

Abstract
We tested the role of Stat5 in dendritic cell (DC) and alveolar macrophage homeostasis in the lung using CD11c-cre mediated deletion (Cre+5f/f). We show that Stat5 is required for CD103+ dendritic cell (DC) and alveolar macrophage development. We found that fetal monocyte maturation into alveolar macrophages was impaired in Cre+5f/f mice, and we also confirmed impaired alveolar macrophage development of progenitor cells using mixed chimera experiments. In the absence of Stat5 signaling in alveolar macrophages, mice developed alveolar proteinosis with altered lipid homeostasis. In addition, loss of Stat5 in CD11c+ cells was associated with exaggerated LPS-induced inflammatory responses and vascular leak. In Cre+5f/f mice, there was loss of immune-dampening effects on epithelial cells, a key source of CCL2 that serves to recruit monocytes/macrophages. These findings demonstrate the critical importance of Stat5 signaling in maintaining lung homeostasis and underscore the importance of resident macrophages in moderating tissue damage and excess inflammation.
Introduction
Pulmonary alveolar proteinosis (PAP) is an uncommon pulmonary disorder characterized by intra-alveolar accumulation of surfactant proteins, impaired lipoprotein processing by macrophages, and increased susceptibility to pulmonary infections (1, 2). Studies in mice led to the discovery of the critical role of GM-CSF and more specifically, the macrophage in disease pathogenesis. Deletion of GM-CSF or its receptor, GM-CSFR, in mice recapitulates the human disease,(3–6) and GM-CSF receptor mutations and autoantibodies to GM-CSF are associated with the clinical disease (7–9).
GM-CSF drives expression of several transcription factors including PU-1 and PPAR-γ. PU-1 is critical for macrophage differentiation and function, and macrophage-specific deletion of PPAR-γ leads to PAP (10, 11). More recently, both GM-CSF and PPAR-γ have been shown to promote the differentiation of fetal monocytes to mature alveolar macrophages further clarifying alveolar macrophage ontogeny (11, 12).
GM-CSF can activate both STAT3 and STAT5 to promote DC and macrophage differentiation via pleiotrophic effects on downstream transcription factors, including IRF8, Spi-B, IRF7, PU-1, and PPAR-γ (13, 14). After cellular maturation, Stat5 can be activated by many additional cytokines, including TSLP, IL-2, and IL-5. We have previously shown using mice lacking Stat5 in CD11c+ cells, a significant role for Stat5 in TSLP-dependent DC activation and Th2 but not Th1 responses (15).
Here, we tested the consequence of CD11c-cre mediated deletion of Stat5 in DC and alveolar macrophage homeostasis in the lung. We show that Stat5 is required for CD103+ DC and alveolar macrophage development. In the absence of Stat5 signaling in alveolar macrophages, mice developed alveolar proteinosis, similar to that reported in the GM-CSF knock-out mouse, with altered lipid homeostasis that could not be rescued with PPAR-γ agonists. In addition, loss of Stat5 in CD11c+ cells was associated with exaggerated inflammatory responses and vascular leak to LPS-induced sterile lung inflammation suggesting the critical importance of Stat5 signaling in maintaining lung homeostasis.
Material and Methods
Mice
Cd11c-Cre on the BALB/c background were crossed with loxP-flanked Stat5 alleles (Stat5f/f) on a BALB/c background as described (15). Mice were maintained under specific pathogen free conditions. Animal procedures were approved by the Institutional Animal Care and Use Committees at the University of Washington and Benaroya Research Institute.
Genotyping and DNA deletion
Mice were genotyped for the presence of the Cre recombinase allele and floxed Stat5 alleles as previously described (16). The detection of the floxed Stat5 and deleted floxed Stat5 in DNA was performed as previously described to determine the deletion efficiency (17).
LPS Challenge
Mice under isoflurane anesthesia received E.coli LPS (1.5 μg/g mouse weight; Sigma-Aldrich, St. Louis, MO) via oropharyngeal aspiration. Mice were weighed and monitored daily until harvest days 2–5. For cell sorting experiments, mice were harvested at day 1.
Bone Marrow chimera
CD45.1/CD45.2 BALB/C host mice (8 weeks old) were irradiated with 900 rads followed by injection of 5 × 106 bone marrow cells from CD45.1 WT BALB/C or CD45.2 Cre+5f/f BALB/C donor mice. Additional mice received a 50:50 mix of WT and Cre+5f/f BALB/C donor cells. Mice were harvested 3 months post-bone marrow transplant for analysis.
Rosiglitazone
Neonate Cre+5f/f and Cre−5f/f mice were injected daily with rosiglitazone (10 mg/kg; Sigma-Aldrich, St. Louis, MO) in 5% DMSO in sterile PBS. Control mice received 5% DMSO in sterile PBS. Injections were given IP in a volume of 10 μl/g mouse weight from DAB2-DAB10, and lungs were harvested on DAB11 for analysis by flow cytometry. Additional Cre+5f/f and Cre−5f/f mice were fed standard chow diet compounded with Rosigiltazone Maleate (20 mg/kg) or standard chow diet alone starting at day 21 for up to 4 weeks.
Tissue Processing
Murine lungs were serially lavaged with 0.8, 0.7, and 0.7 ml PBS + 5 mM EDTA. Bronchoalveolar lavage (BAL) cells were pelleted by centrifugation for analysis. The vascular compartment was perfused with PBS after cutting the hepatic vein, and the lung was removed and stored on ice. Lung tissue was dissociated by chopping and digested using Liberase TM (1 mg/ml; Roche, Indianapolis, IN) and DNase I (1 mg/ml; Sigma-Aldrich) for 20 minutes at 37°C then filtered through a 70uM cell strainer (Fisher Scientific, Hanover Park, IL). Red blood cells were lysed with RBC Lysis Buffer (eBioscience, San Diego, CA) for 8 min at room temperature. Lavage cells and lung digests were washed with 1% FBS+ 2 mM EDTA in PBS and counted on a Cellometer Auto 200 (Nexelcom Bioscience, Lawrence, MA).
Cytospins and Oil Red O Stain
Cytospins were prepared by centrifuging BAL cells onto a slide at 750g for 5 min. Cells were then stained in Diff Quik (Siemens, Malvern, PA) following the manufacture’s protocol. For lipid staining, cells were fixed with 60% isopropanol and stained with Oil Red O (Sigma-Aldrich) for 15 min. Slides were then washed twice with 60% isopropanol for 1 minute, rinsed with water and mounted with Fluoromount-G (Southern Biotech, Birmingham, AL).
