

Abstract
Implanted biomaterials often fail because they elicit a foreign body response (FBR) and concomitant fibrotic encapsulation. To design clinically relevant interference approaches, it is crucial to first examine the FBR mechanisms. Here, we report the development and validation of infrared-excited nonlinear microscopy to resolve the three-dimensional (3D) organization and fate of 3D-electrospun scaffolds implanted deep into the skin of mice, and the following step-wise FBR process. We observed that immigrating myeloid cells (predominantly macrophages of the M1 type) engaged and became immobilized along the scaffold/tissue interface, before forming multinucleated giant cells. Both macrophages and giant cells locally produced vascular endothelial growth factor (VEGF), which initiated and maintained an immature neovessel network, followed by formation of a dense collagen capsule 2–4 weeks post-implantation. Elimination of the macrophage/giant-cell compartment by clodronate and/or neutralization of VEGF by VEGF Trap significantly diminished giant-cell accumulation, neovascularization and fibrosis. Our findings identify macrophages and giant cells as incendiaries of the fibrotic encapsulation of engrafted biomaterials via VEGF release and neovascularization, and therefore as targets for therapy.
Subject terms: Intravital imaging, multiphoton microscopy, higher harmonic generation, foreign body response, foreign body giant cell, vascular endothelial growth factor, M1 macrophage, fibrosis
Tissue exposure to biomaterials triggers a foreign body response (FBR), a step-wise process consisting of inflammation, wound healing and, if not resolved, end-stage tissue fibrosis and scarring1–5. The FBR follows vascular damage and is initiated by absorption of plasma proteins onto the foreign body surface, including fibrinogen and complement2. This local damage response causes the recruitment and activation of monocytes/macrophages, their fusion to form a subset of multinucleated giant cells, and the generation of functional vessels followed by fibrotic encapsulation of the object4, 6. Not unlike in chronic wounds7–9, monocytes/macrophages represent the primary orchestrator of the FBR. Depending on local stimuli, macrophages can be polarized into pro-inflammatory (M1) or anti-inflammatory (M2) macrophages, as well as intermediate types 7, 8, 10. Based on their function profiles, including phagocytosis, adhesion and cytokine release11, the polarization to M1 and M2 macrophages defines the type and outcome of immune defense and tissue regeneration. M1 macrophages are highly phagocytic and produce inflammatory cytokines, which control an early innate damage response, while M2 polarization supports late-stage resolution of inflammation and tissue repair11–14. Both M1 and M2 type macrophages are implicated in organizing the inflammation and fibrous scar formation in response to implant materials9, 15–17, however their mechanistic roles in either enhancing or counteracting fibrotic outcome remain poorly understood18.
Clinically, the FBR reaches relevance when instigated by medical devices and/or implants, including sensors, pacemaker, prostheses (e.g. hip and knee prostheses, breast implants, heart valves), devices for release of bioactive compounds and, more recently, scaffolds used in tissue engineering and regenerative medicine3, 7. As detrimental processes, inflammation and fibrosis compromise implant integration and long-term functionality, impairing transplant bioactivity due to the fibrous barrier or biomaterial degradation2, 7. Therefore, intervention strategies to control the inflammatory phase of the FBR and to limit fibrosis are critical to improve implant integration and long-term function.
Different concepts have been explored to attenuate the FBR and fibrosis experimentally, including improving the biomimetic properties of the biomaterial, e.g. by geometrical and/or biochemical surface modifications19, 20. Likewise, molecular interference has been explored, including local immunosuppression by corticosteroid deposition3; dampening leukocyte and fibroblast activation by anti-transforming growth factor-β antibody or halofunginone2, 21; or stimulating vessel development to improve perfusion and performance of the bioactive implants by pro-angiogenic vascular endothelial growth factor (VEGF)2, 3, 22, 23. These approaches resulted in incomplete and often transient efficacy, with ongoing chronic inflammation, fibrosis, and even graft failure as outcomes2, 3. As potentially stringent approach, depletion of phagocytes by liposomal clodronate bisphosphonate or genetic ablation reduces the onset of inflammation during intraperitoneal FBR19,24, implicating monocytes/macrophages as early effectors and potential targets of ill-fated foreign body response.
Non-linear imaging of biomaterials
To dissect cause-consequence relations of the FBR we here develop an intravital window model and apply nonlinear multiphoton microscopy to monitor the fate of engrafted material in mice and in response to molecular intervention. As currently clinically investigated scaffold design and material platform technology, we applied calcium-coated polycaprolactone (mPCL-CaP) implants fabricated by melt-electrospinning with a fully interconnected honeycomb pore network (average pore size 100 μm, 90% porosity, Fig 1a)25–28. In our previous studies we observed that mPCL-CaP scaffolds combined with bone morphogenetic protein-7 (BMP-7) form bone after ectopic implantation in mice, whereas BMP-7–free scaffolds induce a physiological FBR29, 30. Hence, we hypothesized that these scaffolds would be a suitable porous implant to dissect the mechanism of FBR in 4D. To detect both mPCL-CaP fibers and deriving biological responses by multiphoton microscopy, infrared excitation and higher harmonic generation were used for deep tissue penetration, improved detection of red fluorophores and minimized phototoxicity (Fig S1)31, 32.
Fig. 1. Higher harmonic multiphoton microscopy of 3D printed mPCL-CaP fibers in vitro.

a. Macroscopic overview of melt electrospun PCL scaffold by bright-field microscopy. Bar, 300 μm. b. 3D reconstruction of mPCL-CaP scaffold by multiphoton-excited label-free SHG and THG detection (forward direction). A magnification is shown on the left panel. Box, inset. Arrowhead, calcium deposits. Bar, 200 μm. c. High resolution SHG/THG projection of single fiber in horizontal (xy) and orthogonal (xz) direction and signal intensity analysis. Dotted lines denote calcium coating. Box, detail represented as single channels and merge. d, Co-localization analysis of SHG and THG signal intensity (R2=0.06, p>0.001). e. Forward and backward direction detection of a PCL fiber (SHG and THG) shown as single channels and merge. Bar, 40 μm. f. SHG and THG signal intensity decay in a single fiber in z-direction, comparing forward versus backward detection of orthogonal projections of (e). Bar, 10 μm.
