

Abstract
Purpose of review
About 15–25% of patients with simple steatosis of non-alcoholic fatty liver disease progresses to non-alcoholic steatohepatitis (NASH), and the underlying mechanism for this progression has not been elucidated. NASH ultimately could progress to cirrhosis, an irreversible condition.
Recent findings
Farnesoid X receptor (FXR) has been studied for its role in modulating inflammation, and the expression of FXR is down-regulated during NASH development. FXR deficiency has shown to progress and exacerbate NASH development, and FXR activation has been protective against liver inflammation associated with NASH. The expression of factors in both the adaptive and innate immune response in the liver are regulated in a FXR-dependent and -independent manner.
Summary
Therefore, understanding key signaling pathways of liver inflammation in NASH is important to determine essential components that predispose, progress, or exacerbate NASH. FXR has been identified as a therapeutic target for NASH to prevent liver inflammation.
Keywords: Farnesoid X Receptor, non-alcoholic fatty liver disease, Nonalcoholic Steatohepatitis, Inflammation, Adaptive Immunity, Innate Immunity, Acute Phase Response
Introduction
Classically well reviewed for bile acid regulation, and regulation of lipid and glucose metabolism [1–4], Farnesoid X receptor (FXR) currently has an emerging role in regulating inflammation. Its role in the regulation of inflammation is elucidated through many studies related to liver disorders, cholestasis, and alcoholic liver disease [5, 6]. Most notably the role of FXR in NAFLD and inflammation goes back to the original characterization of FXR knock-out (KO) mice with induced hepatic bile acid levels and liver injury described as steatosis, inflammation, and fibrosis [7]. These results support a role for altered FXR function in NASH development or the progression from simple steatosis to NASH. More recently, the immune pathways, both adaptive and innate responses, have been implicated in liver inflammation and metabolic diseases, specifically NASH [8–11]. Review of the most recent literature will focus specifically on the role of FXR in the development and/or progression and inflammatory response directly related to NASH.
Nonalcoholic Steatohepatitis
Nonalcoholic fatty liver disease (NAFLD) is a worldwide epidemic predominantly in western countries with estimated occurrence to be 20% to 30% of the population, and increased prevalence in populations associated with metabolic syndrome [12]. For example, in the United States patients undergoing bariatric surgery had a prevalence of NAFLD as high as 73% to 97%, with non-alcoholic steatohepatitis (NASH) diagnosis in 25% to 33% of patients [12]. NAFLD is an imminent concern in the field of chronic liver diseases. The prevalence of NAFLD as a cause for chronic liver disease has doubled between 1988 and 2008, according to National Health and Nutrition Examination Surveys [12]. NAFLD was originally considered a benign disease, but due to the spectrum of diseases associated with the initial increased disposition of lipids in the liver it can progress to a chronic inflammatory state that defines NASH. Simple steatosis progresses to NASH in about 7–30% of patients with NAFLD, and a third of those continue to progress to advanced fibrosis or cirrhosis [13]. NASH development is not well elucidated, and even the pathogenesis is not fully determined. The initial two-hit hypothesis of steatosis, followed by inflammatory response, hepatocyte damage, and fibrosis [14], has recently been replaced by rationale supporting a multiparallel hypothesis that suggests many parallel conditions contributing towards the progression of NASH (15). These conditions have been extensively reviewed [13]. NASH is characterized by histopathological features of macrovesicular steatosis, lobular inflammation with mixed cell types (lymphocytes, macrophages, and natural killer T cells), hepatic ballooning, Mallory bodies, and perisinusoidal fibrosis [15]. More recent research has shown the importance of genetic predispositions, as ageing of FXR KO mice displayed histological features of NASH at 10 months without metabolic insult: increased steatosis, perisinusoidal/sinusoidal foam cells, ballooning degeneration, lobular inflammation, elevated activities/levels of plasma ALT, bile acids, and bilirubin, and increased expression of tumor necrosis factor α (TNFα) and toll-like receptor 4 (Tlr-4) [16]. Ageing mimicked the NASH-like phenotype of FXR/LDLr-deficient mice, which was exacerbated on a high-fat diet (HFD) [17]. Chronic inflammation is an encompassing characteristic of conditions associated with NASH development and progression, and understanding key mechanisms will aid in identifying more promising pharmacological targets for the treatment of NASH, since there is currently no drug therapy.
NASH and Liver Inflammation
Initial and key mediators of the development of chronic inflammation in NASH are endogenous danger signals (e.g. damage associated molecular patterns (DAMPs)), and the activation of their cognate sensors or pattern recognition receptors that are responsible for the downstream signaling cascade of the inflammasome. The role of the inflammasome in NASH has recently been reviewed [18]. Briefly, the inflammasome is comprised of the receptors of the danger signals including toll-like receptors (TLRs) and nucleotide-binding oglimerization domain (NOD)-like receptors (NLRs), the adaptor protein apoptosis-associated speclike protein (ASC), and the effector molecule caspase-1. TLRs are responsible for the rapid activation of TNFα, IL-6, and monocyte chemotactic protein 1 (MCP-1), while IL-1β activation requires a highly-regulated two-step mechanism of induction by TLR-activation, followed by the activation of the inflammasome in order for secretion of pro-IL-1β and subsequent activation by caspase-1 cleavage. This process can be a key contributor to chronic inflammatory states due to the ability of cumulative danger signals to activate the inflammasome even at low levels. Major TLRs that have been associated with NASH pathogenesis are TLR2, TLR4, and TLR9 [19–21]. The TLR adaptor molecule, MyD88, was shown to be an upstream regulator for the activation of the NLRP3 inflammasome (NLRP3, ASC, and capsase-1) in an MCD-diet model of NASH [22], and NLRP3, ASC, and caspase-1 levels were higher in steatohepatitis than HFD-steatosis [23]. NLRP3 inflammasome can influence the onset and development of NASH, and can act through the induction of the nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) signaling pathway; subsequently, NLRX1 negatively regulates TNF receptor-associated factor 6 (TRAF6)-mediated activation of NF-κB and the reduction in NLRX1 mRNA and protein levels are associated with the onset of NASH [23]. NLRP3 contributes to NASH inflammation, while NLRX1 acts as a negative regulator of excessive inflammatory response. Lastly, the inflammasome in diet-induced NASH models has been suggested to be induced by fatty acids alone and gut-derived microbial danger signals (e.g. lipopolysaccharide (LPS)) [24], providing support for the role of the inflammasome in the development or progression of key pathological features of liver inflammation associated with NASH.
