

Abstract
Single-molecule protein sequencing is regarded as a promising new method in the field of proteomics. It potentially offers orders of magnitude improvements in sensitivitiy and throughput for protein detection when compared to mass spectrometry. However, the development of such a technology faces significant barriers, especially in the need to chemically derivatize specific amino-acid types with unique labels. For example, fluorescent dyes would be suitable for single-molecule microscopy or nanopore-based sequencing. These emerging single-molecule protein-sequencing technologies suggests a need to develop an amino acid side chain-selective modification scheme that could target several side chains of interest. Current work for modifying residues focuses mainly on one or two side chains. The need to label many side chains, as recent computational modeling suggests, is required for high protein, sequencing coverage of the human proteome. Herein, we report our stragety for modifying two model peptides KYDWEC and KDYWE containing the most reactive residues, using highly opitmized mass labels in a sequential and selective fashion both using solution-phase and solid-phase chemistries, respectively. This will serve as a step towards a modification scheme appropriate for single-molecule studies.
TOC image
Selective modification of side chains on a model peptide was achieved for studies necessary for emerging single-molecule peptide sequencing technologies.
Introduction
Recent theoretical work on single-molecule peptide or protein sequencing suggests that modifying proteins with amino acid side chain specific labels, and determining the order of the subset of modified amino acids (e.g. via single molecule fluorescence microscopy in combination with Edman sequencing1 or nanopore-based sequencing2) can be sufficient to identify proteins in complex mixtures at the single molecule level. Such a technology offers the potential for multiple orders of magnitude improvements in sensitivity and throughput over existing approaches, but still faces barriers for its practical implementation. A key barrier is the development of a side-chain specific labeling scheme capable of modifying multiple amino acid types with specific and detectable labels, such as residue-specific fluorophores. In such a scheme, only the positions of the fluorescent amino acids are determined for a peptide or protein, which is then identified by comparison with the sequences expected based upon that organism’s known genome sequence. Thus, more amino acid-specific modifications translate directly into richer sequence information about a given peptide or protein, ultimately allowing for greater coverage of that organism’s proteome. Computational modelling suggests that schemes incorporating modifications to cysteine, lysine, tryptophan, aspartic, and glutamic acid residues can in principle provide a high coverage of the human proteome.1
Fortunately, work developed for mass spectrometry proteomics has provided optimized chemistries that selectively target residues for modification. These techniques have gained widespread use and are employed to understand biological processes such as expression, post-translation modifications, and protein interactions. These labelling chemistries are highly regarded for their efficiency and low cross-reactivity. Side chain specific protocols are routinely used to tag and modify proteins with high selectivity, where little to no cross-reactivity has previously been established.3 For example, kits are commercially available for targeting cysteine and lysine. The cysteine is modified with an iodoacetamide, followed in the same-pot by selective modification of lysine using O-methylisourea hemisulfate.4 Acylation and reductive alkylation are also employed to modify both Nε-amines and N-termini.5 Cross-modification of threonine, serine, and tyrosine can occur with acylation and alkylating conditions.6 Recently, peptide amines have been modified via reductive methylation preventing cross-reactivity with alcohol and phenol residues. Once these amines were modified, the Smith group achieved global modification of aspartate and glutamate via amidation with amine-containing compounds. Furthermore, studies with less abundant side chains have been explored. For example, Horton, Koshland, and Scoffone demonstrated the modification of tryptophan under acidic conditions using 2-hydroxy-5-nitrobenzyl bromide and dinitrophenylsulfenyl chloride.7–9 More recently, studies of the N-terminus using pyridinecarboxyaldehydes have expanded the chemical repertoire available for selective targeting of functional groups.10 However, missing in these studies is a route integrating all these selective modifications into a sequential protocol. Such a route, taking advantage of these techniques, could have potential applications not only in these emerging single-molecule technologies, but also in protein/peptide mass spectrometry studies. In addition, devising a generalized sequential route consisting of the selective modification of side chains also has applications for synthetic peptide and protein design.11–13 Herein, we describe studies that put this sequential strategy described in Figures 1 and 3 into practice. As described, a series of selective modification steps for KDYWEC (1) and KDYWE (2) was achieved. Modification studies were initially performed in solution-phase targeting cysteine, lysine, the N-terminus, aspartic, glutamic acid, and tryptophan. For ease of identification and purification solution-phase studies were performed. These chemistries were transitioned to the solid-phase. Both peptides proved to be suitable models for the ultimate use in single-molecule peptide/protein sequencing.