Flow Cytometry
All stains were performed in 96 well u-bottom plates in a volume of 100 μl. 1×105–1×106 cells were plated and blocked with Fc receptor block (BD, Biosciences, San Jose, CA) for 15 min at 4°C and subsequently incubated with antibodies for 1 h at 4°C. Cells were then washed and fixed in 5% formalin for 15 min at room temperature. Flow cytometry was acquired using a FACSCanto RUO system (BD Biosciences). Data analysis was performed using FlowJo (Treestar, Ashland OR). Antibodies included: Siglec F PE (BD), Ly6G APC, Ly6G FITC, CD11b PE-Cy7, CD11c PB, CD71 PerCP Cy5.5, CD206 BV605, ICAM1 FITC, CD45 APC-Cy7, MHCII BV510, CD103 APC (Biolegend).
In vitro LPS Stimulation
Murine lungs were serially lavaged with 0.8, 0.7, and 0.7 ml 5 mM EDTA in PBS, and cells were then spun down, suspended in growth medium (RPMI 1640; Gibco, ThermoFisher Scientific) containing 50 μg/ml Penicillin-Streptomycin (Corning, Corning, NY) and 10% FBS (GE Healthcare, Logan, UT) and plated in 96-well flat bottom tissue culture treated plates (Corning, Corning, NY). Alveolar macrophages were allowed to adhere for 1 h at 37°C and non-adherent cells were washed away with medium. Adherent cells were then stimulated with 10 ng/ml E.coli LPS (Sigma-Aldrich) for 4 h at 37°C, washed with warm PBS and then harvested for RNA purification.
Cell Sorting
Lung cells were blocked with Fc block (BD Biosciences) for 20 min 4°C and subsequently incubated with CD45-biotin (eBioscience). Cells were washed with FACS buffer, incubated with anti-biotin microbeads (Miltenyi Biotech, San Diego, CA) and run through magnetic columns per manufacture’s protocol. The CD45pos fraction was collected for RNA. The CD45neg flow through was stained with CD326-FITC, CD31-PE (eBioscience), and CD45-APC-Cy7 (BD Biosciences) antibodies. Cells were sorted using an Aria cell sorter (BD Biosciences).
For sorting of macrophage and DC populations, lung cells were blocked with Fc block and incubated with antibodies to CD45, MHCII, Siglec F, Ly6G, CD11b, CD11c, CD103. Cells were then washed twice with FACS buffer and sorted directly into 500μl RA1 Lysis Buffer (Clontech) on an Aria Cell Sorter (BD). Cell populations were defined by the following surface expression patterns. “CD103+ DCs” were defined as CD45+/Ly6G−/SigF−/CD11c+/CD103+, aAMs” as CD45+/Ly6G−/SigFint/CD11cint/CD11bhigh, and “mAMs” were defined as CD45+/Ly6G−/SigFhigh/CD11chigh/CD11blow, and “CD11b DCs” defined as CD45+/Ly6G−/CD11c+/CD103neg/CD11bhigh/MHCIIhigh.
qRT-PCR
RNA was collected and purified using a NucleoSpin RNA kit (Clontech, Mountain View, CA) and RNA quantity and quality was measured with a spectrophotometer (NanoDrop Inc., Wilmington, DE). Reverse transcription was performed using High Capacity Reverse Transcription (Applied Biosciences, Carlsbad, CA), following manufacturers instructions. Real-time PCR was performed using Sensimax II Hi-ROX kit (Bioline, Taunton, MA) according to manufacturers instructions and run on a 7900HT thermal cycler (Applied Biosystems). The threshold cycle (Ct) was obtained from duplicate samples and averaged. The ΔCt was the difference between the average Ct for the target gene and the housekeeping gene, Hprt. The ΔΔCt was the average ΔCt for a given sample point minus the average ΔCt of control samples. The data are expressed as relative quantification calculated as 2−ΔΔCt.
Protein Measurements
Cytokines and chemokines concentrations for CXCL1/KC, CXCL2/MIP-2α, CCL2/JE, CCL4/MIP-1β, CCL5/RANTES, IP-10, IL-12, and G-CSF were measured in BAL samples using a magnetic bead Luminex assay per manufacture instructions (R&D Systems, Minneapolis, MN) and analyzed on Bioplex 200 (Bio-Rad Laboratories, Raleigh, NC). BAL surfactant D levels were determined by ELISA (Mouse SP-D Quantikine ELISA Kit; R&D systems, Minneapolis, MN), following manufacturers instructions. IgM levels were determined using an IgM ELISA Quantification Kit (Bethyl Labs, Montgomery, TX) following the manufacturer’s protocol.
Electron Microscopy
Murine lungs were inflated with 0.8ml ½ Karnovsky’s fixative (2% Paraformaldehyde/2.5% Glutaraldehyde buffered in 0.2 M Cacodylate buffer) and immersed in fixative overnight. Lungs were then washed in 0.1M Cacodylate buffer and incubated in 2% aqueous OsO4/0.2 M Cacodylate buffer. Lungs were dehydrated through an ethanol gradient and incubated overnight in 1:1 Propyleneoxide/Epon 812 resin solution. The following day fresh resin was applied for 2 hours and then sections were embedded in fresh resin. 70-nm sections were cut and put on grids then stained with uranyl acetate for 2 h and lead citrate for 5 min. Microscopy and imaging was performed on a JEM-1400TEM microscope (Joel, Peabody, MA) with a Ultrascan 1000XP 2K × 2K camera (Gatan, Pleasanton, CA).
Statistics
Results are expressed as means ± SEM. Statistical significance was determined using Student’s t-test after passing normality assumption (Shapiro-Wilk normality test), or Mann Whitney test for non-normally distributed data using Prism software (GraphPad Software, Inc, La Jolla, CA). Differences were considered significant if the P-value was <0.05.
Results
Stat5 is required for dendritic cell homeostasis and alveolar macrophage development
To investigate the role of Stat5 in lung homeostasis, we first measured the effect of CD11c-cre mediated deletion of Stat5f/f in lungs of CD11c-Cre- × Stat5f/f (Cre−5f/f) and CD11c-Cre+ × Stat5f/f (Cre+5f/f) on pulmonary immune cells in naïve mice. We collected alveolar and lung compartment cells from Cre−5f/f and Cre+5f/f mice (ages 8–12 weeks) and assessed cell counts and myeloid populations by FACS. In the alveolar compartment, there was an overall reduction in leukocytes (CD45+ cells) in Cre+5f/f mice, which was due largely to a marked reduction in alveolar macrophages (CD11chighSigFhighCD11blow) (Fig 1A,B). However, in Cre+5f/f mice, there was an additional F4/80+ population characterized by CD11cintSigFintCD11bhigh expression (CD11bhigh cells) (Fig 1A–C). In Cre−5f/f mice, alveolar cells were homogeneous in size and morphology (Fig 1D). In contrast, alveolar cells from Cre+5f/f mice showed heterogeneity in size, granularity and morphology, containing a population of strikingly large and foamy macrophages (Fig 1D). The larger cells expressed markers of alveolar macrophages, whereas the smaller cells were CD11bhigh cells (not shown). In both populations of macrophages, efficiency of Stat5 deletion was > 90% (Supplemental Figure 1).