The role of liver inflammation in the development and progression of NASH has been addressed by several recent reviews, focusing on pathological features of the innate immune response and inflammatory signaling cascade [3, 8–10, 13, 25]. Briefly summarizing, specific key features of cellular stress and response are: increased low-density lipoprotein (LDL)-cholesterol, neutrophil infiltration, pro-inflammatory mediators including TNFα and NF-κB, and subsequent cytokine and chemokine release. Two features of NASH are cholesterol crystals in hepatocytes and foamy Kupffer cell generation. Both features are induced by unrestricted hepatic uptake of naïve or modulated LDL-cholesterol contributing to an inflammatory response, and the oxidation of LDL-cholesterol is clinically significant in serum of NASH patients correlating to proinflammatory cytokine secretion [13]. Patients with NAFLD/NASH have been characterized by decreased expression of ATP-binding cassette sub-family G member 8 (ABCG8) cholesterol transporter, and decrease in CYP7A1 and CYP27A1, enzymes responsible for cholesterol metabolism, resulting in decreased free cholesterol efflux [13]. Decreased cholesterol efflux could contribute to the inflammasome of NASH and result in the dysregulation of bile acid metabolism, directly relating FXR to liver inflammation. Excess neutrophil recruitment is another prominent histological feature of NASH, in which increased infiltration of neutrophils compared to lymphocytes can mediate NASH progression, and directly contribute to hepatocyte damage, inflammation, and proinflammatory cytokine release [8, 13]. TNFα, an important cytokine in hepatic diseases, was induced in patients with NAFLD, and could lead to NF-κB activation and pro-inflammatory signaling [10]. Lastly, cytokines and chemokines have been linked or directly correlated to the inflammatory state of NASH such as: TNFα, interleukin 1 and 6 (IL-1, IL-6), chemokine (C-X-C motif) ligands 1–3 and 8 (CXCL1, CXCL2, CXCL3, CXCL8), and chemokine (C-C motif) ligands 2–4 (CCL2/MCP-1, CCL3/MIP-1α, CCL4 [10]. Most of these specific etiologies related to the progression of NASH resulting in increased liver inflammation have been most recently studied as FXR-mediated responses.
Adaptive Immune Response
The adaptive immune response, also referred to as acquired immune responses, is required for immunological memory and involves antigen-specific lymphocyte recruitment to antigen, or pathogen specific responses. Adaptive immunity is well-characterized and understood in diseased states, and is recognized as a mediator of inflammation in models of NASH [26]. Specifically, the methionine-choline deficient (MCD) model of NASH has increased hepatic recruitment of CD4+ T-lymphocytes that were activated by T helper-1 (Th-1) stimulating macrophage (M1) proinflammatory responses [27]. Kupffer cells, or resident macrophages of the liver, undergo phenotypic changes during steatosis by increased lipid deposition in the cytoplasm (triglycerides, free cholesterol, diacylglycerols, and sphingolipids/ceramides) and altered pro-inflammatory response with increased secretion of interferon gamma (IFNγ), TNFα, and MCP-1/CCL2 [28]. LPS stimulates the release of previous factors along with IL-6, IL-1β, and CXCL10 in HFD-induced Kupffer cells, corresponding to induced recruitment of lymphocytes to the liver in vivo [28]. As mentioned previously, another key mediator of the proinflammatory response is NF-κB, whose prototypical activators are TNFα and IL-1, but due to multiple roles of NF-κB in adaptive immunity and inflammation it is not a clear pharmacological target [29]. Therefore, more specifically, FXR and NF-κB signaling pathways demonstrated negative crosstalk in inflammatory response [30]. Adaptive immunity contributes to the complex immunology and inflammasome associated with NASH, and key factors of these responses have been shown to be directly regulated by FXR.
FXR and MCP-1
MCP-1/CCL2, a protein responsible for the attraction of monocytes and memory T cells to the site of inflammation, is correlated to mononuclear cell infiltration in inflammatory diseases [31]. Recent findings in the field of NAFLD have supported MCP-1 as an essential factor in the development of NASH. HFD-induced steatohepatitis in mice showed upregulation of hepatic MCP-1/CCL2 mRNA prior to induction of TNFα, fibrogenic factors, and steatohepatitis at 50 weeks [32]. In humans, serum TNFα and MCP-1/CCL2 were elevated by 57% and 45%, respectively, in NASH patients (n=25) compared to patients with simple steatosis (n=22) [33]. Therefore, MCP-1 should be researched as a precursor of NASH development and down-regulation of MCP-1 may protect against NASH. MCP-1 mRNA and protein levels are down-regulated by direct FXR binding to an upstream FXR response element (FXRE) of the MCP-1 gene in a dose-dependent manner in macrophage cell lines (ANA-1 and raw 264.7) treated with chenodeoxycholic acid (CDCA, potent bile acid FXR agonist) [34]. In vivo, the FXR agonist WAY-362450 decreased MCP-1 hepatic mRNA expression, and significantly decreased inflammatory cell infiltration in the liver, reducing hepatic inflammation induced by the MCD-diet; all effects of the FXR agonist were lost in FXR KO mice [35]. Liver injury associated with NASH induced by both HFD (28-week) and MCD-diet (3.5 and 11 week) was alleviated by treatment with a bile acid phospholipid conjugate (Ursodeoxycholyl Lysophosphatidylethanolamide, UDCA-LPE), resulting in reduction in inflammatory cell infiltrates specifically in the MCD-diet cohort, and decreased hepatic MCP-1 mRNA and protein levels in both models [36]. Therefore, hepatic and serum MCP-1 levels, and MCP-1 expression in macrophages during low-grade systemic inflammation may play a role in the progression of simple steatosis to NASH. Further studies are needed to identify MCP-1 as a potential biomarker, or to determine the factor’s direct role in the pathogenesis. Currently, in vitro and in vivo treatment models have demonstrated a direct role of FXR in reduction of MCP-1 mediated liver inflammation in NASH.