Scaffold fibers elicited strong second (SHG) and third harmonic generation (THG), using excitation/emission wavelengths of 1090/545 and 1180/393 nm, respectively (Fig 1b). SHG/THG z-scanning resolved both individual fiber geometry in three dimensions (Fig 1b) and an additional weak THG signal from the calcium phosphate coating (Fig 1b, arrowheads). Remarkably, SHG-/THG-positive substructures were present throughout the fiber core, with strong individual signal profile and insignificant co-localization (R2=0.06, p<0.001, Fig 1c, d). Thus, SHG/THG resolves different structural PCL subdomains after 3D printing, such as crystalline phase of the melt electrospun fibers33.
To extend the applicability of non-linear SHG/THG microscopy for other implant types used in clinical applications, we analyzed a series of further non-resorbable materials, including polyester, polymer polyglycolic lactic acid (PGLA) and polysulfone (PSU), and gelatin as resorbable material. All tested materials elicited a strong THG signal predominantly originating from the scaffold interface, independently from their micro-architecture (Fig S2). Likewise, SHG signal was emitted by most materials, except PSU (Fig S2). The results establish particularly THG as universal, interface detection strategy for robust analysis across implant type.
As prerequisite for intravital imaging in animals, we tested whether the SHG and THG signal induced by scaffold fibers was sufficient for reliable detection in backward direction, which is mandatory for thick tissue microscopy32 (Fig 1e, f). Signal intensity was 4-fold decreased when detected in backward as compared to the forward direction, and an additional 50% of signal decayed per 25 μm penetration into the fiber for each detection mode (Fig 1e, f). Despite this scatter-induced signal loss, 40 μm-thick fibers were completely detected in either configuration (Fig 1e). This establishes combined SHG/THG detection for non-destructive analysis of scaffold fiber geometry and substructures of semicrystaline mPCL polymers
Intravital longitudinal imaging of the FBR
To monitor both extracellular matrix and cellular kinetics during the FBR in vivo, a mPCL-CaP scaffold of 5×5×0.2 mm dimension was implanted in parallel to the deep dermis/subcutis interface in GFP-C57BL/6 or nude (nu/nu) mice, and analyzed through an optical imaging window over time (Fig 2a; S3a). Scaffolds integrated without macroscopic tissue trauma, inflammation or bleeding (Fig 2b), comparable to scaffolds in window-free mice25. Fourteen days after implantation, a histologically significant FBR response emerged, characterized by a cell-rich infiltrate and multinucleated giant cells decorating the surface of fibers, high density of neovessels and significant fibrosis between and surrounding the scaffold fibers (Fig 2c). To further delineate the composition of the inflammatory infiltrate by flow cytometry, which indeed may vary with material type and geometry19, 25, 34, 35, macrophages/giant cells represented the predominant of immune infiltrate cell type (47.2%), next to T-cells (20%), granulocytes (9.1%), and B-cells (4%)(Fig S4a). Giant cells and macrophages accumulated with a ratio of about 1:6, retained monocytic differentiation marker CD68 (Fig 2d) and M1 polarization (IRF-5) but lacked the M2 profile marker CD163 overtime (up to 7 weeks; Fig 2e, S4b). Thus, a macrophage-dominated pro-inflammatory M1-type response is induced by mPCL-CaP implants during early and late stages of the FBR, with no evidence for M2 polarization over time, in contrast to other material types9, 15, 16.
Fig. 2. Generation and characterization of an in vivo model to study FBR by longitudinal intravital imaging.

a. Schematic representation of the model. C57BL/6 GFP mice were onplanted with a dorsal skin-fold chamber (DSFC) on their back. A mPCL-CaP scaffold was implanted inside the DSFC within the subcutaneous tissue (x-y and x-z projection of the implantation site). b. Longitudinal macroscopic overview of the FBR after scaffold implantation within the DSFC. Dotted line, position of scaffold. c. Histology of the FBR in response to scaffold implantation within the DSFC at d14. White arrowhead, giant FBR cells; black arrowhead, blood vessels; asterisk, scaffold fiber. Boxes, zoomed details. Bar, 100 μm. d. CD68 expression detected by immunohistochemistry at day 14 post-implantation and representation of all mono- and multi-nucleated cells as percentage of total CD68-positive cells. The frequency of mono-nucleated cells stably attached to the fibers was negligible (4 fields/mouse; 3 mice). Image area, 500 × 700 μm (x 5 μm). e. IRF5 and CD163 expression detected by immunohistochemistry at day 14 post-implantation. Bar, 50 μm. f, g. Longitudinal intravital imaging of FBR at d4, 7 and 14 was performed for different areas of the same lesions chosen randomly. f: merged multi-parameter images representing THG (red); GFP-positive cells (cyan); 70KDa dextran (magenta); SHG (green). g: Single channel representations (top panels) and image-based quantifications of scaffold fibers (THG), GFP-positive infiltrate cells (white arrowhead), dextran-positive blood vessels (magenta arrowhead) and SHG, detecting PCL fibers (d4, 7; white asterisks) and fibrillar collagen (d7, 14; green asterisks). For image analysis, the quantification of the signal-positive area (% of total image) for each channel was obtained from 4–8 independent fields/implant from 4 mice. Bar, 50 μm. (*), p<0.05; (**), p<0.01; (***), p<0.001 by one-way Anova followed by Tukey’s HSD post hoc test.
When monitored longitudinally by intravital microscopy, the tissue surrounding the THG-positive mPCL-CaP scaffold became gradually infiltrated by GFP-positive cells (Fig 2f, g), followed by sprouting and perfused neovessels and deposition of initially loose and later bundled fibrillar collagen (SHG) throughout the initially SHG-free interstitial tissue (Fig 2f, g; S3b, c). SHG signal intensity in scaffold fibers was diminished in parallel to progressing collagen deposition, whereas THG intensity remained largely unperturbed (Fig 2g), establishing THG as most robust detection mode in vivo to monitor the positioning and integrity of the scaffold over time. Whereas the FBR kinetics in GFP-C57BL/6 and nude mice was similar, neovascularization was enhanced in nu/nu mice, consistent with angiogenic strength depending on the genetic background of mouse strains36, 37. Despite severe inflammatory tissue remodelling, mPCL-CaP fibers were neither resorbed nor infiltrated by cells (Fig S3d, e), confirming the documented stability of this material in vivo38–40.