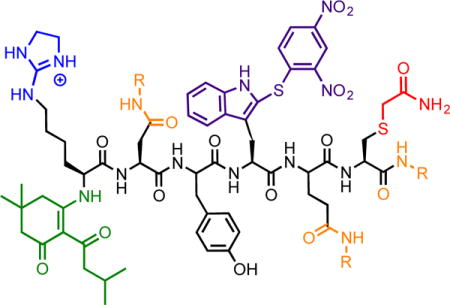
Figure 1.
Selective modification route in solution. (a) – (b) Labeling of cysteine and lysine are done consecutively in the same vial. (c) Labeling of N-terminums occurs with 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl diethyl phosphate formed in situ. (d) Labeling of carboxylates were done using three different amines. (e) Labeling of tryptophan.
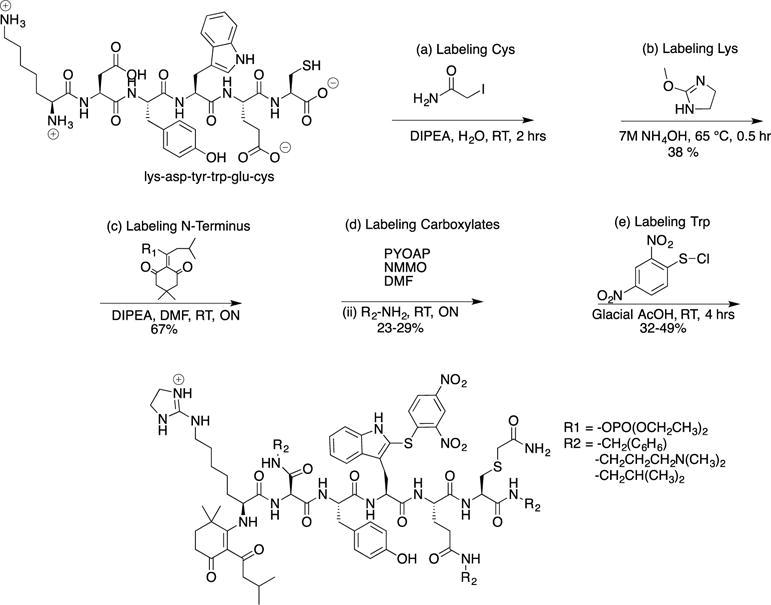
Figure 3.
Solid-phase modification of KDYWE. (a) Modification of lysine was done similarly for immobilized KDYWEC. (b) Two repetitions were performed to drive reaction to completion. (c) Modification of tryptophan. (d–;e) Cleavage with water releases C-terminus as an acid. (f) Cleavage with amine functionalized C-terminus with an alkyne.
Results and Discussion
We figured that global modification of several amino acids of different classes should be possible if the appropriate conditions and the right sequence of derivatization steps were used. For example, by using iodoacetamide, guanidination reagents, amide-coupling reagents, and aromatic-targeting electrophilic reagents, we may selectively functionalize cysteine, lysine, aspartic and glutamic acids, and tryptophan, respectively, if used in the proper protocol. Minimizing cross-reactivity between each step was to be achieved if the nucleophilicity and pKa of each side chain, as well as the reactivity of modification of reagents, reactions times, and temperature are all considered. Good nucleophiles, such as the thiol in cysteine or the amine in lysine, and the N-terminus, should be targeted first. Selective modification of cysteine between pH 7–8 is possible, while modification of amines is possible at a higher pH.14 Because guanidinating reagents are selective for amines, distinguishing between the Nε-amine of lysine and the terminal α-amine was possible. As described herein, a different modification of reagent is required for each kind of amine. Once the most nucleophilic sites were modified, we tested if the carboxylate side chains could then be targeted followed finally by modification of tryptophan.
Characterization of the resulting peptides in nearly all these examples is performed only with HPLC for purity and mass spec for identity due to the minimal quantity of material produced. As is the case with the studies reported herein, the quantity of material is not amenable to 1H and/or 13C NMR spectroscopy. Our goals herein were to create a protocol for sequential modification of numerous side chains using known selective derivitizations, and to verify that each gave the expected products after step-by-step linear implementation.