FXR and NF-kB-mediated inflammation
FXR KO mice are more sensitive to NF-κB activation (TPA (12-o-tetradecanoyl-phorbol-13-acetate), TNFα, or LPS treatment) and ageing (12 months), revealed by increased TNFα and IL-1α expression, as well as a significant increase in hepatic cytokine signaling molecules, inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2), in stimulated primary hepatocytes or in vivo [30]. Most importantly, it was demonstrated that pretreatment with FXR agonists (GW4064 and 6ECDCA) suppressed NF-κB agonist-induced inflammatory gene expression in an FXR-dependent manner in HepG2 cells and mouse primary hepatocytes [30]. NF-κB binding sites were identified on FXR and FXR target genes BSEP, SHP, MRP2, MDR3, and ABCG5/G8; specifically, NF-κB bound to the promoter of BSEP and actively suppressed transcription [37]. Overexpression of NF-κB recruited nuclear receptor co-repressor 2 (SMRT), and blocked FXR binding to its FXRE [37]. Direct interaction between FXR and NF-κB was further confirmed in vivo, NF-κB recruitment to FXR and BSEP promoter was increased in models of disease and inflammation (bile-duct ligation and LPS-injection) [37], confirming previous observations of suppressed FXR signaling during inflammatory response [30, 38]. NF-κB and SMRT recruitment to promoters of FXR and FXR-regulated genes may be a predominant adaptive mechanism in chronic inflammatory disease states characterized by decreased FXR and FXR-dependent signaling cascades.
Post-transcriptional modifications of FXR have recently been proposed, which suggests a more specific interaction between FXR and NF-κB. Existing evidence support increased acetylation of transcription factors in models of nutrient excess, but specifically acetylated-FXR is elevated in diet-induced obesity [39]. Kemper et al. has demonstrated over the years that FXR acetylation is dynamically regulated under normal feeding, but remains constitutively activated in diet-induced obesity, that was characterized by decreased FXR binding at FXREs of inflammatory genes in obese mice compared to wild-type mice (WT) [39–41]. Acetylation of FXR at lysine 217 (K217) was shown to promote hepatic inflammation through inhibition of sumoylation at K277 in obese mice; proinflammatory gene expression, macrophage infiltration, and increased levels of TNFα and IL-1β were increased in K217Q mutant (FXR acetylation-mimic) overexpressed mice [41]. SUMO2-modified FXR levels were increased by treatment with FXR agonists (GW4064, CDCA), and expression of SUMO2-FXR was decreased with HFD feeding to nearly undetectable levels after 12 weeks [41]. SUMO2-FXR bound selectively to NF-κB inhibiting transactivation and subsequent inflammatory signaling pathway [41]. Sumoylation of FXR did not alter the expression of SHP or BSEP due to substantially decreased interaction of SUMO2-FXR to RXRα, and SUMO2-FXR was shown to not be recruited to FXR/RXRα target genes [41]. Targeting the dysregulation of the acetyl/SUMO switch associated with FXR regulation of NF-κB signaling and inflammatory gene expression can be beneficial with future research in NASH-specific expression of acetylated FXR verse SUMO2-FXR. Further characterization of the regulation of inflammatory responses related to adaptive immunity is essential for inhibiting the exacerbation or progression of a disease characterized by increased inflammatory state, such as NASH.
Acute Phase Response (APR, Innate Immunity)
The APR is a systemic reaction to systemic or local disturbances induced by infection, tissue injury, trauma, surgery, or immune dysfunction [42]. Acute phase proteins (APP) are regulated by the hepatic APR, manifested as the upregulation (positive APPs) or downregulation (negative APPs) of these secretory proteins [42]. Cytokines stimulate the production and release of positive APPs from hepatocytes that are known to mediate inflammatory responses locally or systemically, and are associated with a decrease production in negative APPs or normal blood proteins, including: transthyretin, retinol binding protein (RBP), cortisol binding globulin, transferrin and albumin [42]. The APR can become chronic with repeated activation [42], therefore innate immunity can play a role in models of chronic inflammation, such as NASH. A key publication in 2003 demonstrated that FXR was downregulated during the APR, in which down-regulation of hepatic FXR was achieved by LPS administration after 8 hours in a tissue-specific manner, secondary to reduction in RXR (~2 hrs), resulting in decreased mRNA expression of downstream targets SHP and apoCII [38]. Cytokine treatment (TNF and IL-1) of Hep3B cells resulted in decreased gene expression of FXR and its target genes; also, TNF and IL-1 abolished CDCA-stimulated luciferase activity of a FXRE-luciferase construct [38]. These key findings have helped to suggest that FXR expression and function is affected by APR and inflammation. The effects of FXR in regulating APPs are emerging, and FXR has been shown to regulate APPs including C reactive protein (CRP), and more recently lesser characterized family of APPs, such as the lipocalin family (LCN2, LCN13).