To demonstrate in vivo applicability of THG imaging for an independent material, PSU implants were efficiently monitored, including early recruitment of GFP-positive cells, robust neovescaularization and collagen deposition (Fig S5), similar to the response to mPCL-CaP.
Kinetic and mapping of infiltrating cells
Unlike their composition, the dynamics of FBR infiltrate cells remain unknown. We therefore mapped infiltrate kinetics and the accumulation of foreign body giant cells along the scaffold surface and performed time-lapse recordings and single-cell tracking of infiltrating GFP-positive cells. Relative to the implant position, we identified two kinetic subpopulations which were recruited over time, including interstitially moving and fiber-attached resident cells (Fig 3a–c; Movies S1–3). A population of mononuclear interstitial cells retained mostly kinetic behaviours and moved along and between collagen fibrils, without directly contacting scaffold fibers (Fig 3a, d; speed up to 3 μm/min). This subset represents interstitial leukocytes also detected by histology (Fig 2c). Conversely, scaffold-attached cells were typically multi-nucleated, as detected by intravital injection of Hoechst dye (Fig 3b, arrowhead), with high positional stability despite vigorous cytoplasmic dynamics (Fig 3d; Movie S4). This identifies giant cells indeed as stably anchored resident cells, as predicted7, which nonetheless retain dynamic behaviours and slow dynamics locally exploring the material surface by ruffling lamellipodia (Fig 3b, d; average speed at day 14: 0.07 μm/min)4.
Fig. 3. Dynamics of infiltrate cells of the FBR monitored by intravital microscopy.

a, b. Dynamics of GFP-positive infiltrate cells at day 7 monitored by time-lapse intravital microscopy and analyzed by single-cell tracking. Sequential frames obtained at different time points from Movie 2 (see Supplementary information) used for single-cell tracking classified as interstitial (a) or scaffold-associated cells (b). White arrowhead, single-cell positioning over time. GFP-positive cells (green); SHG and Hoechst (red and as single channel, grayscale). Lower panels, speed representation as heatmaps from 10 representative cells. Bar, 20 μm. c. Number (mean±SD) of interstitial or mPLC-CaP fiber-associated infiltrate cells per imaging field over time. Area, 360 × 360 (x 40 μm). (*), p<0.05; (***), p<0.01; (n.s.), non significant, by Student t-test, unpaired, two tails. d. Frequency distribution of the average speed for interstitial and scaffold-associated cells over time. 3 mice/time point and 2 fields/mouse were analyzed. Sc-A, scaffold-associated cells; Interst, interstitial cells. Data show one representative image sequence per time point from 3 independent experiments.
Neoangiogenesis and fibrotic capsule development during FBR
Whether neovessels contribute to enhance or resolve the FBR remains unclear2, 3, 22, 23. Both immunodeficient and immunocompetent mice carrying mPCL-CaP material developed a dense neovascular network (Fig 2f, g; S3b, c), with irregular-shaped morphology, enhanced number of branch points and decreased inter-branch length, compared to the linear organization of normal dermal blood vessels (Fig 4a, b). These features are similar to those of tumor neovessels41, 42, where dysfunctional angiogenesis supports cancer lesion growth, progression and metabolic deregulation43. Neovessels preferentially oriented in parallel or perpendicular to the scaffold fibers, defined by angle distribution favoring 0 and 90 degrees (Fig 4c). This indicates that neovessels sense and undergo guidance by the pre-defined architecture of the scaffold but thereafter fail to normalize over time, similar to dysfunctional vessels in tumor-associated desmoplastic tissue43.
Fig. 4. Neovessel development and collagen deposition in scaffold-elicited FBR monitored by intravital microscopy.

a. Regular skin and scaffold-elicited vasculature. Bar, 100 μm. b. Quantification of neo-vessel organization, including: number on branching points, inter-branching distance, and total vessel length. Data (mean±SD) represent 3 fields/mouse from 3 mice. c. Frequency distribution of vessel-to-scaffold angle. Data represent 3 fields/mouse from 3 mice. d. Collagen staining by Masson’s Trichrome at end point (d14). Dotted line, scaffold; red boxes, details; asterisk, scaffold fiber Bar, 1 mm. e. Collagen-wrapped mPLC-CaP fibers at different imaging depth (SHG, green; THG, red). Bar, 100 μm. f. Frequency distribution of the angle of collagen fibrils relative to scaffold fiber orientation (3 fields/mouse, 3 mice). (**), p<0.01; (***), p<0.001 (unpaired two tailed Student t-test).
In parallel to neoangiogenesis, collagen deposition occurred in an incremental fashion (Fig 2g, f) histologically forming a fibrillar capsule surrounding and penetrating the scaffold architecture by day 14 and thereafter (Fig 4d). When reconstructed three-dimensionally, two subsets of SHG-positive collagen fibers and bundles were detected, including fibers wrapping around (60–90 degree orientation) or aligning in parallel (0–15 degrees) to scaffold fibers (Fig 4e, f). This indicates fibrous scarring as mechanically precise process leading to functionally distinct subsets of collagen bundles. In aggregate, giant cell development is accompanied by a desmoplasia-like process consisting of leukocyte influx, primordial vascularization, and progressive collagen deposition and encapsulation of the porous implant.