In the experimental design we implement here, we are exploiting reagents that have already been proven to have high selectivity for their individual amino acid side chains. Our goal was to direct the derivitizations by modifying experimental conditions and then use each derivitization step sequentially, which, to our knowledge, has not been previously accomplished. The order of steps in Figure 1 took into consideration the nucleophilicity and acid/base-dependent reactivity of the target side chains in KDYWEC. This peptide was synthesized to contain the most reactive natural amino acids. The sulfhydryl group in cysteine is the most nucleophilic, and is prone to oxidation and disulfide bond formation. To ensure selectivity in future modification steps, cysteine was first alkylated with iodoacetamide, forming a stable thioether. Maintaining a pH between 7–8 ensured the amines remained protonated, thus limiting the possibility of undesired alkylation. Subsequently, the pH was raised to 11 and 2-methylthio-2-imidazoline hydroiodide (MDI) was introduced. Modification of the Nε-amine occurred in 24 minutes when heated to 50 °C. Longer reaction times increased the extent of N-terminal modification. The Cys and Lys modification steps were performed in one-pot. The yield of peptide 3 after purification was 38%. Because the lysine was modified while heating under basic conditions, the thioether and guanidinium group were considered to be stable in future derivatization steps.
Of the remaining nucleophilic sites, the N-terminus was first targeted. Protection of the N-terminus was required prior to modification of aspartate, glutamate, and the C-terminus. If not, concatenation of peptides could occur during amidation. The modification conditions of the N-terminus also required a group compatible to both basic and acidic conditions in subsequent derivatization steps. Literature accounts have reported using 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl to protect amines during peptide synthesis. This protecting group is stable to highly basic and acidic conditions, and is removed under hydrazinolysis conditions.15
However, refluxing overnight to efficiently add the protecting group is common. Heating overnight was undesired so as to minimize observed degradation. Thus, we devised the use of 5,5-dimethyl-2-(3-methylbutanoyl)-3-oxocyclohex-1-en-1-yl diethyl phosphate (phos-DOD) as an alternative. Diethyl phosphate was anticipated to be a better leaving group, thereby facilitating the reaction. This compound was formed with chloro diethyl phosphate in situ, followed by incubation with a basic solution of peptide 3 overnight. Post-purification the yield of peptide 4 was 67%. Protection of the N-terminus thus occurred as shown in equation eq. 1.
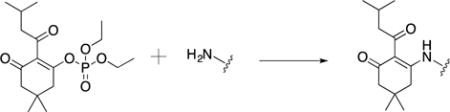 |
eq. 1 |
Once the most nucleophilic sites in the model peptide were modified, the carboxyl groups were targeted. Amidation has been used for derivatization of aspartate, glutamate, and the C-terminus.16 Unlike the modification of lysine and the N-terminus, distinguishing among these target side chains was not possible. Also, because there were three sites for reaction, an efficient modification approach was necessary. Highly efficient, global modification using (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) and N-methylmorpholine (NMM) has been reported.6 Peptide 5, amidated with benzylamine dissolved in MeCN/H2O mixtures, and purification of the resulting modified peptides was possible. Yield for peptide 5 was 29%. To improve the solubility of the model peptide, 3-dimethylaminopropylamine (DMAPA) was used to increase the overall positive charge under acidic conditions. Purity checks of the peptide demonstrated an approximate 10-minute difference in elution when comparing peptide 5 and 6. For peptide 7, isobutylamine was explored as an alternative, believed to form a peptide less hydrophobic than peptide 5, but more hydrophilic than peptide 6. Peptide 7 readily dissolved in MeCN/H2O mixtures, but coeluted with an impurity characterized by LCMS as m/z 313.4. The impurity was removed after synthesizing peptide 8.
Tryptophan was the remaining target. As a less abundant amino acid in nature, the ability to identify this side chain can be informative for determining the protein origin of peptides in proteomic studies.17 In synthetic peptide design, incorporating an additional site for derivatization increases the repertoire of side chains to modify. Therefore, devising a selective modification strategy incorporating modification of tryptophan was seen as important. Cysteine reacting with sulfenyl chlorides has been reported.18 As with the other modification steps, modification of cysteine prior to that of tryptophan was chosen. However, under acidic conditions the tryptophan could be selectively targeted in the presence of unprotected N-terminus and lysine. The advantage to modification of the tryptophan last was the relative ease of the reaction. Peptides 5 and 6 readily dissolved in glacial acetic acid, and the reaction occurred in 4 hrs at RT. 2,4-Dinitrobenzenesulfenyl chloride (DBSC) was a chromophore and the peptides could also be monitored at 330 nm (SI). Yields were 32% and 49% for peptides 8 and 9, respectively. The ultimate application in single molecule sequencing of our derivitization protocol will employ immobilization of the peptides on solid supports, and subsequent attachment of the peptides to surfaces via the thiols on cysteines. Thus, in a protocol that employs solid supports for derivitization, cysteines do not need to be derivitized, because they will be the points of attachment to the surface. Hence, we removed the cysteine of the peptide we studied in solution, resulting in KDYWE, for our solid-phase synthesis studies.