FXR and CRP
CRP, is an APP that has been reported to be the most robust feature and predictive factor of future cardiovascular events and outcomes. CRP is considered an inflammatory biomarker that represents systemic inflammation, and high-sensitivity (hs)-CRP assays allow for diagnosis of low grade inflammation [43]. Ndumele et al. (2011) determined that hs-CRP levels were significantly higher among both obese and non-obese individuals [44]. All three metabolic conditions (hepatic steatosis, obesity, metabolic syndrome) were independently associated with increased hs-CRP levels, and an additive increase in hs-CRP levels (≥3mg/dL, high levels) was observed in individuals with all 3 conditions (4-times higher odds of having ≥3mg/dL hs-CRP levels) [44]. The additive effect of metabolic diseases supports previous findings that serum hs-CRP levels can distinguish between patients with NASH verses those with simple steatosis, and specifically, hepatic mRNA of CRP was induced in patients with NASH [43]. Therefore, hs-CRP remains an essential biomarker in NAFLD inflammation but may also contribute to the progression of NAFLD. Zhang et al. demonstrated that FXR agonism (WAY-362450 and GW4064) inhibits IL-6 induced CRP release and mRNA expression in Hep3B cells in a dose-dependent manner, and siRNA knockdown of FXR (85% suppression) resulted in IL-6 stimulated CRP levels [45]. Serum amyloid P component (SAP) and serum amyloid A3 (SAA3) are the major APPs in murine models, and LPS-stimulated expression of hepatic SAP and SAA3 were significantly decreased by WAY-362450 treatment compared to vehicle-control; FXR KO mice were more sensitive to LPS-stimulated APP induction, which was not decreased by pre-treatment with WAY-362450 [45]. Therefore, in vitro human modeling measuring CRP, and in vivo mouse modeling measuring SAP and SAA3, demonstrated a direct role of FXR agonists in the regulation of hepatic APPs across species.
FXR and Lipocalins
Lipocalins are small secreted proteins and many members of the lipocalin (LCN) family have been identified as APPs. It was first shown that hepatic expression of members of the LCN family are dysregulated in liver-specific FXR KO mice (lFXR-KO), specifically 13 LCNs are upregulated [46]. The study focused on ORM1 regulation by FXR/RXR binding and identified FXREs due to the FXR-dependent induction of orosomucoids (ORMs) in lFXR-KO mice treated with taurocholic acid (TCA) [46]. It was further demonstrated that ORM1 induction by FXR agonist was tissue-specific, with induction of ORM1 only in the liver (39). The activity of ORMs in inflammation has shown potential immunosupressive effects that can be correlated to FXR activation in intestinal diseases [46], but ORM1 expression in states of chronic inflammation has not been studied. More importantly, TCA also induced LCN2 and LCN9 gene expression. The regulation of other lipocalins by FXR supports our laboratory’s recent findings regarding FXR’s role in the APR. ChIP-seq analysis comparing in vivo exposure of mouse hepatocytes and in vitro exposure of primary human hepatocytes to a FXR agonist (GW-4064) identified APR induction as an FXR-dependent pathway via GO-BP analysis [47]. Further investigation via RNA microarray has identified two LCNs of interest: LCN2 and LCN13.
LCN2 had a significant induction in lFXR-KO mice, but remained induced in TCA-treated WT and lFXR-KO mice suggesting an alternative regulation potentially through STAT3 and/or TGR5 signaling [46]. Our current research supports a potential role of FXR-regulation of LCN2 expression in mice treated with GW-4064. LCN2 expression followed the same pattern as previously reported [46], LCN2 was upregulated by GW-4064, was induced by liver-specific and whole-body FXR KO and further induced by GW-4064 treatment (unpublished data). The role of LCN2 has been documented in three NASH models in mice, and correlated to human NAFLD and NASH patients [48–50]. The Fatty liver Shionogi (FLS) mouse model, known for development of fatty livers in a setting of insulin resistance progressing to hepatocellular carcinoma via steatohepatitis, were evaluated for their transcriptome compared to control strain (DS) mice; after 19-weeks histopathological analysis identified FLS-mice to have a NASH phenotype, and DS mice were considered to have a fatty liver phenotype [50]. Microarray analysis identified 14 genes up-regulated in NASH compared to steatosis, including ORM2, LCN2, CXCL1, and CXCL9 [50]. Due to the heterogeneity of inflammatory cell populations noted in NASH livers, LCN2, CXCL1, and CXCL9 were studied as biomarkers of NASH; all three genes were overexpressed in NASH livers [50]. LCN2 was induced at the protein level and immunohistological staining showed a positive correlation between the number of LCN2-positive cells and inflammatory cell clusters, with LCN2 concentrated in parenchymal hepatic cells of FLS mice [50]. Another study implicated LCN2 as an inflammatory marker of NASH in both a high-fat high-cholesterol (HFHC) and MCD-diet model of NASH, and was able to further characterize direct downstream mechanisms [48]. These two models also demonstrated an induced infiltration of neutrophils in mouse livers with NASH, and elevation of both plasma and liver LCN2 levels; a significant difference was made by Ye et al. demonstrating that LCN2 enrichment was found in non-parenchymal cells, specifically LCN2 staining was colocalized with the neutrophil marker Ly6G [48]. ApoE KO mice backcrossed with LCN2 KO mice were protected from HFHC diet-induced liver damage (60% and 40% decrease in ALT and AST, respectively), and neutrophil accumulation was reduced by 50%, followed by reductions in CXCL2, CXCR2, TNFα, IL-1β, and MCP-1; the same results were achieved in LCN2 KO on an MCD-diet [48]. Further characterization of the models identified LCN2 enhances neutrophil migration via CXCR2, and CXCR2 expression is down-regulated by 70% in LCN2 KO mice therefore blocking CXCL2-induced neutrophil migration [48]. P42/44 extracellular signal-related kinase (ERK) was also inhibited in LCN2 KO neutrophils by 60% suggesting that LCN2-induced phosphorylation of ERK results in CXCR2-mediated neutrophil infiltration and inflammation in NASH [48]. Lastly, LCN2 serum levels were induced in patients with NASH compared to subjects with steatosis, and LCN2 was colocalized with neutrophil marker CD66b in humans [48]. A cohort of severely obese women also demonstrated, at the protein level, LCN2 was upregulated in livers of patients with NASH compared with simple steatosis or normal livers, moreover, a significant correlation between LCN2 expression and TNFα was noted [49]. The role of LCN2 in neutrophilic inflammation is not specific to NASH, but has also been shown to be necessary for alcohol induced-steatohepatitis (ASH) [51]. In a model of ASH, it has been suggested that FXRα and FXRβ, β-klotho, and Cyp7A1 are down-regulated upon ethanol-feeding in a LCN2 dependent-manner [52]. A direct role of LCN2 in disease progression and correlation to NASH phenotype needs to be further determined, and clarification whether FXR plays a role in its regulation during NASH, or an alternative upstream mediator is responsible still needs to be investigated.