Therapeutic targeting of macrophages and neoangiogenesis
To test whether infiltrating macrophages and multinucleated giant cells are cause or rather consequence of neovascularization and late-stage fibrosis, we depleted the macrophage lineage of scaffold-bearing mice with clodronate liposomes prior to graft implantation. After internalization, clodronate induces apoptosis of phagocytes but no other cells44. Clodronate treatment abrogated the recruitment of infiltrate cells and giant cell development, as previously reported24, and further ablated scaffold-associated neo-vascularization and fibrosis (Fig 5a, b; S6a, upper panels). To directly test whether neovessel development and late-stage scarring were linked and both dependent on the presence of macrophages and giant cells, lesions containing the scaffold were stained for VEGF-A, the predominating proangiogenic cytokine in regenerating and cancerous tissue45 and an important cytokine produced by M1-type macrophages8, 9. VEGF-A was prominently expressed in mononucleated stromal cells as well as giant cells (Fig 5c, left panel, arrowheads) and both VEGF-positive cell subsets were absent after clodronate treatment, without compensatory up-regulation of VEGF-A in other tissue regions (Fig 5c, right panel) together with the absence of neoangiogenesis after clodronate treatment (Fig 5a). This identifies myeloid and giant cells as main source for VEGF-A and drivers of neo-vessel formation.
Fig. 5. Therapeutic targeting of the FBR by macrophage and VEGF depletion.

a. Histology (H&E staining) of FBR to PCL scaffolds 14d post-implantation in control mice and mice after clodronate liposome (Cl), VEGF Trap (VT) and VEGF Trap+clodronate liposome (Cl+VT) treatment. White arrowhead, giant FBR cell; black arrowhead, blood vessels. Inset, magnification of giant cell. Bar, 100 μm. b. Quantification of giant cell and vessel number represented in panel. Analysis was performed for multi-image representations from entire scaffolds from 3 mice/group. Image area, 500 × 700 μm (x 5 μm). c. VEGF-A expression in control- and clodronate liposome-treated mice, detected by immunohistochemistry and counted as percentage of VEGF-A positive cells of all mono- and multi-nucleated cells (4 fields/mouse; 3 mice). Image area, 500 × 700 μm (x 5 μm). Bar, 100 μm. d. Longitudinal intravital MPM analysis and 3D reconstruction of FBR in untreated and treated mice (d 14). Diagrams show analysis of signal area (mean±SD) at different time points. 3 fields/mouse; 3 mice; 2 experiments. Bar, 50 μm. (**), p<0.01; (***), p<0.01 by one-way Anova followed by Tukey’s HSD post hoc test.
We next aimed to clarify whether neo-vascularization is an independent mechanism enhancing or counteracting the severity of the FBR and scarring. To minimize neo-vessel establishment directly, without targeting giant cell, we administered VEGF Trap to prevent VEGF availability46, using a dosing scheme effective for anti-angiogenic therapy in patients47, 48. VEGF Trap abrogated vessel formation and further significantly diminished giant cell number and collagen deposition (Fig 5a, b; S3a, upper panels). Whereas VEGF Trap monotherapy was slightly less efficient than clodronate treatment, combining both agents resulted in near-complete ablation of giant cell and fibrosis development, albeit without further improving the already profound effect reached by clodronate treatment alone (Fig 5a, b).
When addressed longitudinally by intravital microscopy, clodronate-mediated cell depletion, effective by day 7, preceded ablation of neovascularization and fibrous encapsulation of scaffold fibers by d14 (Fig 5d, S6b). This failure to mount both neovessels and fibrosis after macrophage/giant cell targeting indicates myeloid cells as non-redundant drivers of FBR-associated tissue remodelling. VEGF Trap inhibited tissue infiltration by GFP-positive cells after, but not before, day 7 (Fig 5d, S6b), suggesting a two-step process with early-onset inflammation mediated by VEGF-independent pre-existing vessels followed by secondary exacerbation via VEGF-dependent neovessels.
Whereas cell accumulation near the implant was strongly diminished by clodronate and/or VEGF Trap (Fig S7a, b), the positional stability of residual scaffold-associated cells and the mobility distribution of interstitial cells remained unperturbed, compared to untreated lesions (Fig S7c–e). These results indicate that both clodronate and VEGF targeting act via limiting cell entry into the tissue, rather than mobility within the lesion.
To investigate the long-term efficacy of macrophage and neovessel depletion on the FBR and fibrotic encapsulation beyond the 2-week observation period achievable in the window system, window-free mice bearing scaffold were treated with clodronate liposomes and/or VEGF Trap and assessed by ex-vivo 3D SHG/THG microscopy and histology at day 28 (Fig 6a–c; S6a, lower panels). Suppression of the FBR, including diminished inflammation, vascularization and fibrosis, was sustained by anti-angiogenesis therapy and, with stronger effects, by clodronate and combined clodronate and VEGF Trap (Fig 6b, c).
Fig. 6. Long-term efficacy of macrophage and VEGF depletion treatment.

a. Long-term treatment schedule. b. Histological analysis (H&E; left) and quantification (right graphs) of end-stage FBR in untreated and treated mice at d28. White arrowhead, giant cell; solid arrowhead, blood vessels. Analysis was performed for multi-image representations from entire scaffolds from 3–5 mice/group. Bar, 100 μm. Image area, 500 × 700 μm (x 5 μm). c. Ex vivo MPM analysis and 3D reconstruction of scaffold 28 d post-implantation after different treatments and quantification of SHG surface area. Bar, 50 μm. d. Therapy withdrawal schedule. Mice were treated for 21 days and analyzed 35 or 49 days after scaffold implantation. e. Histological analysis (H&E) of end-stage FBR in untreated and treated mice at d 49. White arrowhead, giant cell; solid arrowhead, blood vessels. f. Ex vivo MPM analysis and 3D reconstruction of scaffold at day 49 after different treatments. Bar, 50 μm g, h. Quantification of SHG surface area, vessel and giant cell number at day 35 (g) or 49 (h) post-therapy withdrawal. Analysis was performed for multi-image representations from entire scaffolds from 3–5 mice/group. Image area, 500 × 700 μm (x 5 μm). (*), p<0.05; (**), p<0.01; (***), p<0.01 by one-way Anova followed by Tukey’s HSD post hoc test.