Efforts to modify the amino acids on solid phase supports were explored after the sequence of targeting side chains had been successfully demonstrated in solution. Synthetic peptides have been commonly modified when immobilized on a solid support, usually at reactive side chains such as lysine.19 Requirements for successful solid-phase reactions include making sure each step is highly selective. Further, the reagents must be able to diffuse into the resin to reach sites for reaction. A high concentration of starting material in the bulk solution ensures a concentration gradient is formed for reactants to diffuse.20 Inherent in this study was devising an approach that selectively modified target side chains in a sequential fashion. Therefore, high specificity was required. Literature, and the work presented here, has demonstrated that excess reagent can be used while maintaining selectivity. The final requirement for solid-phase studies was using a resin that would not cleave with acid or base. 4-Fmoc-hydrazinobenzoyl resin AM was selected, because literature accounts describe the stability towards strong acids and bases. Peptides immobilized on this resin were isolated after oxidative cleavage with Cu(II) and base.21–22
Figure 3 summarizes the modification of reactions performed on the solid support for peptide KDYWE. The first side chain targeted was the lysine. Two changes were made from the solution approach. The reaction time was longer. The immobilized peptide was incubated overnight with MDI. A solution of MeOH/DIPEA/H2O (7:2:1) (v/v/v) was used instead of a solution of NH4OH. Overnight incubation and the use of DIPEA have been reported in the literature.23 The doubly modified peptide was not observed after an overnight reaction at RT, however, extending the reaction time to 48 hrs led to doubly modified peptide. Selectivity for the Nε-amine can be explained due to inductive and steric effects. The Nε amine in lysine is part of a hydrocarbon side chain and not adjacent to an electron-withdrawing amide group, and thus, the lysine side chain amine is more nucleophilic than the α-amine. Furthermore, the N-terminal amine is closer to the amide backbone, impeding MDI due to sterics. The same inductive and steric effects played a role when modifying KDYWEC in solution phase. However, lowering the reaction temperature from 60°C to RT made these effects more pronounced. A protection step of the N-terminus was not performed. One reason was to discover if in the presence of excess amine, the carboxylates would be modified without concatenation to this terminal amine. A second goal was to check if the number of modification of steps could be reduced, leaving the terminal-amine unmodified for future reactions. This approach could provide synthetic flexibility by diversifying the kinds of reactions performed at the N-terminus once the peptide is cleaved from the resin. The amine used in solid-phase synthesis differed from that of the solution-phase studies. 1-Amino-3-butyne had an alkyne group that could also provide sites for derivatization via Huigen-Sharpless.24 The same coupling reactants PyAOP and NMM were employed for solid-phase studies. Two repetitions ensured all carboxylates were modified. Cleavage of the peptide was performed using a catalytic amount of Cu(II) and a mixture of MeCN/H2O/Pyr. To a different batch of resin, the lysine and carboxylates were also modified. Tryptophan was modified in a similar fashion as in solution, four hours at RT.25
Two different cleavage conditions were tested for the model peptide after the target side chains were modified. The first condition was water, liberating a carboxylate at the C-terminus. Peptide 11 was thereby isolated with a HPLC purified yield of 4 %. Additionally, a nonaqueous condition in the presence of a nucleophile could also be employed to cleave the peptide. 1-Amino-3-butyne was the nucleophile used, liberating peptide 12 with an HPLC purified yield of 5 %. The peptide could also have been cleaved with a different nucleophile diversifying the functional groups, further differentiating between the C-terminus and carboxylate side chains. Isolating peptide 12 required extra washes with DMF, because solubility in H2O/MeCN was reduced once an alkyne was introduced at the C-terminus. Initially, the peptide was rinsed with MeCN, and LCMS data of the extract did not indicate the presence of desired product. Once rinsed with DMF and the solvent removed, peptide 12 was observed.
