzi infection. Trans R Soc Trop Med Hyg. 1979;73:524–527. [DOI] [PubMed] [Google Scholar]
- 19. Gouy M, Delmotte S. Remote access to ACNUC nucleotide and protein sequence databases at PBIL. Biochimie. 2008;90:555–562. [DOI] [PubMed] [Google Scholar]
- 20. Zerbino DR, Birney E. Velvet: algorithms for de novo short reads assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carels N, Frias D. Classifying coding DNA with nucleotide statistics. Bioinform Biol Insights. 2009;3:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carels N, Frias D. A statistical method without training step for the classification of coding frame in transcriptome sequences. Bioinform Biol Insights. 2013;7:35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Díaz S, Villavicencio B, Correia N, Costa J, Haag KL. Triatomine bugs, their microbiota and Trypanosoma cruzi: asymmetric responses of bacteria to an infected blood meal. Parasite Vector. 2016;9:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hypsa V, Dale D. In vitro culture and phylogenetic analysis of “Candidatus Arsenophonus triatominarum,” an intracellular bacterium from the triatomine bug, Triatoma infestans. Int J Syst Bacteriol. 1997;47:1140–1144. [DOI] [PubMed] [Google Scholar]
- 26. Zheng W, Tsompana M, Ruscitto A, et al. An accurate and efficient experimental approach for characterization of the complex oral microbiota. Microbiome. 2015;3:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hildebrand F, Meyer A, Eyre-Walker A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010;6:e1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ponce de, Leon M, de Miranda A, Alvarez-Valin F, Carels N. The purine bias of coding sequences is determined by physicochemical constraints on proteins. Bioinform Biol Insights. 2014;8:93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carels N, Ponce de, Leon M. An interpretation of the ancestral codon from Miller’s amino acids and nucleotide correlations in modern coding sequences. Bioinform Biol Insights. 2015;9:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tavares P, Pereira AS, Moura JJ, Moura I. Metalloenzymes of the denitrification pathway. J Inorg Biochem. 2006;100:2087–2100. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Strous M. Denitrification and aerobic respiration, hybrid electron transport chains and co-evolution. Biochim Biophys Acta. 2013;1827:136–144. [DOI] [PubMed] [Google Scholar]
- 32. Segers FH, Kešnerová L, Kosoy M, Engel P. Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood-borne pathogen. ISME J. 2017;11:1232–1244. doi: 10.1038/ismej.2016.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kwon O, Bhattacharyya DK, Meganathan R. Menaquinone (vitamin K2) biosynthesis: overexpression, purification, and properties of o-succinylbenzoyl-coenzyme A synthetase from Escherichia coli. J Bacteriol. 1996;178:6778–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayaishi O. An odyssey with oxygen. Biochem Bioph Res Co. 2005;338:2–6. [DOI] [PubMed] [Google Scholar]
- 35. Waterman MR. Professor Howard Mason and oxygen activation. Biochem Bioph Res Co. 2005;338:7–11. [DOI] [PubMed] [Google Scholar]
- 36. Junker F, Field JA, Bangerter F, et al. Dioxygenation and spontaneous deamination of 2-aminobenzene sulphonic acid in Alcaligenes sp. strain O-1 with subsequent meta ring cleavage and spontaneous desulphonation to 2-hydroxymuconic acid. Biochem J. 1994;300:429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kobayashi S, Hayaishi O. Anthranilic acid conversion to catechol (Pseudomonas). Methods Enzymol. 1970;17:505–510. [Google Scholar]
- 38. Fetzner S. Ring-cleaving dioxygenases with a cupin fold. Appl Environ Microb. 2012;78:2505–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pastrorova I, de Koster CG, Boom JJ. Analytical study of free and ester bound benzoic and cinnamic acids of gum benzoin resins by GC-MS and HPLC-frit FAB-MS. Phytochem Anal. 1997;8:63–73. [Google Scholar]
- 40. Ziegler D. An overview of the mechanism, substrate specificities, and structure of FMOs. Drug Metab Rev. 2002;34:503–511. [DOI] [PubMed] [Google Scholar]
- 41. Eswaramoorthy S, Bonanno JB, Burley SK, Swaminathan S. Mechanism of action of a flavin-containing monooxygenase. Proc Natl Acad Sci U S A. 2006;103:9832–9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilks A, Schmitt MP. Expression and characterization of a heme oxygenase (Hmu O) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle. J Biol Chem. 1998;273:837–841. [DOI] [PubMed] [Google Scholar]
- 43. Kikuchi G, Yoshida T, Noguchi M. Heme oxygenase and heme degradation. Biochem Bioph Res Co. 2005;338:558–567. [DOI] [PubMed] [Google Scholar]
- 44. Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. [DOI] [PubMed] [Google Scholar]
- 45. Evans JP, Niemevz F, Buldain G, de Montellano PO. Isoporphyrin intermediate in heme oxygenase catalysis. Oxidation of alpha-meso-phenylheme. J Biol Chem. 2008;283:19530–19539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol. 2004;58:611–647. [DOI] [PubMed] [Google Scholar]