Lastly, we tested whether the therapeutic effects persisted after discontinuation of clodronate and/or VEGF Trap administration, by monitoring FBR giant cells, vessels and collagen deposition 14 and 28 days after the treatment was suspended (Fig 6d). For all therapy settings, the FBR severity remained significantly diminished until at least 4 weeks post therapy withdrawal, including stable collagen levels and vessel number, and a decreased but partly relapsing density of giant cells (Fig 6e, f). This indicates that dampening the initial inflammatory phase is crucial to reach sustained anti-angiogenic and -fibrotic response.
Discussion
In summary, by implementing nonlinear intravital 3D microscopy to monitor 3D porous mPCL-CaP scaffolds and related cell and tissue dynamics, we identify the connection between giant-cells and VEGF-induced neovessels as central pathogenic axis driving the FBR and late-stage fibrosis. By a reciprocal process, implant-associated myeloid cells, including giant cells, locally release VEGF-A required to induce neovessels which, in turn, maintains additional cell recruitment and enhances myeloid inflammation and local encapsulation of the graft site by collagen fibers (Fig 7). These results suggest that an immature neovessel network aggravates the FBR, not unlike tumor-associated neovessels supporting inflammation, metabolic deregulation, and desmoplastic remodelling of the tumor stroma43. Because macrophage recruitment, giant cell development and emerging neovascularization cooperate in series to entertain the fibrosis response, therapeutic interference with either process is suited to limit tissue remodelling and scar formation. 24
Fig. 7. FBR and late-stage scarring mediated by macrophages, giant cells and neovessels.
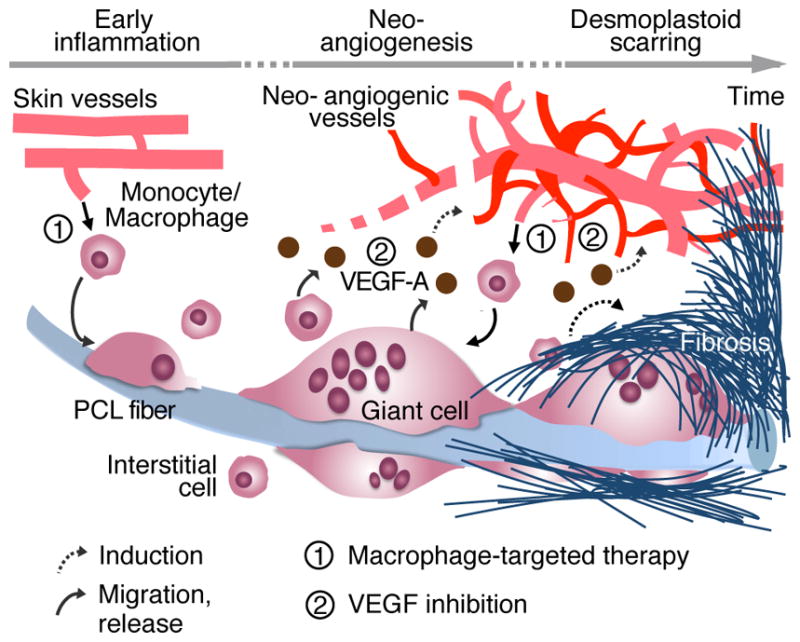
Implantation of mPCL-CaP scaffold into vascularized tissue triggers a FBR. Early influx of monocytes from normal skin vessels is followed by differentiation to macrophages and multinucleated giant cells decorating the foreign material. By releasing VEGF, macrophages/giant cells induce the formation of a primitive neovasculature, that resembles primordial vessels of tumor and desmoplastic tissues, which supports further cell recruitment, inflammation and accumulation of fibrillar collagen to border both graft material and site of inflammation. Continuous arrow represents migratory events or release; dashed arrow represents induction of a phenomenon; ➀ and ➁ highlight steps of interference with macrophage recruitment and VEGF functions, respectively.
A rich set of cytokines is released during a FBR, including IL-1b, IL-4, IL-6, IL-8, IL-10, IL-13, MCP-1, TGF-b and VEGF 2, 7, 9, 49, 50, however their pathogenic impact on whether they aggravate or limit particularly late stages of the FBR is poorly understood2. Besides confirming macrophages and giant cells as central drivers of FBR, we identify a major role for VEGF-releasing M1 polarized cells as important exacerbators of the FBR to mPCL-CaP, with persisting and likely dysfunctional neovascularization and fibrosis as outcomes. Targeting macrophages and giant cells, either by non-specific depletion using clodronate liposome treatment, or by targeted molecular therapy to remove specific subsets (e.g. M1- or M2-skewed cells51), thus represents a promising strategy to modulate both early inflammation (days), as shown recently19, as well as late-stage tissue remodeling (weeks) to minimize a chronic FBR and detrimental scarring.
Our results further establish anti-VEGF therapy as yet unappreciated intervention principle49 to efficiently reduce both neovescularization and subsequent macrophage influx. Thus, anti-VEGF therapy might be indicated for grafts which do not require sustained vascularization for full functionality but suffer from inflammation and fibrosis, including implanted prosthesis, valves, pacemakers, catheters and defibrillators3, 7.
As end-result of depleting myeloid cells and neovascularization by targeted (combination) therapy, a largely avascular and non-inflamed integration site may be critical to reach scar-free long-term integration. Given their proven safety profile in clinical application, generally well tolerated with no long-term adverse effects toward the immune system52–54, individual or combined administration of bisphosphonates and anti-angiogenesis regimens represent promising systemic, or local, topical strategies to improve long-term integration and functionality of material-based grafts.
Methods
Scaffold design and fabrication
Medical grade poly(ε-caprolactone) (mPCL; Purac Biochem, Gorinchem, The Netherlands) was 3D printed by means of melt electrospinning writing (MEW), as described in details elsewhere55. Briefly, PCL was melted at 94°C in a glass syringe and extruded through a 23G blunt-end needle at 10 μL/h and an electrical field of 11–12 KV with a 20 mm distance from a rotating metallic collector, which was mounted on a x-y stage. Scaffolds were surface-treated with NaOH etching and/or calcium phosphate (CaP) coating to reduce the hydrophobicity of PCL. For NaOH etching, scaffolds were soaked in 70% ethanol for 15 min, washed with distilled water and subjected to a 3M NaOH solution (1h, 37°C), and washed with distilled water until pH 7.0 was reached. Calcium phosphate coating was performed as described55. After surface treatment, scaffolds were sterilized by immersion in 70% ethanol and air drying under exposure to UV-light.