Figure 1. Loss of Stat5 in CD11c+ cells impairs alveolar macrophage development.
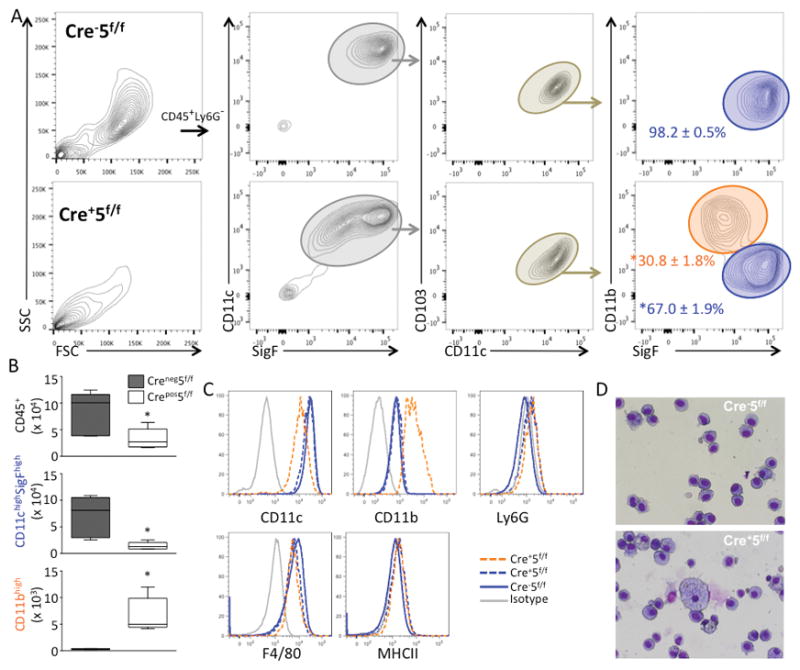
Naïve Cre−5f/f and Cre+5f/f murine lungs were lavaged and cell counts, morphology, and differential assessed. (A) FACS gating strategy of BAL cells from Cre−5f/f (top row) and Cre+5f/f (bottom row) mice. In Cre+5f/f mice, there was a significant shift in macrophage populations, with a reduction of alveolar macrophages (blue gate) and a new population of CD11bhigh cells (orange gate). Results presented as % ± SEM of cells within the CD11c+CD103− sub-gate. (B) Quantification of the total leukocytes (CD45+) and alveolar macrophages (CD11chighSigFhigh), and CD11cintCD11bhigh cells. (C) Histograms demonstrating differential expression of CD11c, CD11b, similar expression of F4/80, and negative staining for MHCII and Ly6G across the three populations (solid blue: Cre−5f/f alveolar macrophages; solid orange: Cre+5f/f alveolar macrophages; dashed orange: Cre+5f/f CD11bhigh cells). (D) Diff-Quick stained cytospins demonstrating heterogeneity of Cre+5f/f BAL cells (bottom) compared to Cre−5f/f cells. *p value <0.05 (n=5 mice/genotype; 3–5 experimental replicates).
In the Cre+5f/f lung digests, we found a similar reduction in tissue macrophages and an increase in CD11bhigh cells within the CD11c+SigF+ gate (Fig 2A,B). Since Stat5 has been reported to be important in DC development, we investigated the changes to CD103+ and CD11b+ DC populations in the naïve lung (Fig 2A). We found a significant reduction in CD103+ DCs (2.3±0.2 × 105 versus 0.4±0.1 × 105 cells in Cre− and Cre+ lungs; p value < 0.005) and a no change in CD11b+ DCs (2.5±0.3 × 105 versus 2.4±0.1 × 105 cells in Cre− and Cre+ lungs) suggesting Stat5 is required for CD103+ DC development but not that of other DC populations. In CD11b+ DCs and CD103+ DCs isolated from the lung from Cre+5f/f mice, efficiency of Stat5 deletion was 100% (Supplemental Figure 1).
Figure 2. Loss of Stat5 in CD11c+ cells reduces alveolar macrophage and CD103+ DC populations in the lung.
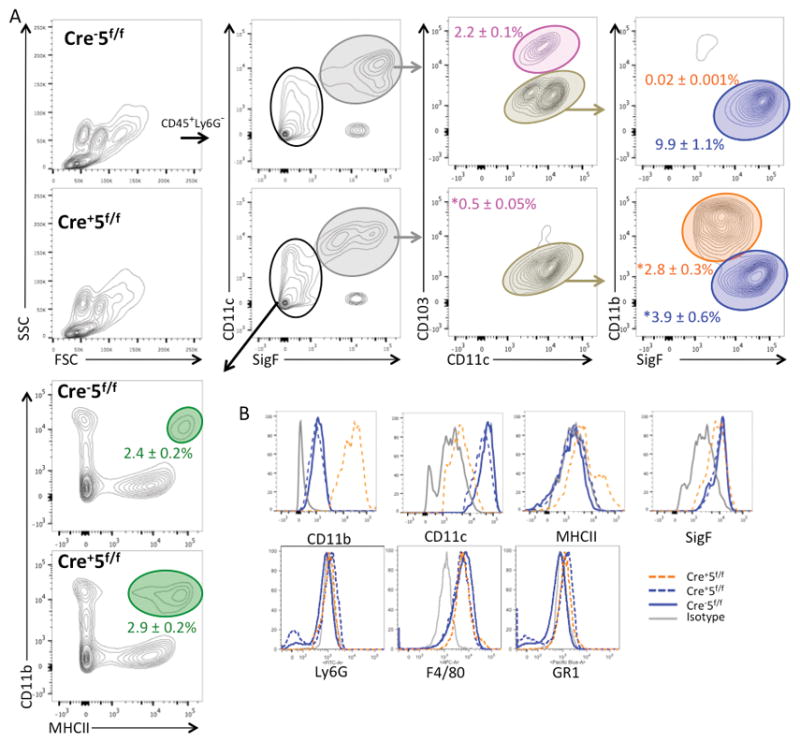
Naïve Cre−5f/f and Cre+5f/f murine lungs were mechanically and enzymatically digested for FACS analysis of leukocyte subsets. (A) FACS gating strategy of lung cells demonstrating a reduction of alveolar macrophages (blue gate) and CD103+ DCs (pink gate), and an increase in CD11bhigh cells (orange gate), and no change in CD11b+ DCs (green gate). Percentages shown represent % of CD45+ cells. *p value <0.05 (n=5 mice/genotype; 3–5 experimental replicates).