LCN13 was first identified as part of a lipocalin gene cluster on mouse chromosome 2 within the epididymis [53]. This lipocalin has since been identified to be expressed in liver, skeletal muscle, and pancreas, secreted into circulation, and expressed in the bloodstream during fed-states [54]. LCN13 plasma levels were reduced in the fasting state and in db/db mouse model of obesity, and LCN13 hepatic expression was suppressed upon HFD feeding, suggesting this secreted factor is sensitive to glucose levels and nutrient status [54]. LCN13 is suggested to have a role in glucose metabolism through an autocrine, paracrine, and endocrine manner by acting as an insulin sensitizer. Glucose intolerance and insulin resistance were reversed by LCN13 overexpression through adenovirus or recombinant protein in HFD-fed and db/db mice [54]. Glucose metabolism was regulated by LCN13 in an insulin-dependent manner via enhanced insulin-signaling in adipocytes and stimulation of glucose-uptake, and in an insulin-independent manner by suppression of hepatic glucose production [54]. LCN13 not only regulates glucose metabolism, but regulates lipid metabolism endogenously by suppressing lipogenesis and key transcriptional regulators, FAS and Srebp1c, and promotes fatty acid β-oxidation and increased Cpt1α expression in primary mouse hepatocytes [55]. Furthermore, mice with transgenic overexpression of LCN13 were resistant to diet-induced hepatic steatosis, and in a genetic model of obesity (ob/ob mice) LCN13 transgenic overexpression ameliorated hepatic steatosis through identical changes in gene expression, reduction in plasma and liver triglycerides, and reduced liver weight [55]. Furthermore, anit-LCN13 antibody increased lipogenesis and decreased β-oxidation, demonstrating regulation of key genes by LCN13 signaling [55]. Identifying putative bioactive small molecule or cognate receptors related to LCN13 regulation is essential to understand LCN13’s role in models of obesity and diabetes. Most recent data from our lab suggest LCN13 as a putative hepatic FXR target gene through gene microarray data. LCN13 can contribute to NAFLD through dysregulation of glucose and lipid metabolism with decreased expression in models of obesity, but is currently being studied for its role in liver inflammation due to its potential role as an APP. Further characterization of LCN13 as an APP, will demonstrate a greater role of LCN13 in NASH.
Small heterodimer partner (SHP) role in NASH
Small heterodimer partner (SHP/NR0B2) is an established target gene of FXR directly involved in suppression of bile acid homeostasis via transcriptional repression of Cyp7a1 and Slc10a1 [56]. SHP has been demonstrated to act as a downstream mediator of other FXR-dependent, suppressive responses, e.g. 1) FXR activation via bile acids induces SHP, which suppresses sterol regulatory element-binding protein 1 (Srebp1c) to decrease fatty acid synthesis in the liver [57], and 2) FXR-dependent up-regulation of SHP represses apical sodium-dependent bile acid transporter (ASBT) expression, therefore maintaining negative feedback regulation of bile acids [58]. Most recently, a genome-wide transcriptome analysis identified new genes and gene signatures regulated by SHP in human chronic liver diseases, including NASH [59]. Differentially expressed genes (DEGs) were determined in SHP knock-out (SHP KO) mice and a cohort of human livers with NASH, there were 68 shared DEGs in SHP KO and NASH livers, including peptidoglycan-recognition protein 2 (PGLYRP2) that was identified as a SHP direct or indirect target in NASH and demonstrated to be involved in inflammation (LPS treatment) [59]. SHP’s role in hepatic inflammation has also been addressed in a vertical sleeve gastrectomy (VSGx) study in mice to improve NAFLD, in which SHP KO mice had a pro-inflammatory phenotype and hepatic inflammation developed despite weight loss following VSGx [60]. This study suggests that SHP may have a direct role in the repression of liver inflammation, and prevent liver injury after VSGx in SHP KO obese mice [60]. Therefore, downstream effects of FXR activation in the prevention/treatment of NASH must not only be evaluated for direct FXR regulation, but mechanisms of SHP-dependent repression must be considered.