Dorsal skinfold chamber (DSFC) model and mPCL-CaP scaffold implantation
Animal studies were approved by the Institutional Animal Care and Use Committee of The University of Texas, M.D. Anderson Cancer Center and performed according to the institutional guidelines for animal care and handling. Dorsal skin-fold chambers were transplanted onto 10 to 12 week-old C57BL/6-Tg(UBC-GFP) 30Scha/J (Jackson Lab) or athymic nu/nu mice (ERO, MD Anderson), as described56, 57. During surgery, a 5×5×0.2 mm scaffold was surgically implanted into the subcutaneous tissue of either imaging window bearing or window-free mice and monitored for up to 14 and 28 days, respectively. For surgery, mice were anesthetized using Ketamine/Xylazine and the wound closed by 4.0 polypropilene sutures.
Intravital microscopy, scaffold imaging and image analysis
For intravital microscopy, mice were anesthetized with isofluorane and stably mounted onto a temperature-controlled platform (37°C). FBR elicited by an implanted mPCL-CaP scaffold was monitored using a custom intravital multiphoton microscope (LaVision BioTech)31 with three Ti:Sapphire lasers (Chameleon-XR, Coherent) and two Optical Parametric Oscillators (APE/Coherent), resulting in a tuneable excitation range from 800 to 1300 nm (Fig S1). Multi-spectral detection was performed using up to 5 backward or 2 forward photomultipliers (PMTs) using up to three excitation wavelengths in two consecutive scans, to separate the following excitation and emission channels: GFP (920 nm; 525/50 nm); Hoechst 33342 (920 nm; 450/60nm), Rhodamine (1090 nm; 595/40 nm), SHG (1090 nm; 525/50 nm), THG (1180 nm; 387/15 nm) and AlexaFluor750 (1180 nm; 810/90 nm).
For intravital detection, long-working distance 16x NA 0.8 water or 25x NA 1.05 multi-immersion oil/water objectives (Olympus) were used. Sequential 3D stacks were obtained with 5–10 μm step-size reaching up to 200 μm penetration depth. Images were acquired in a random fashion within the subcutaneous tissue up to the dermis. Perfused blood vessels were visualized by i.v. injection of Rhodamine- or AlexaFluor750-conjugated dextran (70 kD; Invitrogen; 1 mg/mouse). mPCL-CaP scaffolds, in vitro, were analyzed using SHG and THG imaging.
Digital image processing, segmentation and quantitative analysis
Images were reconstructed, stitched and analyzed using FIJI (W. Rasband, NIH). Individual 3D scan fields representing z-projections of 50 to 300 μm were stitched to large-field montages for both overview and detail analysis. Quantitative analysis of THG, SHG and fluorescent channels was performed on 3D stacks of 360 × 360 μm and 10 μm step interval in z-direction. For standardized enumeration of the local density of GFP-positive cells or vascular density, single channel z-stacks were masked, thresholded (default or Li algorithm), converted to binary images, and the signal-positive area was obtained. For each sample, the relative fluorescence density was obtained from 10 slices per z-stack, averaged and represented as percentage of the total area. 4–8 independent fields were averaged per implant from at least 4 mice/condition. The experiment was repeated 3 times.
For topography-controlled analysis of fibrillar collagen density in association with scaffold fibers, the SHG intensity of 40 × 40 μm regions were quantified as an average from 2 adjacent slices preceding the appearance of the PCL fiber identified by the THG signal (3–5 fibers/sample; 3 mice/group). Each region was masked, thresholded (Li algorithm) and quantified for the signal-positive area fraction. Single cell velocities from time-lapse sequences were obtained by computer-assisted cell tracking (Autozell software; Univ. of Bremen and Würzburg, Germany). For giant cell count, any cell displaying >=2 nuclei was counted as multinucleated. The mean number of nuclei/giant cell was 5.95±2 as quantified on 60 random giant cells, and only a minority of events retained only 2 nuclei.
Therapeutic treatment
Mice bearing mPCL-CaP scaffold in the subcutaneous tissue received VEGF Trap (500 ng/week, once/week) starting 4 days after scaffold implantation. VEGF Trap is a recombinant fusion protein combining the VEGFR 1 and 2 binding portion with the Fc region of IgG1 with VEGF-A-neutralizing activity 46. Clodronate liposomes (200μl/mouse) were administered every 2–3 days, starting 3 days before scaffold implantation to deplete macrophages by the day of implantation. For combination therapy, both regimens were combined. For intravital microscopy, 3 independent fields were averaged per implant, 3 mice/group. The experiment was repeated 2 times.
Histological analysis
Mice were euthanized 14 or 28 days after implantation of the scaffold. Scaffold-bearing skin was excised, fixed (4% buffered formaldehyde) and embedded in paraffin for hematoxylin and eosin or Masson’s Trichrome staining (5 sections/sample, 5μm thick, 3–4 samples/treatment; the experiment was repeated 2 times).
Flow cytometry
Infiltrate cells associated with the FBR in vivo were isolated and phenotyped by flow cytometry (LSRII FACS, Becton-Dickinson;Diva Software). To reach sufficient cell numbers, 20 scaffolds were recovered 14 days post-implantation, mechanically disaggregated to generate a single-cell suspension and pooled. Phenotyping was performed using the following rat monoclonal antibodies (BD Pharmingen): PE-Cy 7 anti-CD45; PerCP-Cy 5.5 anti-CD11b; Pacific Blue anti-Ly6G, APC-conjugated anti-F4/80 and anti-CD8b. Subset gating and analysis were performed using the FLowJo10.2 software.