Stat5-deficiency leads to early failure of alveolar macrophage development
Since GM-CSF is required for maturation of alveolar macrophages from fetal monocytes, we assessed macrophage maturation in post-natal lungs (PND11), when fetal monocytes differentiate to alveolar macrophages. We found reduced alveolar macrophage maturation and an increase in arrested alveolar macrophages (aAM) in the Cre+5f/f lungs (Fig 3). At PND11, aAM represented 40.3 ± 7.6% of F4/80+CD11c+ cells in Cre−5f/f mice and 60.9 ± 1.2% in Cre+5f/f mice. In contrast, mature alveolar macrophages (mAM) represented an increased proportion in Cre−5f/f compared to Cre+5f/f mice (51.6 ± 7.9% vs. 30.6 ± 1.1%, respectively.) In 30 day old mice, there was complete maturation of fetal monocytes in Cre−5f/f lungs, whereas they remained in significant numbers in Cre+5f/f lungs (Fig 3B).
Figure 3. Stat5 is required for alveolar macrophage development from fetal monocytes.
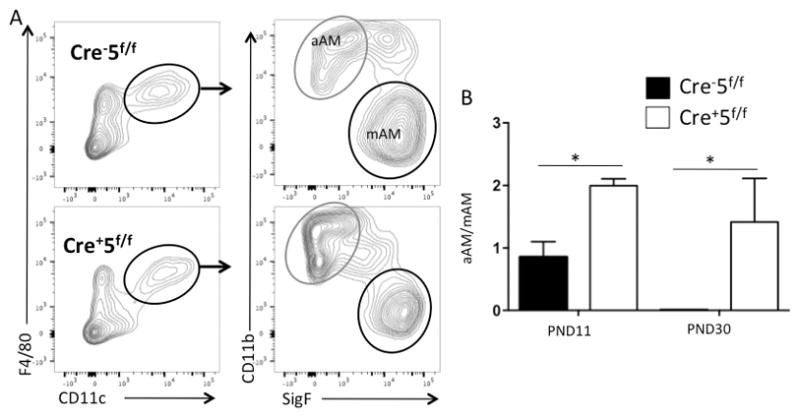
Cre−5f/f (top row; black bar) and Cre+5f/f (bottom row; white bar) murine lungs were harvested at PND11 and PND30 for analysis of alveolar macrophages maturation by FACS. (A) F4/80+/CD11c+ cells were identified and among those cells, mature alveolar macrophages (mAM) identified as CD11b−/Siglec F+ and arrested alveolar macrophages (aAM) identified as CD11b+/Siglec F−. (B) Expressed as a ratio of mAM/aAM, there was impairment of fetal monocyte maturation in Cre+5f/f mice. *p value <0.05 (n=3–5 mice/genotype).
Since PPAR-γ deficient CD11c+ cells develop a similar phenotype of impaired maturation of monocytes into alveolar macrophages,(11) we assessed if treatment with a PPAR-γ agonist could rescue alveolar macrophage development. To test this hypothesis, we administered the PPAR-γ agonist, rosiglitazone, from PND2-11 via intraperitoneal injection or from 2–4 weeks via diet supplementation. In both groups, rosiglitazone had no effect on alveolar macrophage development (not shown). These negative findings suggest that either PPAR-γ levels are too low for rescue by an agonist or there are other Stat5-dependent mechanisms required for macrophage maturation.
As another assessment of the ability of Stat5-deficient cells to reconstitute alveolar macrophages in the lung, we generated chimeras using CD45.1 Cre+5f/f, CD45.2 Cre−5f/f or 50:50 mixed donor marrow into CD45.1/CD45.2 wild type recipients. At 3 months, we assessed the BAL cell populations by FACS (Supplemental Fig 2). Mice that received 100% Cre+5f/f bone marrow failed to reconstitute alveolar macrophage with donor cells, with the majority of the alveolar macrophages representing remaining host cells (Supplemental Fig 2). In contrast, mice that received wildtype bone marrow had nearly entire reconstitution of their alveolar macrophages with donor cells. Recipients of the 50:50 mixed marrow demonstrated that a majority of alveolar macrophages were derived from WT cells (78%), with fewer than 1% of Cre+5f/f origin (Supplemental Fig 1B). In mice reconstituted with either 50% or 100% Cre+5f/f donor cells, there was a population of CD11c+ cells representing a population of immature macrophages (Supplemental Fig 2C).
Cre+5f/l alveolar macrophages have increased lipid accumulation and altered transcriptional profiles
It is well established that GM-CSF is required for phospholipid processing by alveolar macrophages and disruption of this pathway leads to pulmonary alveolar proteinosis.(18, 19) Since GM-CSF signals via Stat5, we investigated if loss of Stat5 in alveolar macrophages would cause pulmonary alveolar proteinosis. By 12 weeks of age, there was a significant increase in lipid-loaded macrophages and material in the alveolar fluid (Fig 4A-C), and surfactant protein D was markedly increased in the alveolar fluid from Cre+5f/f mice consistent with development of pulmonary alveolar proteinosis (Fig 4D).
Figure 4. Loss of Stat5 results in pulmonary alveolar proteinosis and abnormal lipid clearance and homeostasis.
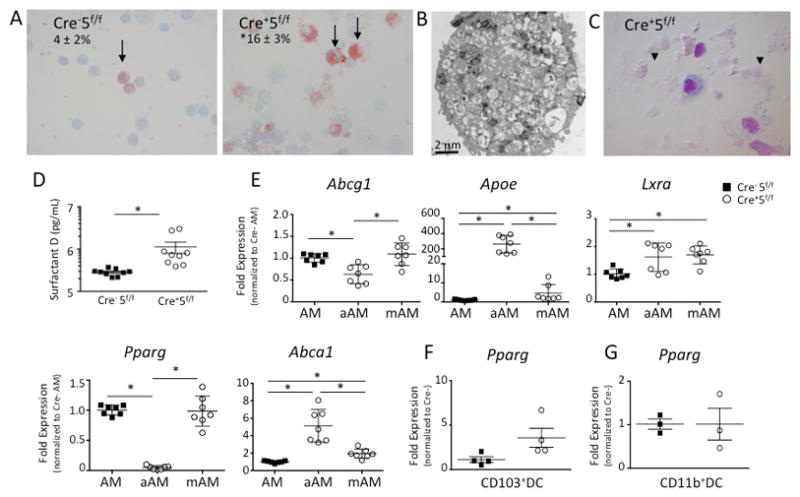
Naïve Cre−5f/f and Cre+5f/f murine lungs were lavaged for surfactant protein D measurements and assessment of cells by cytospins. (A) Cytospins of BAL cells were stained with Oil Red O revealing increased percentage of stained cells (arrows) from Cre+5f/f mice. (B) Electron microscopy of a Cre+5f/f alveolar macrophage demonstrating extensive lipid loaded vacuoles. (C) Representative Cytospin from Cre+5f/f mice revealing amorphous, acellular material (arrow heads). (D) Surfactant protein D levels were increased in the BAL fluid of Cre+5f/f mice. *p value <0.05 (n=5–9 mice/genotype; 2 experimental replicates). (E) Abcg1, Apoe, Lxra, Pparg, and Abca1 gene expression was assessed from sorted aAM and mAM from Cre+5f/f lungs and AM from Cre−5f/f lungs. (F,G) Pparg gene expression was determined from sorted CD103+DCs and CD11b+ DC from both genotypes. *p value <0.05 (n=3–4 mice/genotype; 2 experimental replicates).