FXR Agonists and Decreased Liver Inflammation
FXR agonism is proposed as a potential therapeutic target for NASH with two promising FXR activators currently in clinical trials, INT-747 (Obeticholic acid, OCA) [61] and Px-104 (non-steroidal isoxazole, GW4064 derivative) [62]. Many FXR agonists are currently being studied in mouse models for further development of the pipeline of drugs for NASH. Protection against hepatic inflammation secondary to hepatic triglyceride accumulation was achieved with the synthetic agonist, WAY-362450 [35]. Although MCD diet is not considered to represent metabolic syndrome-induced NASH in humans, the MCD diet none-the-less leads to markers commonly found in NASH including increased serum AST and ALT activity, induction of serum MCP-1, and elevation in keratinocyte derived chemokine (mKC/IL-8); WAY-362450 treatment (p.o. daily/4 weeks) significantly reduced all serum markers measured, demonstrating beneficial effects on hepatic inflammation and function [35]. A bile acid phospholipid conjugate, UDCA-LPE, was also designed with the goal of increased therapeutic efficacy in NAFLD/NASH due to potent anti-inflammatory properties against TNFα-induced or endotoxin-mediated cytotoxicity in vitro and in vivo, respectively [63]. Liver injury, induced by two models of metabolic NAFLD/NASH development (28-week HFD and 3.5 or 11 weeks MCD-diet), was alleviated by UDCA-LPE (30 mg/kg three times a week); the treatment achieved near normal ALT and AST serum levels, decreased NAFLD activity score, reduction in inflammatory cell infiltrates specifically in MCD-diet cohort, decreased triglyceride and cholesterol levels, reduced hepatic MCP-1 and TNFα in both models, and significantly decreased proinflammatory lysophosphatidylcholine (LPC) in both models and additionally hydroperoxides in mice fed the MCD diet [36]. Another proposed treatment for the attenuation of diet-induced NASH is the additive effects of omega-3 polyunsaturated fatty acid (omega-3) and UDCA, which both have previously been demonstrated in animal models, but individual evidence within clinical trials is lacking or no apparent improvement was noted, respectively [64]. In mice fed a HFD for 24 weeks prior to treatment, omega-3 alone decreased ALT levels and resulted in reduced hepatic TNFα and iNOS expression [64]. Omega-3 in combination with UDCA treatment resulted in a significant decrease in histological scoring and further decrease in TNFα and iNOS expression [64]. Therefore, Omega-3 and UDCA have an additive effect to ameliorate diet-induced NASH and improve hepatic inflammation [64]. All three models represent an induction in FXR and FXR signaling pathways, which have been shown in animal models to have dramatic effects on prevention of the NASH phenotype.
Conclusion
NAFLD is a prevalent disease in today’s society, especially in western countries due to rise in obesity and metabolic syndrome. NASH remains an elusive disease state of NAFLD that can only be diagnosed by biopsy, and its progression is not well understood. The only key features of human NASH are derived from histopathology, and the goal within research is to recapitulate these histological features in murine models. The multiparallel hypothesis has evolved from the simple ‘two-hit’ hypothesis and directly demonstrates the complexity of the disease. Major findings have demonstrated that FXR signaling is involved in both the adaptive and innate immune response associated with chronic inflammation of NASH. FXR agonism have been shown to be a promising pharmacological target, avoiding complicated NF-κB signaling cascades while acting on essential pathways inhibiting neutrophil accumulation and pro-inflammatory cytokine signaling. Further research is needed to fully characterize tissue/cell-specific actions of FXR as a nuclear factor that can combat NASH development and progression, while maintaining liver homeostasis decreasing potential adverse signaling cascades. Lastly, APPs have begun to be identified as potential biomarkers for disease prevention, and FXR-regulation of the APR is another beneficial effect of FXR agonists that can potentially be correlated to humans and measured within clinical settings to assess disease and treatment. Further research is needed to characterize FXR regulation of the APR, specifically lipocalins, and distinguish the role of the APR in the chronic inflammation associated with NASH. The inflammasome of NASH is complex, but the established role of FXR in regulating inflammation, lipid metabolism, and glucose metabolism currently makes it a promising drug target for the treatment of NASH.
Figure 1. NASH is a disease-state characterized by decreased FXR expression, and FXR agonism has been shown to inhibit molecular pathways directly related to NASH.
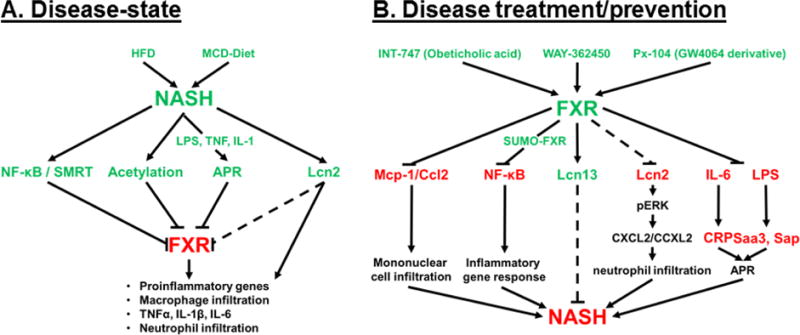
A) The disease-state schematic depicts the contributing factors of diet-induced NASH that result in decreased FXR and FXR-signaling resulting in the inflammatory phenotype that characterizes NASH. B) The disease treatment/prevention pathway represents the role of FXR agonism, via experimental FXR-agonists, in inhibiting key factors in the inflammatory pathways (Mcp-1, NF-κB, Lcn2, Il-6-mediated CRP, and LPS-mediated Saa3 and Sap) or the induction of Lcn13 to inhibit proinflammatory pathways that contribute to NASH. (ˇ) represents induction, (−) represents inhibition of subsequent factors, and (---) represents proposed regulation
Acknowledgments
The authors would like to acknowledge the NIH Grant, GM104037, to support Dr. Grace L. Guo’s research and the NIH/NIEHS Training Grant, T32ES007148, to support Dr. Laura E. Armstrong’s post-doctoral training.