Data availability
The authors declare that all the relevant data supporting the findings of this study are available within the paper and its supplementary information and from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgments
We thank Steve Alexander for maintenance and support with the multiphoton microscope; Michael Starbuck, Carol Johnston, Yuan Xiaoqing, Roberto Jimenez and Dr. Joseph Douglas for histological processing of the samples; Dr. Elena De Juan Pardo for the manufacturing of the mPCL-CaP scaffolds. E.D. is supported by the Cancer Prevention and Research Institute of Texas (RP140482), Texas, USA and the Prostate Cancer Foundation (16YOUN24), CA, USA; P.F. is supported by NWO-VICI (918.11.626), European Research Council (ERC-CoG DEEPINSIGHT, Project No. 617430) and the Cancer Genomics Cancer, The Netherlands; D. W. H. is supported by the Australian Research Council Training Centre in Additive Biomanufacturing, the National Health and Medical Research Council of Australia, National Breast Cancer Foundation (IN-15-047) and World Cancer Foundation. The Genitourinary Cancers Program of the CCSG shared resources at MD Anderson Cancer Center was supported by NIH/NCI award number P30 CA016672.
Footnotes
Author Contributions: E.D., B. M. H., S. A., D. W. H. and P.F. designed research; E.D., B. M. H., S. A. S. F. and D.W. H. performed research; E.D., S.F., D.W. H. and P.F. analyzed data; E.D., B. M. H., D. W. H and P.F. wrote the paper.
Competing Financial Interests: the authors do not have competing financial interest to declare.
References
- 1.Babensee JE, Anderson JM, McIntire LV, Mikos AG. Host response to tissue engineered devices. Advanced drug delivery reviews. 1998;33:111–139. doi: 10.1016/s0169-409x(98)00023-4. [DOI] [PubMed] [Google Scholar]
- 2.Ward WK. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. Journal of Diabetes Science and Technology. 2008;2:768–777. doi: 10.1177/193229680800200504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morais JM, Papadimitrakopoulos F, Burgess DJ. Biomaterials/tissue interactions: possible solutions to overcome foreign body response. The AAPS journal. 2010;12:188–196. doi: 10.1208/s12248-010-9175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheikh Z, Brooks PJ, Barzilay O, Fine N, Glogauer M. Macrophages, Foreign Body Giant Cells and Their Response to Implantable Biomaterials. Materials. 2015;8:5671–5701. doi: 10.3390/ma8095269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sussman EM, Halpin MC, Muster J, Moon RT, Ratner BD. Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Ann Biomed Eng. 2014;42:1508–1516. doi: 10.1007/s10439-013-0933-0. [DOI] [PubMed] [Google Scholar]
- 6.Anderson JM. Inflammatory response to implants. ASAIO transactions/American Society for Artificial Internal Organs. 1988;34:101–107. doi: 10.1097/00002480-198804000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu TT, VJ, Spiller K. The Role of Macrophages in the Foreign Body Response to Implanted Biomaterials. Biomaterials in Regenerative Medicine and the Immune System. 2015:17–34. [Google Scholar]
- 9.Spiller KL, et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35:4477–4488. doi: 10.1016/j.biomaterials.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore LB, Kyriakides TR. Molecular Characterization of Macrophage-Biomaterial Interactions. Adv Exp Med Biol. 2015;865:109–122. doi: 10.1007/978-3-319-18603-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 12.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrante CJ, Leibovich SJ. Regulation of Macrophage Polarization and Wound Healing. Advances in wound care. 2012;1:10–16. doi: 10.1089/wound.2011.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rostam HM, et al. The impact of surface chemistry modification on macrophage polarisation. Immunobiology. 2016;221:1237–1246. doi: 10.1016/j.imbio.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Palmer JA, Abberton KM, Mitchell GM, Morrison WA. Macrophage phenotype in response to implanted synthetic scaffolds: an immunohistochemical study in the rat. Cells, tissues, organs. 2014;199:169–183. doi: 10.1159/000363693. [DOI] [PubMed] [Google Scholar]
- 17.Yu T, et al. Temporal and spatial distribution of macrophage phenotype markers in the foreign body response to glutaraldehyde-crosslinked gelatin hydrogels. Journal of biomaterials science. Polymer edition. 2016;27:721–742. doi: 10.1080/09205063.2016.1155881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miron RJ, Bosshardt DD. OsteoMacs: Key players around bone biomaterials. Biomaterials. 2016;82:1–19. doi: 10.1016/j.biomaterials.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 19.Veiseh O, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nature materials. 2015;14:643–651. doi: 10.1038/nmat4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Major MR, Wong VW, Nelson ER, Longaker MT, Gurtner GC. The foreign body response: at the interface of surgery and bioengineering. Plastic and reconstructive surgery. 2015;135:1489–1498. doi: 10.1097/PRS.0000000000001193. [DOI] [PubMed] [Google Scholar]
- 21.Zeplin PH, Larena-Avellaneda A, Schmidt K. Surface modification of silicone breast implants by binding the antifibrotic drug halofuginone reduces capsular fibrosis. Plastic and reconstructive surgery. 2010;126:266–274. doi: 10.1097/PRS.0b013e3181dbc313. [DOI] [PubMed] [Google Scholar]
- 22.Klueh U, Dorsky DI, Kreutzer DL. Enhancement of implantable glucose sensor function in vivo using gene transfer-induced neovascularization. Biomaterials. 2005;26:1155–1163. doi: 10.1016/j.biomaterials.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Kastellorizios M, Papadimitrakopoulos F, Burgess DJ. Multiple tissue response modifiers to promote angiogenesis and prevent the foreign body reaction around subcutaneous implants. J Control Release. 2015;214:103–111. doi: 10.1016/j.jconrel.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 24.Mooney JE, et al. Cellular plasticity of inflammatory myeloid cells in the peritoneal foreign body response. Am J Pathol. 2010;176:369–380. doi: 10.2353/ajpath.2010.090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao H, McHugh K, Chew SY, Anderson JM. The topographical effect of electrospun nanofibrous scaffolds on the in vivo and in vitro foreign body reaction. Journal of biomedical materials research. Part A. 2010;93:1151–1159. doi: 10.1002/jbm.a.32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S, et al. Characterization of topographical effects on macrophage behavior in a foreign body response model. Biomaterials. 2010;31:3479–3491. doi: 10.1016/j.biomaterials.2010.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damanik FF, Rothuizen TC, van Blitterswijk C, Rotmans JI, Moroni L. Towards an in vitro model mimicking the foreign body response: tailoring the surface properties of biomaterials to modulate extracellular matrix. Scientific reports. 2014;4:6325. doi: 10.1038/srep06325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maria Ann Woodruff DWH. The return of a forgotten polymer—Polycaprolactone in the 21st century. Progress in Polymer Science. 2010;35:1217–1256. [Google Scholar]