- 47. Hider RC, Kong X. Chemistry and biology of siderophores. Nat Prod Rep. 2010;27:637–657. [DOI] [PubMed] [Google Scholar]
- 48. Miethke M, Marahiel M. Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev. 2007;71:413–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Létoffé S, Ghigo JM, Wandersman C. Iron acquisition from heme and hemoglobin by a Serratia marcescens extracellular protein. Proc Natl Acad Sci U S A. 1994;91:9876–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Létoffé S, Ghigo JM, Wandersman C. Secretion of the Serratia marcescens HasA protein by an ABC transporter. J Bacteriol. 1994;176:5372–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Poole K, Braun V. Iron regulation of Serratia marcescens hemolysin gene expression. Infect Immun. 1988;56:2967–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fukushima T, Allred BE, Sia AK, Nichiporuk R, Andersen UN, Raymond KN. Gram-positive siderophore-shuttle with iron-exchange from Fe-siderophore to apo-siderophore by Bacillus cereus YxeB. Proc Natl Acad Sci U S A. 2013;110:13821–13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsui T, Furukawa M, Unno M, Tomita T, Ikeda-Saito M. Roles of distal Asp in heme oxygenase from Corynebacterium diphtheriae, HmuO: a water-driven oxygen activation mechanism. J Biol Chem. 2005;280:2981–2989. [DOI] [PubMed] [Google Scholar]
- 54. Lai W, Chen H, Matsui T, et al. Enzymatic ring-opening mechanism of verdoheme by the heme oxygenase: a combined X-ray crystallography and QM/MM study. J Am Chem Soc. 2010;132:12960–12970. [DOI] [PubMed] [Google Scholar]
- 55. Unrean P, Srienc F. Metabolic networks evolve towards states of maximum entropy production. Metab Eng. 2011;13:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. De Martino D, Capuani F, De Martino A. Growth against entropy in bacterial metabolism: the phenotypic trade-off behind empirical growth rate distributions in E. coli. Phys Biol. 2016;13:036005. doi: 10.1088/1478-3975/13/3/036005. [DOI] [PubMed] [Google Scholar]
- 57. Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD(+) involvement in Bacillus subtilis. J Bacteriol. 2006;188:5460–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Manjasetty BA, Powlowski J, Vrielink A. Crystal structure of a bifunctional aldolase-dehydrogenase: sequestering a reactive and volatile intermediate. Proc Natl Acad Sci U S A. 2003;100:6992–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mirwaldt C, Korndörfer I, Huber R. The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2.5 A resolution. J Mol Biol. 1995;246:227–239. [DOI] [PubMed] [Google Scholar]
- 60. Wolanin PW, Thomason PA, Stock JB. Histidine protein kinases: key signal transducers outside the animal kingdom. Genome Biol. 2002;3:reviews3013. doi: 10.1186/gb-2002-3-10-reviews3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fraher MH, O’Toole PW, Quigley EM. Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev Gastroenterol Hepatol. 2012;9:312–322. [DOI] [PubMed] [Google Scholar]
- 62. Mesquita RD, Vionette-Amaral RJ, Lowenberger C, et al. Genome of Rhodnius prolixus, an insect vector of Chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc Natl Acad Sci U S A. 2015;112:14936–14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Markowitz VM, Chen IM, Palaniappan K, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jonasson J, Olofsson M, Monstein HJ. Classification, identification and subtyping of bacteria based on pyrosequencing and signature matching of 16S rDNA fragments. APMIS. 2002;110:263–272. [DOI] [PubMed] [Google Scholar]
- 65. Clarridge JE., III Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev. 2004;17:840–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Spiegelman D, Whissell G, Greer CW. A survey of the methods for the characterization of microbial consortia and communities. Can J Microbiol. 2005;51:355–386. [DOI] [PubMed] [Google Scholar]
- 67. Yun JH, Roh SW, Whon TW, et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microb. 2014;80:5254–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gentry TJ, Wickham GS, Schadt CW, He Z, Zhou J. Microarray applications in microbial ecology research. Microb Ecol. 2006;52:159–175. [DOI] [PubMed] [Google Scholar]
- 69. DeSantis TZ, Brodie EL, Moberg JP, Zubieta IX, Piceno YM, Andersen GL. High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb Ecol. 2007;53:371–383. [DOI] [PubMed] [Google Scholar]
- 70. Segata N, Huttenhower C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS ONE. 2011;6:e24704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Newton ILG, Sheehan KB, Lee FJ, Horton MA, Hicks RD. Invertebrate systems for hypothesis-driven microbiome research. Microbiome Sci Med. 2013;1:1–9. doi: 10.2478/micsm-2013-0001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.