Footnotes
Conflict of Interest
Laura E. Armstrong and Grace L. Guo declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance •• Of major importance
- 1.Li T, Apte U. Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer. Adv Pharmacol. 2015;74:263–302. doi: 10.1016/bs.apha.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368(1–2):17–29. doi: 10.1016/j.mce.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Y, et al. Fatty liver diseases, bile acids, and FXR. Acta Pharm Sin B. 2016;6(5):409–412. doi: 10.1016/j.apsb.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Y, Li F, Guo GL. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol Res. 2011;63(4):259–65. doi: 10.1016/j.phrs.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan ZQ, Li KW. Role of farnesoid X receptor in cholestasis. J Dig Dis. 2016;17(8):501–509. doi: 10.1111/1751-2980.12378. [DOI] [PubMed] [Google Scholar]
- 6.Manley S, Ding W. Role of farnesoid X receptor and bile acids in alcoholic liver disease. Acta Pharm Sin B. 2015;5(2):158–67. doi: 10.1016/j.apsb.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinal CJ, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 8.Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int. 2013;7(Suppl 2):771–81. doi: 10.1007/s12072-013-9468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meli R, Raso GMattace, Calignano A. Role of innate immune response in non-alcoholic Fatty liver disease: metabolic complications and therapeutic tools. Front Immunol. 2014;5:177. doi: 10.3389/fimmu.2014.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bieghs V, Trautwein C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 2013;34(9):446–52. doi: 10.1016/j.it.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Sutti S, Bruzzi S, Albano E. The role of immune mechanisms in alcoholic and nonalcoholic steatohepatitis: a 2015 update. Expert Rev Gastroenterol Hepatol. 2016;10(2):243–53. doi: 10.1586/17474124.2016.1111758. [DOI] [PubMed] [Google Scholar]
- 12.Satapathy SK, Sanyal AJ. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin Liver Dis. 2015;35(3):221–35. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 13.Caligiuri A, Gentilini A, Marra F. Molecular Pathogenesis of NASH. Int J Mol Sci. 2016;17(9) doi: 10.3390/ijms17091575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day CP, James OF. Steatohepatitis: a tale of two “hits’? Gastroenterology. 1998;114(4):842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 15.Ibrahim SH, et al. Animal Models of Nonalcoholic Steatohepatitis: Eat, Delete, and Inflame. Dig Dis Sci. 2016;61(5):1325–36. doi: 10.1007/s10620-015-3977-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bjursell M, et al. Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH. PLoS One. 2013;8(5):e64721. doi: 10.1371/journal.pone.0064721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong B, et al. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;328(1):116–22. doi: 10.1124/jpet.108.144600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szabo G, Iracheta-Vellve A. Inflammasome activation in the liver: Focus on alcoholic and non-alcoholic steatohepatitis. Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S18–23. doi: 10.1016/j.clinre.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 19.Miura K, et al. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57(2):577–89. doi: 10.1002/hep.26081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivera CA, et al. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47(4):571–9. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miura K, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139(1):23–34 e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Csak T, et al. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014;34(9):1402–13. doi: 10.1111/liv.12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YG, et al. The involvement of NLRX1 and NLRP3 in the development of nonalcoholic steatohepatitis in mice. J Chin Med Assoc. 2013;76(12):686–92. doi: 10.1016/j.jcma.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Ganz M, et al. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J Transl Med. 2015;13:193. doi: 10.1186/s12967-015-0552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magee N, Zou A, Zhang Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed Res Int. 2016;2016:5170402. doi: 10.1155/2016/5170402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutti S, et al. Is there a role for adaptive immunity in nonalcoholic steatohepatitis? World J Hepatol. 2015;7(13):1725–9. doi: 10.4254/wjh.v7.i13.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutti S, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59(3):886–97. doi: 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- 28.Leroux A, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57(1):141–9. doi: 10.1016/j.jhep.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YD, et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48(5):1632–43. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daly C, Rollins BJ. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: therapeutic opportunities and controversies. Microcirculation. 2003;10(3–4):247–57. doi: 10.1038/sj.mn.7800190. [DOI] [PubMed] [Google Scholar]
- 32.Ito M, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37(1):50–7. doi: 10.1111/j.1872-034X.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 33.Haukeland JW, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44(6):1167–74. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 34*.Li L, et al. Activation of farnesoid X receptor downregulates monocyte chemoattractant protein-1 in murine macrophage. Biochem Biophys Res Commun. 2015;467(4):841–6. doi: 10.1016/j.bbrc.2015.10.056. This manuscript provides a direct role of FXR in the adpative immune response and anti-inflammatory pathways with the down-regulation of Mcp-1. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S, et al. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51(2):380–8. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 36.Pathil A, et al. Ursodeoxycholyl lysophosphatidylethanolamide improves steatosis and inflammation in murine models of nonalcoholic fatty liver disease. Hepatology. 2012;55(5):1369–78. doi: 10.1002/hep.25531. [DOI] [PubMed] [Google Scholar]