Conclusion
A sequential and selective scheme using common mass-modifications was developed for derivatizing peptides as a model for ultimate use in single-molecule sequencing studies, helping to overcome a key barrier identified by previous computational models of single molecule protein sequencing.1,2 Selective targeting of side chains in KDYWEC was achieved in solution-phase. Most of the side chains, as well as the N-terminus and C-terminus were modified. No heating was required to target N-terminal amine with 4,4-dimethyl-2,6-dioxocyclohex-1-ylidene when using Phos-DOD. Selective modification also occurred in solid-phase studies for KDYWE. Modification of all the target side chains was possible while omitting any reaction at the α-amine. Oxidative cleavage of the resin provided flexibility to choose between releasing modified or unmodified C-terminus. The use of 1-amino-3-butyne as the carboxylate-modification of reagent introduced further functionality that could be exploited in future reactions. Such an approach can also have applications for peptide modification studies and novel synthetic peptide design. Other tags, like fluorescent probes, can be designed to have the same functional handles presented in this paper. Work on the design, synthesis, and selective modification of peptides using these dyes will be discussed in future publications.
Methods
General
For automated, Fmoc amino solid-phase peptide synthesis, OtBu (Asp, Glu), Boc (Lys, Trp), tBu (Tyr) were used. Fmoc-protected amino acids were purchased from Novabiochem (USA) and AAPPTec (USA). Fmoc-Cys(Trt)-Wang resin (100–200 mesh) and 4-Fmoc-hydrazinobenzoyl resin AM Novagel™ were purchased from Novabiochem (USA). Other chemicals used for automated, solid-phase peptide synthesis were purchased from Fisher Scientific and Sigma-Aldrich. Reagents used for selective modification of studies were iodoacetamide (IA), 2-methylthio-2-imadazoline hydroiodide (MDI), sodium methoxide, diethylchlorophosphate, 2-(3-Methylbutyryl)-5,5-dimethyl-1,3-cyclohexandione, benzylamine (BA), isobutylamine, 3-dimethylaminopropylamine (DMAPA), 1-amino-3-butyne (AB), (7-Azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP), N-methylmorpholine (NMM), and 2,4-Dinitrobenzenesulfenyl chloride (DBSC). All chemicals were purchased from Sigma-Aldrich.
A Prelude peptide synthesizer (Protein Technologies, Inc.) was used for automated-solid phase synthesis. Preparative HPLC purification of peptides was performed using an Agilent Zorbax SB-C18 Prep HT column 21.2 × 250 mm; 10 ml/min, 5–95% MeCN (0.1% TFA) in 90 min. Analytical HPLC characterization of peptides was performed using an Agilent Zorbax column 4.6 × 250 mm; 1 ml/min, 5–95% MeCN (0.1 % TFA) in 40 min (RT). An Agilent Technologies 6530 Accurate Mass QTofLC/MS was used for high-resolution mass spectra of purified peptides. Solvents used were HPLC grade.
KDYWEC was synthesized using Fmoc-Cys(Trt)-Wang Resin (0.57 mmole/g, 100 μmole) by sequential coupling of Nα-Fmoc-amino acid (0.1 M, 1.5 ml) in DMF in the presence of N,N,N,N-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 0.15 M, 1.0 ml) and DIPEA (0.2 M, 0.5 ml) with a reaction time of 30 minutes at room temperature. A total of three repetitions were performed for each amino acid building block. DMF (3 ml, 3 min, 3×) and DCM (3 ml, 3 min, 3×) washes were done before each repetition. After incorporation of the third amino acid, a 0.8 M LiCl wash step was performed after swelling with DCM (3 ml, 3 min, 3×). Post synthesis, resin was washed with glacial AcOH (5 ml, 3×), DCM (5 ml, 3×), and MeOH (5 ml, 3×). The resin was placed under vacuum overnight. The peptide was cleaved from the resin using trifluoroacetic acid (TFA), triisopropylsilane (TIS), 1,2-ethanedithiol (EDT), and nanopure water (94: 1.0: 2.5: 2.5), and precipitated with diethyl ether at 0°C. No HPLC purification of the crude peptide was necessary. KDYWE was synthesized using 4-Fmoc-hydrazinobenzoyl resin AM Novagel™. TFA, TIS, and nanopure water were used (95: 2.5: 2.5) to deprotect the side chains, and the peptide remained immobilized on the solid support.
Solution-phase Modification Studies of KDYWEC
Modification of cysteine with iodoacetamide
Peptide 1 (75 μmole) was dissolved in 0.4 ml of nanopure water. A solution consisting of 0.37 ml of MeOH/Pyr/TEA/nanopure H2O (7/1/1/1) (v/v/v/v) was introduced (adjusting to pH 8), followed by addition of iodoacetamide (97 μmole). The reaction was incubated for 2 hrs at RT.