Given these marked increases in lipid-loaded macrophages, we assessed the gene expression of several genes involved in lipid homeostasis in the macrophage populations from Cre−5f/f and Cre+5f/f mice. We sorted CD11chighSigFhighCD11blow (mAM) and CD11cintSigFintCD11bhigh (aAM) from Cre+5f/f lungs and CD11chighSigFhighCD11blow (AM) from Cre−5f/f lungs. Using qRT-PCR, we found that aAM from Cre+5f/f mice expressed less Abcg1 and Pparg but greater levels of Apoe and Abca1 compared to Cre+5f/f mAM and Cre−5f/f AM (Fig 4E). Cre+5f/f mAM gene expression more closely resembled that of Cre−5f/f AM, with similar levels of Pparg and Abcg1, but with increased Lxra and Apoe expression (Fig 4E). We also assessed if loss of Stat5 in CD103+DCs and CD11b+ DCs altered Pparg expression. Unlike that observed for aAM, the expression of Pparg from Cre+5f/f and Cre−5f/f CD103+ DCs and CD11b+ DCs was similar (Fig 4F,G) indicating that Stat5 effects on Pparg are cell-specific. Overall, the absence of Stat5 was associated with altered pathways involving lipid homeostasis, most notably in the aAM population.
Inflammatory responses in the lung are increased in Cre+5f/f mice
To determine the consequences of Stat5 deletion on inflammatory responses in the lung, we challenged mice with LPS delivered via oropharyngeal aspiration post-LPS.
At the peak of inflammation on day 2, Cre+5f/f mice lost more weight than Cre−5f/f mice (Fig 5A), and there was a greater increase in recruited PMNs and CD11b+ macrophages in Cre+5f/f compared to Cre−5f/f along with a persistent reduction in alveolar macrophages (Fig 5B). In addition, pro-inflammatory cytokines and chemokines were markedly higher in LPS-treated Cre+5f/f mice (Fig 5C).
Figure 5. Acute inflammatory responses are increased in Stat5-deficient mice.
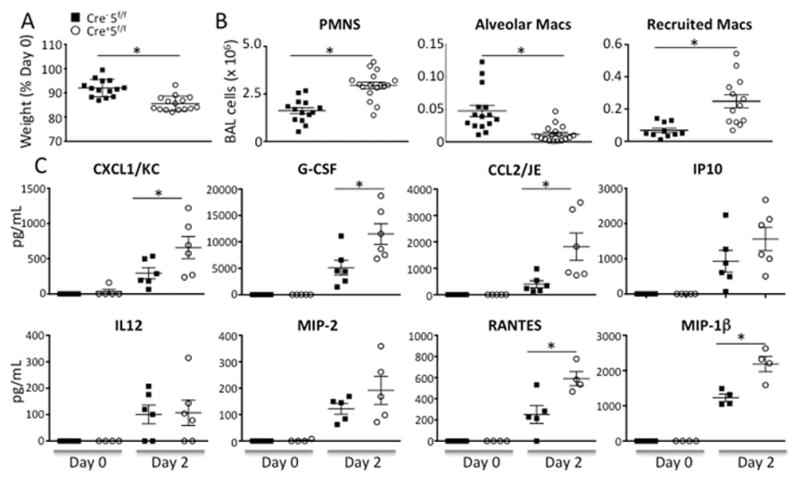
Cre−5f/f and Cre+5f/f mice received LPS via oropharygeal aspiration, and the inflammatory cells and cytokines in the alveolar compartment were quantified at day 2. (A) There was greater weight loss in LPS-treated Cre+5f/f. (B) Number of recruited neutrophils and CD11bhighCD11clowSigFlow macrophages (recruited Macs) were increased in LPS-treated Cre+5f/f mice compared to control. Resident alveolar macrophage remained reduced in Cre+5f/f mice. (C) Several pro-inflammatory cytokines and chemokines were increased in LPS-treated Cre+5f/f mice compared to LPS-treated control mice. *p value <0.05 (n=4–6 mice/genotype x 4 experimental replicates).
By day 5, Cre+5f/f mice started to recover their weights (not shown) and BAL neutrophilia was less (~ 50 fold) at d5 as compared to d2, suggesting resolution of lung injury. However, compared to LPS-treated Cre−5f/f mice, there were increased numbers of neutrophils and reduced numbers of resident alveolar macrophages in the alveolar compartment (Fig 6A, B). Most striking was a marked increase in vascular leak with elevated IgM that was detected at baseline (day 0) with markedly greater increases at days 2 and 5 post-LPS in Cre+5f/f mice compared to control Cre−5f/f mice (Fig 6C). Along with greater IgM levels, increased RBCs in BALF were also observed in LPS-treated Cre+5f/f mice (not shown). In the lung compartment, there was also increased numbers of neutrophils and reduced numbers of resident alveolar macrophages and CD103+ DCs in Cre+5f/f mice (Fig 7A,B). Together, these results suggest that perturbations in DC or alveolar macrophage function are critical for tempering the induction of lung inflammation and for regulating vascular integrity at both baseline and during acute lung injury.
Figure 6. Late inflammatory responses and lung injury are increased in Stat5-deficient mice.
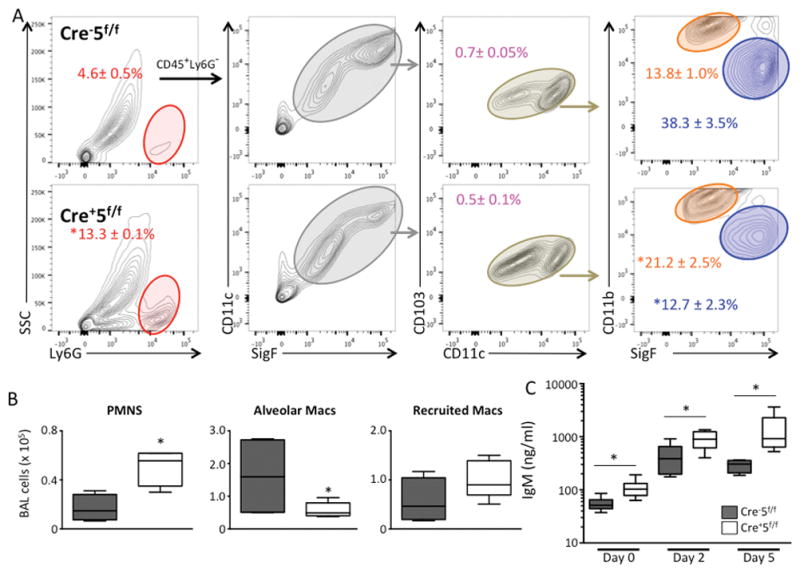
Cre−5f/f and Cre+5f/f mice received LPS via oropharygeal aspiration, and at day 5, the inflammatory cells in the alveolar compartment were quantified and analyzed by FACS. (A) Gating strategy to identify leukocyte subpopulations in BAL. (B) Quantification of BAL leukocytes demonstrating an increase in BAL neutrophils and a reduction in alveolar macrophages in Cre+5f/f mice compared to control Cre−5f/f mice. (C) BAL IgM concentrations from BALF from day 0 (uninjured), and days 2 and 5 post-LPS demonstrating a significant increase in Cre+5f/f mice compared to Cre−5f/f mice at all time points. *p value <0.05 (n=4–6 mice/group; 3 experimental replicates).