- 29.Holzapfel BM, et al. Tissue engineered humanized bone supports human hematopoiesis in vivo. Biomaterials. 2015;61:103–114. doi: 10.1016/j.biomaterials.2015.04.057. [DOI] [PubMed] [Google Scholar]
- 30.Holzapfel BM, et al. Species-specific homing mechanisms of human prostate cancer metastasis in tissue engineered bone. Biomaterials. 2014;35:4108–4115. doi: 10.1016/j.biomaterials.2014.01.062. [DOI] [PubMed] [Google Scholar]
- 31.Andresen V, et al. Infrared multiphoton microscopy: subcellular-resolved deep tissue imaging. Current opinion in biotechnology. 2009;20:54–62. doi: 10.1016/j.copbio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Weigelin B, Bakker GJ, Friedl P. Third harmonic generation microscopy of cells and tissue organization. J Cell Sci. 2016;129:245–255. doi: 10.1242/jcs.152272. [DOI] [PubMed] [Google Scholar]
- 33.Ho RM, Chiang YW, Lin CC, Huang BH. Crystallization and melting behavior of poly(epsilon-caprolactone) under physical confinement. Macromolecules. 2005;38:4769–4779. [Google Scholar]
- 34.Rodriguez A, Macewan SR, Meyerson H, Kirk JT, Anderson JM. The foreign body reaction in T-cell-deficient mice. Journal of biomedical materials research. Part A. 2009;90:106–113. doi: 10.1002/jbm.a.32050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez A, Voskerician G, Meyerson H, MacEwan SR, Anderson JM. T cell subset distributions following primary and secondary implantation at subcutaneous biomaterial implant sites. Journal of biomedical materials research. Part A. 2008;85:556–565. doi: 10.1002/jbm.a.31562. [DOI] [PubMed] [Google Scholar]
- 36.Marques SM, et al. Genetic background determines mouse strain differences in inflammatory angiogenesis. Microvascular research. 2011;82:246–252. doi: 10.1016/j.mvr.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Rohan RM, Fernandez A, Udagawa T, Yuan J, D’Amato RJ. Genetic heterogeneity of angiogenesis in mice. FASEB J. 2000;14:871–876. doi: 10.1096/fasebj.14.7.871. [DOI] [PubMed] [Google Scholar]
- 38.Middleton JC, Tipton AJ. Synthetic biodegradable polymers as orthopedic devices. Biomaterials. 2000;21:2335–2346. doi: 10.1016/s0142-9612(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 39.Gunatillake PA, Adhikari R. Biodegradable synthetic polymers for tissue engineering. European cells & materials. 2003;5:1–16. doi: 10.22203/ecm.v005a01. discussion 16. [DOI] [PubMed] [Google Scholar]
- 40.Woodruff MAH, DW The return of a forgotten polymer—Polycaprolactone in the 21st century. Progress in Polymer Science. 2010;35:1217–1256. [Google Scholar]
- 41.Wang P, et al. Quantitative analysis of tumor vascular structure after drug treatment. Conference proceedings: … Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual Conference; 2010. pp. 726–729. [DOI] [PubMed] [Google Scholar]
- 42.Wild R, Ramakrishnan S, Sedgewick J, Griffioen AW. Quantitative assessment of angiogenesis and tumor vessel architecture by computer-assisted digital image analysis: effects of VEGF-toxin conjugate on tumor microvessel density. Microvascular research. 2000;59:368–376. doi: 10.1006/mvre.1999.2233. [DOI] [PubMed] [Google Scholar]
- 43.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 44.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. Journal of immunological methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 45.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 46.Holash J, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang TF, Lockhart AC. Aflibercept in the treatment of metastatic colorectal cancer. Clinical Medicine Insights. Oncology. 2012;6:19–30. doi: 10.4137/CMO.S7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stewart MW. Aflibercept (VEGF Trap-eye): the newest anti-VEGF drug. The British journal of ophthalmology. 2012;96:1157–1158. doi: 10.1136/bjophthalmol-2011-300654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwee BJ, Mooney DJ. Manipulating the intersection of angiogenesis and inflammation. Ann Biomed Eng. 2015;43:628–640. doi: 10.1007/s10439-014-1145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones JA, et al. Proteomic analysis and quantification of cytokines and chemokines from biomaterial surface-adherent macrophages and foreign body giant cells. Journal of biomedical materials research. Part A. 2007;83:585–596. doi: 10.1002/jbm.a.31221. [DOI] [PubMed] [Google Scholar]
- 51.Hristodorov D, et al. Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies. mAbs. 2015;7:853–862. doi: 10.1080/19420862.2015.1066950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kennel KA, Drake MT. Adverse effects of bisphosphonates: implications for osteoporosis management. Mayo Clinic proceedings. 84:632–637. doi: 10.1016/S0025-6196(11)60752-0. quiz 638. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McClung M, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13–20. doi: 10.1016/j.amjmed.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 54.Do DV, et al. The DA VINCI Study: phase 2 primary results of VEGF Trap-Eye in patients with diabetic macular edema. Ophthalmology. 2011;118:1819–1826. doi: 10.1016/j.ophtha.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 55.Thibaudeau L, et al. A tissue-engineered humanized xenograft model of human breast cancer metastasis to bone. Disease models & mechanisms. 2014;7:299–309. doi: 10.1242/dmm.014076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P. Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochemistry and cell biology. 2008;130:1147–1154. doi: 10.1007/s00418-008-0529-1. [DOI] [PubMed] [Google Scholar]
- 57.Guba M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the relevant data supporting the findings of this study are available within the paper and its supplementary information and from the corresponding authors upon reasonable request.