Modification of lysine with 2-methoxy-4,5-dihydro-1H-imidazole (3)
In the same pot, 0.5 ml of a 7 N solution of NH4OH was added, followed by introduction of MDI (SI) (750 μmole). The reaction mixture was incubated for 24 mins at 65°C, followed by introduction of TFA (0.3 ml) at 0°C. The crude peptide was prepared for preparative HPLC using an Extract Clean™ C18 500 mg /4 ml solid phase extraction column (SI). The peptide was purified using preparative HPLC, and the organic solvent in the peptide fraction was removed via rotary evaporation. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (29 μmole) 38%. High-res MS: found m/z 968.39360, calcd. 968.39310 (M+H)+; found m/z 966.37880, calcd. 966.37850 (M-H)−.
Modification of the N-terminus with 5,5-dimethyl-2-(3-methylbutanoyl)-3-oxocyclohex-1-en-1-yl diethyl phosphate (Phos-DOD) (4)
Peptide 3 (12 μmole) was dissolved in 0.1 ml of nanopure water, followed by dilution with 0.2 ml of MeCN. To the solution, 0.12 ml of 7/2/1 MeOH/TEA/H2O (v/v/v) was introduced. A solution of Phos-DOD (SI) (18 μmole) was introduced. The solution was incubated overnight at RT. The peptide was purified using preparative HPLC. Organic solvent in peptide fraction was removed via rotary evaporator. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (8 μmole) 67%. High-res MS: found m/z 1174.52380, calcd. 1174.52380 (M+H)+; found m/z 1172.50750, calcd. 1172.50920 (M-H)−.
Modification of the carboxylate side chains and C-terminus with benzylamine (BA) (5)
Peptide 4 (51 μmole) was dissolved in 0.2 ml of 3/1 MeOH/H2O (v/v). In a separate vial, benzylamine (1.3 mmole) was dissolved in 0.1 ml of MeCN, followed by addition of NMM (1.0 mmole). The BA/NMM solution was introduced to the peptide solution, followed by addition of solid PyAOP (0.51 mmole) and anhydrous HOBt (0.56 mmole). 0.1 ml of MeCN was introduced to improve the solubility of PyAOP/HOBt. The solution was incubated for a total of 4 hrs at RT. The peptide was purified using preparative HPLC. The organic solvent in the peptide fraction was removed via rotary evaporation. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purifed yield: (15 μmole) 29%. High-res MS: found m/z 1441.71230, calcd. 1441.71260 (M+H)+; found m/z 1439.69600, calcd. 1439.69800 (M-H)−.
Modification of the carboxylate side chains and C-terminus with 3-dimethylaminopropylamine (6)
Peptide 4 (11 μmole) was dissolved in 0.2 ml of dry DMF. DMAPA (1.6 mmole) and NMM (1.4 mmole) were combined in a separate vial. The amine/NMM solution was introduced to the peptide solution, followed by addition of solid PyAOP (1.9 mmole). The solution was incubated for 24 hrs at RT. The sample was placed in a centrifugal evaporator for 21 hrs at 35°C. The resulting oil was dissolved in 1.5 ml of 2/1 H2O/DMF (v/v), and purified by prep HPLC. The organic solvent in the peptide fraction was removed via rotary evaporation. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (2.4 μmole) 23%. High-res MS: found m/z 1426.83900, calcd. 1426.83920 (M+H)+.
Modification of the carboxylate side chains and C-terminus with isobutylamine (7)
Isobutylamine (60 μmole) and NMM (excess) were combined in a separate vial to make 0.1 ml solution in DMF. Amine/NMM solution was introduced to peptide 4 (20 μmole), followed by introduction of solid PyAOP. The solution was incubated for 3 hrs at RT, followed by quenching with 1 ml of H2O. The solution was placed in centrifugal evaporator for 14 hrs at 35°C. The residual oil was dissolved in 1.5 ml of 1/1 H2O/MeCN (v/v) and purified via prep HPLC. An impurity and the desired compound both eluted at the same time. The peptide was therefore taken directly to the subsequent modification of tryptophan without further purification.
Modification of tryptophan in peptide 6 (8)
Peptide 6 (19 μmole) was dissolved in 1 ml of glacial acetic acid, followed by introduction of 2,4-dinitrobenzenesulfenyl chloride (57 μmole). The reaction was shaken for 4 hrs at RT. Glacial acetic acid was removed by rotary evaporation. The residual film was dissolved in 1/1 MeCN/H2O (v/v), and purified via preparative HPLC. The organic solvent in the peptide fraction was removed via rotary evaporation. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (6.4 μmole) 32%. High-res MS: found m/z 812.91050, calcd. 812.91000 (M+2H)2+; found m/z 1622.79650, calcd. 1622.79810 (M-H)−.