Figure 7. Recruitment of CD103+ DCs is diminished in Stat5-deficient mice.
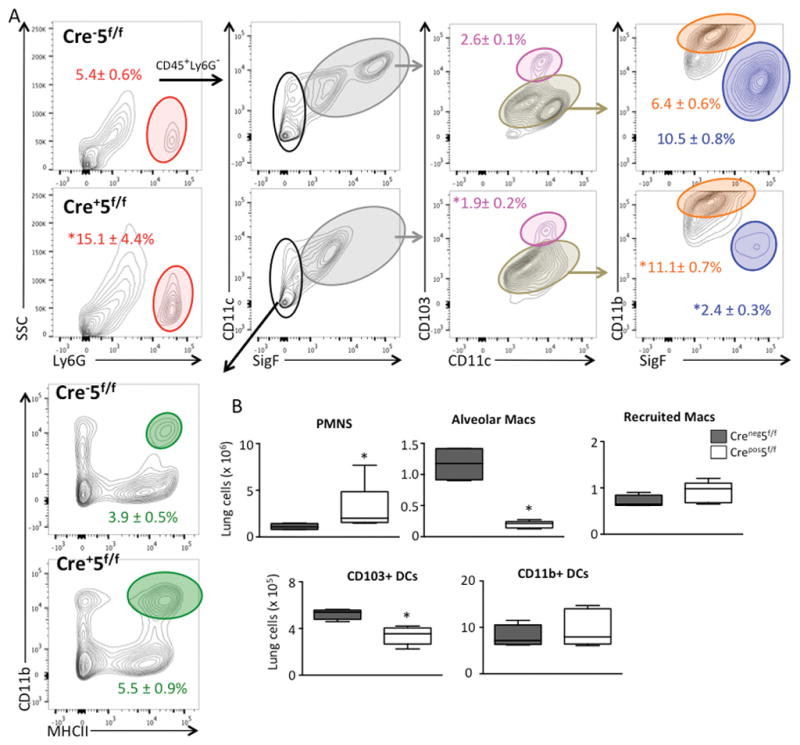
Cre−5f/f and Cre+5f/f mice received LPS via oropharygeal aspiration, and at day 5, the inflammatory cells in the lung compartment were quantified and analyzed by FACS. (A) Gating strategy to identify leukocyte subpopulations. (B) Quantification of lung leukocytes demonstrating an increase in neutrophils, intact recruitment of CD11b+ macrophages, a reduction in alveolar macrophages and CD103+ DCs in Cre+5f/f mice compared to control Cre−5f/f mice.
Increased Inflammatory responses in Cre+5f/f lung are localized to leukocytes and epithelial cells
Given the early inflammatory changes to LPS, we hypothesized that the source of increased inflammatory cytokines/chemokines (Fig 5C) was the macrophage. We removed the alveolar cells from mice and stimulated these cells with LPS to assess the inflammatory cytokine expression by qRT-PCR. In contrast to what we observed in vivo, the cells from Cre+5f/f mice were less responsive to LPS and generated less Cxcl1, Tnfa, and Il1b mRNA (Fig 8A), suggesting that other cells sources are responsible for the increased levels observed in vivo and may highlight an indirect anti-inflammatory role for either CD103+ DCs, CD11b+ DCs, or alveolar macrophages. To test this hypothesis, we sorted CD31+ endothelial cells, CD326+ epithelial cells, and CD45+ leukocytes from the lung at 24 h post-LPS treated Cre−5f/f and Cre+5f/f mice. We found that endothelial cells had similar expression of inflammatory cytokines/chemokines across both genotypes (Fig 8B). However, epithelial cells from Cre+5f/f mice expressed significantly more CCL2 and G-CSF (Fig 8C), and leukocytes from Cre+5f/f mice expressed significantly more CXCL1, G-CSF, and MIP-1β (Fig 8D). These results indicate that there was a loss of immune-dampening effects on epithelial cells, a key source of CCL2 that serves to recruit monocytes/macrophages. In addition, there was evidence of altered leukocyte activation or composition that contributes to enhanced expression of neutrophil chemokines.
Figure 8. Inflammatory cytokine expression in cellular populations reveals immune-dampening roles of Stat5-expressing cells.
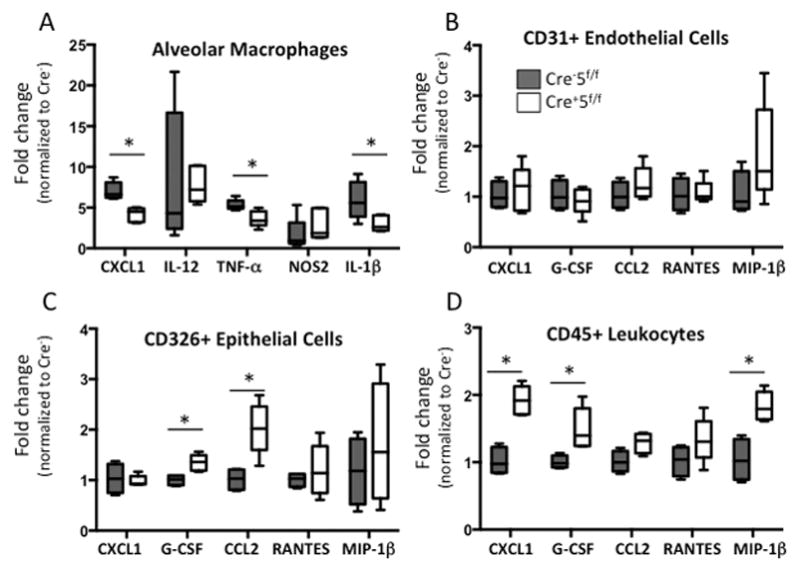
(A) Alveolar macrophages from uninjured Cre−5f/f and Cre+5f/f mice were isolated and stimulated with LPS in vitro. Gene expression for Cxcl1, Il12, Tnfa, Nos2, and Il1b was measured using qRT-PCR and shown as fold change over untreated Cre−5f/f cells. There was a significant blunting of Cxcl1, Tnfa, and Il1b expression in alveolar macrophages from Cre+5f/f mice. *p value <0.05 (n=4–6 mice/group; 2 experimental replicates). (B–D) CD31+ endothelial, CD326+ epithelial cells, and CD45+ leukocytes from lung digests were isolated by magnetic bead selection and FACS sorting 24 h post LPS treatment. Gene expression for Cxcl1, Gcsf, Ccl2, Rantes, Mip1b were determined and expressed as fold change relative to Cre−5f/f cells. (B) Endothelial cell inflammatory responses were similar between genotypes. (C) Epithelial cells from Cre+5f/f mice had significantly greater Gcsf and Ccl2 expression, and (D) CD45+ leukocytes from Cre+5f/f mice expressed higher levels of Cxcl1, Gcsf, and Mip1b. *p value <0.05 (n=5–6 mice/group).