- 37.Balasubramaniyan N, Ananthanarayanan M, Suchy FJ. Nuclear factor-kappaB regulates the expression of multiple genes encoding liver transport proteins. Am J Physiol Gastrointest Liver Physiol. 2016;310(8):G618–28. doi: 10.1152/ajpgi.00363.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim MS, et al. Repression of farnesoid X receptor during the acute phase response. J Biol Chem. 2003;278(11):8988–95. doi: 10.1074/jbc.M212633200. [DOI] [PubMed] [Google Scholar]
- 39.Kemper JK, et al. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 2009;10(5):392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J, et al. Genomic analysis of hepatic farnesoid X receptor binding sites reveals altered binding in obesity and direct gene repression by farnesoid X receptor in mice. Hepatology. 2012;56(1):108–17. doi: 10.1002/hep.25609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41*.Kim DH, et al. A dysregulated acetyl/SUMO switch of FXR promotes hepatic inflammation in obesity. EMBO J. 2015;34(2):184–99. doi: 10.15252/embj.201489527. The manuscript demonstrates that acetylation/sumolyation of FXR alters transcriptional pathways related to inflammation. SUMO-modified FXR is identified to be involved in the transrepression of NF-κB and inhibits pro-inflammatory gene expression thus showing FXR has a role in anti-inflammation. This created a direct connection between FXR, NF-κB, and inflammation, which supports emerging research in FXR protection against inflammation associated with NASH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruys E, et al. Acute phase reaction and acute phase proteins. J Zhejiang Univ Sci B. 2005;6(11):1045–56. doi: 10.1631/jzus.2005.B1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoneda M, et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J Gastroenterol. 2007;42(7):573–82. doi: 10.1007/s00535-007-2060-x. [DOI] [PubMed] [Google Scholar]
- 44.Ndumele CE, et al. Hepatic steatosis, obesity, and the metabolic syndrome are independently and additively associated with increased systemic inflammation. Arterioscler Thromb Vasc Biol. 2011;31(8):1927–32. doi: 10.1161/ATVBAHA.111.228262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang S, et al. Suppression of interleukin-6-induced C-reactive protein expression by FXR agonists. Biochem Biophys Res Commun. 2009;379(2):476–9. doi: 10.1016/j.bbrc.2008.12.117. [DOI] [PubMed] [Google Scholar]
- 46**.Porez G, et al. The hepatic orosomucoid/alpha1-acid glycoprotein gene cluster is regulated by the nuclear bile acid receptor FXR. Endocrinology. 2013;154(10):3690–701. doi: 10.1210/en.2013-1263. This is the first publication to demonstrate that members in the lipocalin gene family are putative FXR target genes, specifically Lcn13 was the most downregulated gene in FXR KO mice and Orm3 was the most up-regulated gene in FXR KO mice. This manuscript supports further research into FXR-mediated regulation of lipocalins involved in the acute phase response. [DOI] [PubMed] [Google Scholar]
- 47.Zhan L, et al. Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS One. 2014;9(9):e105930. doi: 10.1371/journal.pone.0105930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48*.Ye D, et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol. 2016;65(5):988–997. doi: 10.1016/j.jhep.2016.05.041. This manuscript demonstrates the direct role of lipocalin 2 in NASH, and mechanistically supports previous publications with obeservations of the role of lipocalin 2 in NASH of murine and human models. In addition, it demonstrates the importance of acute phase proteins in inflammation related to NASH. [DOI] [PubMed] [Google Scholar]
- 49.Auguet T, et al. Liver lipocalin 2 expression in severely obese women with non alcoholic fatty liver disease. Exp Clin Endocrinol Diabetes. 2013;121(2):119–24. doi: 10.1055/s-0032-1331696. [DOI] [PubMed] [Google Scholar]
- 50.Semba T, et al. The FLS (fatty liver Shionogi) mouse reveals local expressions of lipocalin-2, CXCL1 and CXCL9 in the liver with non-alcoholic steatohepatitis. BMC Gastroenterol. 2013;13:120. doi: 10.1186/1471-230X-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wieser V, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol. 2016;64(4):872–80. doi: 10.1016/j.jhep.2015.11.037. [DOI] [PubMed] [Google Scholar]
- 52.Cai Y, et al. The Detrimental Role Played by Lipocalin-2 in Alcoholic Fatty Liver in Mice. Am J Pathol. 2016;186(9):2417–28. doi: 10.1016/j.ajpath.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki K, et al. Molecular evolution of epididymal lipocalin genes localized on mouse chromosome 2. Gene. 2004;339:49–59. doi: 10.1016/j.gene.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 54.Cho KW, et al. Lipocalin-13 regulates glucose metabolism by both insulin-dependent and insulin-independent mechanisms. Mol Cell Biol. 2011;31(3):450–7. doi: 10.1128/MCB.00459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheng L, et al. Lipocalin 13 protein protects against hepatic steatosis by both inhibiting lipogenesis and stimulating fatty acid beta-oxidation. J Biol Chem. 2011;286(44):38128–35. doi: 10.1074/jbc.M111.256677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodwin B, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–26. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe M, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. Journal of Clinical Investigation. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen F, et al. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem. 2003;278(22):19909–16. doi: 10.1074/jbc.M207903200. [DOI] [PubMed] [Google Scholar]
- 59.Smalling RL, et al. Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am J Physiol Gastrointest Liver Physiol. 2013;305(5):G364–74. doi: 10.1152/ajpgi.00077.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Myronovych A, et al. The role of small heterodimer partner in nonalcoholic fatty liver disease improvement after sleeve gastrectomy in mice. Obesity (Silver Spring) 2014;22(11):2301–11. doi: 10.1002/oby.20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neuschwander-Tetri BA, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–65. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016;15(4):249–74. doi: 10.1038/nrd.2015.3. [DOI] [PubMed] [Google Scholar]
- 63.Pathil A, et al. The synthetic bile acid-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide suppresses TNFalpha-induced liver injury. J Hepatol. 2011;54(4):674–84. doi: 10.1016/j.jhep.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 64.Kim JK, et al. Omega-3 polyunsaturated fatty acid and ursodeoxycholic acid have an additive effect in attenuating diet-induced nonalcoholic steatohepatitis in mice. Exp Mol Med. 2014;46:e127. doi: 10.1038/emm.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]