Modification of tryptophan in peptide 7 (9)
Peptide 7 (6.2 μmole) was dissolved in 1 ml of glacial acetic acid, followed by introduction of 2,4-dinitrobenzenesulfenyl chloride (19 μmole). The reaction was shaken for 4 hrs at RT. The peptide was purified using preparative HPLC. The organic solvent in the peptide fraction was removed via rotary evaporator. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (6.4 μmole) 49%. High-res MS: found m/z 769.37050, calcd. 769.37020 (M+2H)2+; found m/z 1535.71420, calcd. 1535.71850 (M-H)−. B Headings should always be subordinate to A headings e.g. Synthetic procedures, Materials and methods, Crystallography.
Solid-phase Modification Studies of KDYWE
Before and after each modification step, the resin was washed with DMF and DCM (3 mL, 3 mins, 3×). Resins were placed under high vacuum overnight before cleavage at each step. Cu(II)Ac (0.3 mmole) was dissolved in 3 ml 45/45/10 MeCN/H2O/Pyr (v/v/v). The copper acetate solution was introduced to the dried resin and incubated for 4 hrs at RT to cleave peptide. This solution was removed from the resin and collected, followed by washing with 1/1 MeCN/H2O (v/v) (1 ml, 3 mins, 3×); washes were collected.
Modification of the lysine with 2-methoxy-4,5-dihydro-1H-imidazole in 2
To the swollen resin (130 mg, 0.66 mmole g−1), 3 ml of a 200 mM solution of 2-methoxy-4,5-dihydro-1H-imidazole in 7/2/1 MeOH/DIPEA/H2O (v/v/v) was added. The resin was incubated overnight at RT. The peptide was cleaved from the resin using copper acetate solution, and the MeCN and pyridine were removed by rotary evaporation. The remaining aqueous solution was frozen at −78°C and lyophilized overnight. The resulting solid was dissolved in 1.5 ml of 1/1 MeCN/H2O (v/v) and purified by prep HPLC. The organic solvent in the peptide fraction was removed via rotary evaporator. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (1.4 μmole) 2%. High-res MS: found m/z 910.45740, calcd. 910.45700 (M+H)+; found m/z 908.44300, calcd. 908.44240 (M-H)−.
Modification of the carboxylates and C-terminus (10)
1-Amino-3-butyne (0.61 mmole) was dissolved in NMM (0.45 mmole), and the mixture was diluted with 1 ml of DMF. PyAOP (0.40 mmole) was separately dissolved in 2 ml DMF. The amine/NMM solution was introduced to the resin, followed by introduction of the PyAOP solution. The resin was incubated overnight at RT, followed by rinsing with MeOH (3 ml, 3mins, 3×). The peptide was cleaved with 55 μmole of Cu(OAc)2, and the MeCN and pyr were removed by rotary evaporation. The remaining aqueous solution was frozen at −78°C and lyophilized overnight. The solid was dissolved in 1.5 ml of 1/1 MeCN/H2O (v/v) and purified by prep HPLC. The organic solvent in the peptide fraction was removed via rotary evaporation, and aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (1.4 μmole) 2%. High-res MS: found m/z 910.45740, calcd. 910.45700 (M+H)+; found m/z 908.44300, calcd. 908.44240 (M-H)−.
Tryptophan modification of immobilized peptide (11)
Immobilized peptide 10 was prepared as described using 193 mg of the same resin. 2,4-Dinitrobenzenesulfenyl chloride (0.30 mmole) was dissolved in 3 ml of glacial acetic acid. This solution was introduced to the swollen resin, and incubated for 4 hrs at RT. The solution was removed from the resin and 6 ml of DMF was passed through the resin.
Cleavage of peptide 11 from hydrazinobenzoyl resin using H2O
Cleavage of the peptide was performed as described with copper acetate (0.3 mmole) dissolved in 3 ml of 45/45/10 MeCN/H2O/Pyr (v/v/v). MeCN and pyr were removed by rotary evaporation, and the remaining aqueous solution was frozen at −78°C and lyophilized overnight. The solid was dissolved in 1.5 ml of 1/1/MeCN/H2O (v/v) and purified by prep HPLC. The organic solvent in the peptide fraction was removed via rotary evaporation. Aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (5.4 μmole) 4%. High-res MS: found m/z 1108.42840, calcd. 1108.43050 (M+H)+; found m/z 1106.41400, calcd. 1106.41600 (M-H)−.