Discussion
We tested the role of Stat5 in DC and alveolar macrophage homeostasis in the lung using CD11c-cre mediated deletion. Here, we show that Stat5 is required for alveolar macrophage development in the lung. We found that fetal monocyte maturation into alveolar macrophages was impaired in Cre+5f/f mice, and we also confirmed impaired alveolar macrophage development of progenitor cells using mixed chimera experiments. These findings are consistent with the known role of GM-CSF in alveolar macrophage development,(12) which is known to signal through Stat5,(14) and further confirms dependency on Stat5 as the key transcription factor involved in their development.
In addition to alveolar macrophage development, we also found a role for Stat5 in pulmonary CD103+ DC development. In hematopoietic Stat5−/− mice, tissue development of CD103+ DC has been shown to be Stat5-dependent,(13) and GM-CSF has been shown to control the development of dermal CD103+ DCs but not that of other DC populations (20). Interestingly, previous work in these mice revealed that Stat5 was dispensable in DC homeostasis in other tissues,(15) and these differences likely reflect the unique cytokine milieu in the lung. To our knowledge, deficiency of CD103+ DCs has not been reported in other murine models of pulmonary alveolar proteinosis (PAP) or in human PAP; however, CD103+ DC development has been shown to be dependent on GM-CSF (21).
In the absence of Stat5 in alveolar macrophages, mice developed pulmonary alveolar proteinosis as evidenced by increased surfactant protein in the alveolar compartment and presence of lipid-laden macrophages. We also observed altered expression of genes involved in lipid homeostasis, including reduced PPAR–γ expression in aAM. Reduced PPAR-γ has been reported in alveolar macrophages from patients with PAP,(22) and deletion of PPAR-γ in CD11c+ cells results in a similar phenotype to Cre+5f/f mice in terms of impaired alveolar macrophage maturation and development of pulmonary alveolar proteinosis (11). Hence, our findings implicate Stat5 as being an upstream regulator of PPAR-γ in progenitors of alveolar macrophages. Since PPAR–γ was reduced in aAM from Cre+5f/f mice, we tested whether a PPAR-γ agonist could rescue alveolar macrophage development. Despite two delivery strategies, we were unable to pharmacologically rescue alveolar macrophage development, suggesting that additional pathways may be involved.
Despite effective deletion of Stat5 in both arrested and mature alveolar macrophage populations in the Cre+5f/f mice, we cannot explain why some alveolar macrophages mature and others remain in an arrested state. However, Pparg levels were unaffected by Stat5 deletion in mAMs, suggesting that these cells may escape the early effects of Stat5 deletion on alveolar macrophage maturation.
We also tested the inflammatory responses in Cre+5f/f mice using a sterile model of inflammation. Loss of Stat5 in CD11c+ cells was associated with exaggerated inflammatory responses and vascular leak to LPS-induced acute lung injury. Although the overall inflammatory response was increased in Cre+5f/f mice compared to Cre−5f/f mice at days 2 and 5, there was a significant decrease in neutrophils over time in Cre+5f/f, suggesting that resolution was intact. These findings may reflect the fact that Cre+5f/f mice were still able to recruit macrophages from the circulation that participate in resolution of lung injury.
More impressive was the prominent vascular leak observed by both increased RBCs and IgM in the alveolar compartment. By scanning EM, we did not observe any defects in endothelial cell morphology in naïve mice (not shown), although there was a small increase in IgM at baseline suggesting some degree of endothelial cell injury. It is compelling to hypothesize that there is a population of macrophages in the lung that helps support endothelial cell health. In CD11b+DTR mice, ablation of macrophages in the neonate led to a marked vascular phenotype, suggesting there may exist populations that directly modulate endothelial cell biology (23).
The increased recruitment of neutrophils and macrophages in LPS-treated Cre+5f/f mice was likely due to augmented expression of CXCL1 and CCL2 chemokines. Although inflammatory cytokines and chemokines were markedly increased in Cre+5f/f mice, we found no evidence that Cre+5f/f macrophages were directly contributing, as their in vitro response to LPS was not enhanced. Similarly, we found no evidence that the pulmonary DCs were contributing, as sorted CD103+ and CD11b+ DCs from LPS-treated Cre+ and Cre− mice did not differ in their cytokine expression (not shown). These findings suggest either of two possibilities -there is loss of critical cell populations that have anti-inflammatory effects, or other sequelae, such as aberrant lipid accumulation, may lead to low-level injury and susceptibility to acute lung injury.
CD103 DCs have known roles in resolution of lung injury; (21, 24) however, findings of an early difference at 2 days post-LPS challenge does not reflect a resolution phenotype. To our knowledge, CD103+ DC’s role in modulating initial inflammatory responses in the lung have not been shown; however, in a colitis model, selective depletion of Clec9A+CD103+CD11b− DCs resulted in more severe disease with increased neutrophil recruitment and rectal bleeding (25). There was also decreased epithelial integrity and protein leak, and the authors found that Clec9A+CD103+CD11b− DCs increased lymphocyte expression of IFN-γ that then resulted in IFN-γ inducible gene expression in the epithelium.
However, dissecting a role for CD103+ DCs in Cre+5f/f is more challenging given the other perturbations in these mice, and rescue experiments by adoptive transfer of such small populations of cells remain a limitation. In addition, alveolar macrophages have been shown to dampen inflammatory responses of epithelial cells to LPS (26). Hence, loss of alveolar macrophages or aberrant macrophage function may lead to over-exuberant inflammatory responses by the epithelium. Lastly, others have shown that Pparg deletion in CD11c cells led to defects in mitochondrial H2O2 and increased NF-κβ activation in response to inhaled antigens (27). However, we found that Pparg expression was intact in CD103+ DCs, CD11b+ DCs, and mAMs from Cre+5f/f mice. Although there are similarities between mice that lack Pparg in CD11c+ cells (Cre+Ppargf/f) and Cre+5f/f mice, such as defects in alveolar macrophage development, Pparg expression does not appear to be Stat5-dependent in all cell types. Given these findings, it is not surprising that these mice would also differ in phenotypes. In fact, the Cre+5f/f mice have reduced Th2 responses in the lung, (15) whereas Cre+Ppargf/f mice have impaired tolerance and increased pro-inflammatory cytokines in CD11c+ cells (27, 28). Since we show that Pparg expression is unaltered in DCs and mature AMs in Cre+Ppargf/f, there may be other factors or cell sources contributing to augmented inflammation in Cre+5f/f mice.
To dissect out the cellular source of the increased pro-inflammatory cytokines/chemokines in LPS-treated Cre+5f/f mice, we compared gene expression of CXCL1/KC, G-CSF, CCL2/MCP-1, RANTES, and CCL4/MIP-1β from sorted leukocytes, epithelial, and endothelial cells. We found that loss of Stat5 resulted in greater epithelial expression of CCL2/MCP-1, consistent with our finding of increased macrophage recruitment in these mice. In contrast, the leukocytes were the source of greater CXCL1/KC, G-CSF, and CCL4/MIP-1β, and the endothelial cells did not contribute to cytokine differences between genotypes. These findings suggest one consequence of Stat5 deletion is loss of an immune-dampening effect on the epithelium. Since LPS-treated alveolar macrophages from Cre+5f/f mice did not express more pro-inflammatory cytokines compared to control alveolar macrophages, it is likely that the increased neutrophil chemokine expression by Cre+5f/f pulmonary leukocytes may reflect changes in leukocyte recruitment and/or activation.
Overall, these findings implicate the importance of Stat5 on alveolar macrophage and pulmonary CD103+DC development. In the absence of Stat5, there was aberrant accumulation of surfactant protein in the alveolar compartment, and alveolar macrophages accumulated excess lipids with marked alteration of pathways involving lipoproteins and cholesterol efflux. At baseline there was evidence of mild vascular injury, and post-LPS, there was enhanced inflammation and more prominent vascular injury, highlighting novel roles for Stat5 in lung homeostasis and acute injury responses. Our studies also support an immune-dampening role via epithelial-mediated expression of CCL2. Future studies will be needed to determine the relationship of myeloid cell populations in vascular integrity and immune-dampening properties.
Supplementary Material
Acknowledgments
This work was supported by R01 HL116514 (AMM), P30-ES-007033-19-6363 (AMM), Howard Hughes Medical Research Institute (AMM).
References
- 1.Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M, Epaud R, Crestani B. Pulmonary alveolar proteinosis. European respiratory review: an official journal of the European Respiratory Society. 2011;20:98–107. doi: 10.1183/09059180.00001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol. 2009;21:514–521. doi: 10.1016/j.coi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 5.Robb L, Drinkwater CC, Metcalf D, Li R, Kontgen F, Nicola NA, Begley CG. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc Natl Acad Sci U S A. 1995;92:9565–9569. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishinakamura R, Wiler R, Dirksen U, Morikawa Y, Arai K, Miyajima A, Burdach S, Murray R. The pulmonary alveolar proteinosis in granulocyte macrophage colony-stimulating factor/interleukins 3/5 beta c receptor-deficient mice is reversed by bone marrow transplantation. J Exp Med. 1996;183:2657–2662. doi: 10.1084/jem.183.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest. 1996;97:649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, Smolarek T, Dishop MK, Wert SE, Whitsett JA, Grabowski G, Carey BC, Stevens C, van der Loo JC, Trapnell BC. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 11.Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 2014;15:1026–1037. doi: 10.1038/ni.3005. [DOI] [PubMed] [Google Scholar]
- 12.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li HS, Yang CY, Nallaparaju KC, Zhang H, Liu YJ, Goldrath AW, Watowich SS. The signal transducers STAT5 and STAT3 control expression of Id2 and E2-2 during dendritic cell development. Blood. 2012;120:4363–4373. doi: 10.1182/blood-2012-07-441311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol. 2002;71:511–519. [PubMed] [Google Scholar]
- 15.Bell BD, Kitajima M, Larson RP, Stoklasek TA, Dang K, Sakamoto K, Wagner KU, Kaplan DH, Reizis B, Hennighausen L, Ziegler SF. The transcription factor STAT5 is critical in dendritic cells for the development of TH2 but not TH1 responses. Nat Immunol. 2013;14:364–371. doi: 10.1038/ni.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF regulates protein and lipid catabolism by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2001;280:L379–386. doi: 10.1152/ajplung.2001.280.3.L379. [DOI] [PubMed] [Google Scholar]
- 19.Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annual review of physiology. 2002;64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847. [DOI] [PubMed] [Google Scholar]
- 20.Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, Bogunovic M, Gautier EL, Miller J, Leboeuf M, Lu G, Aloman C, Brown BD, Pollard JW, Xiong H, Randolph GJ, Chipuk JE, Frenette PS, Merad M. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36:1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unkel B, Hoegner K, Clausen BE, Lewe-Schlosser P, Bodner J, Gattenloehner S, Janssen H, Seeger W, Lohmeyer J, Herold S. Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. J Clin Invest. 2012;122:3652–3664. doi: 10.1172/JCI62139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, Kavuru MS, Thomassen MJ. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol. 2003;29:677–682. doi: 10.1165/rcmb.2003-0148OC. [DOI] [PubMed] [Google Scholar]
- 23.Eldredge LC, Treuting PM, Manicone AM, Ziegler SF, Parks WC, McGuire JK. CD11b(+) Mononuclear Cells Mitigate Hyperoxia-Induced Lung Injury in Neonatal Mice. Am J Respir Cell Mol Biol. 2016;54:273–283. doi: 10.1165/rcmb.2014-0395OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manicone AM, Huizar I, McGuire JK. Matrilysin (Matrix Metalloproteinase-7) regulates anti-inflammatory and antifibrotic pulmonary dendritic cells that express CD103 (alpha(E)beta(7)-integrin) Am J Pathol. 2009;175:2319–2331. doi: 10.2353/ajpath.2009.090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muzaki AR, Tetlak P, Sheng J, Loh SC, Setiagani YA, Poidinger M, Zolezzi F, Karjalainen K, Ruedl C. Intestinal CD103(+)CD11b(−) dendritic cells restrain colitis via IFN-gamma-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 2016;9:336–351. doi: 10.1038/mi.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, Bhattacharya J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. 2014;506:503–506. doi: 10.1038/nature12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khare A, Raundhal M, Chakraborty K, Das S, Corey C, Kamga CK, Quesnelle K, St Croix C, Watkins SC, Morse C, Oriss TB, Huff R, Hannum R, Ray P, Shiva S, Ray A. Mitochondrial H2O2 in Lung Antigen-Presenting Cells Blocks NF-kappaB Activation to Prevent Unwarranted Immune Activation. Cell reports. 2016;15:1700–1714. doi: 10.1016/j.celrep.2016.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khare A, Chakraborty K, Raundhal M, Ray P, Ray A. Cutting Edge: Dual Function of PPARgamma in CD11c+ Cells Ensures Immune Tolerance in the Airways. J Immunol. 2015;195:431–435. doi: 10.4049/jimmunol.1500474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.