Cleavage of peptide 12 from hydrazinobenzoyl resin using 1-amino-3-butyne
Copper acetate (0.33 mmole) was dissolved in 3 ml of 9/8.3/1.6 MeCN/Pyr/1-amino-3-butyne (v/v/v). Subsequently, the solution was introduced to swollen resin. The resin was incubated for 4 hrs at RT, followed by filtration to collect the solution, and the MeCN and pyridine were removed by rotary evaporation. Collected washes of the resin with DMF (3 ml, 3 mins, 3×) were used to improve the solubility of the peptide. The solvent was removed by centrifugal evaporation (35 °C, 24 hrs). The solid was dissolved in 1.5 ml of 1/1 MeCN/H2O (v/v) and purified by prep HPLC. The organic solvent in the peptide fraction was removed via rotary evaporation and the aqueous remnants were frozen at −78°C and lyophilized overnight. Purified yield: (5 μmole) 5%. High-res MS: found m/z 1159.47250, calcd. 1159.47780 (M+H)+; found m/z 1157.46220, calcd. 1157.46330 (M-H)−.
Supplementary Material
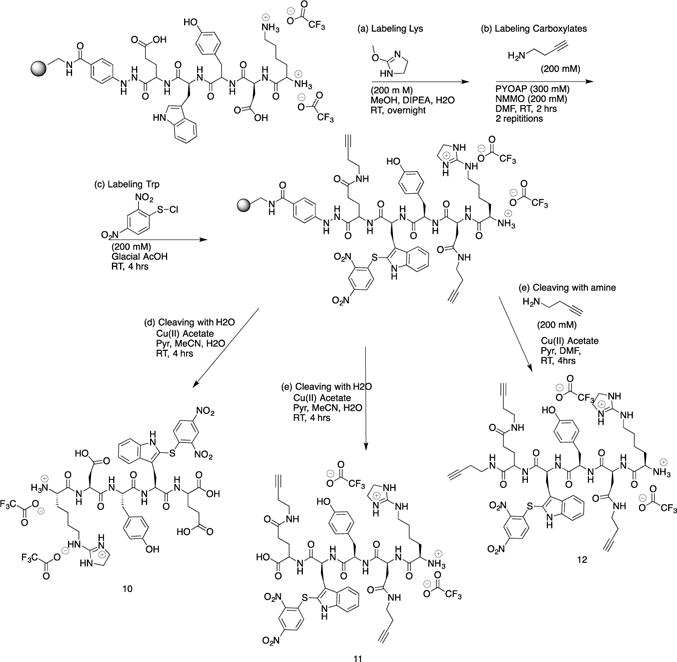
Figure 2.
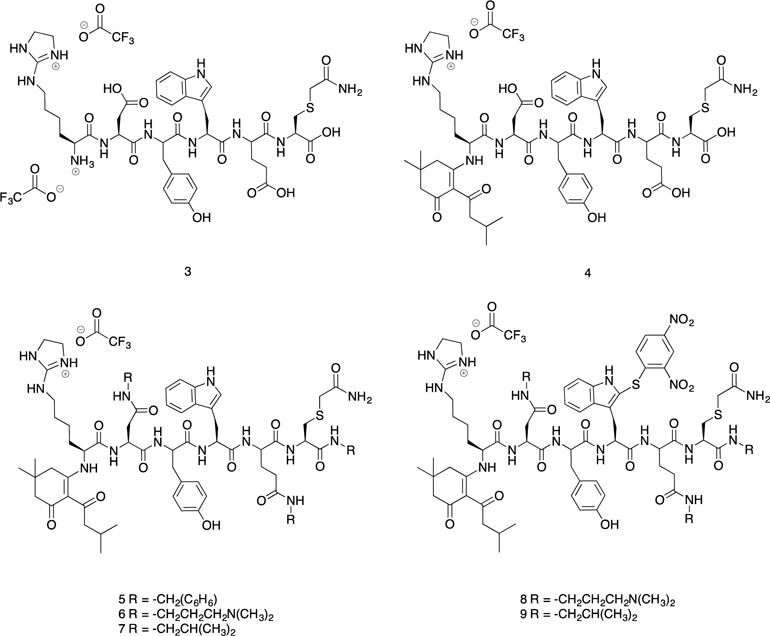
Modified model peptide intermediates for KDYWEC.
Acknowledgments
We gratefully acknowledge our funding sources: the National Institute of Health (5DP1GM106408), the Welch Foundation (F1515 to E.M.M.), the Defense Advanced Research Projects Agency (DARPA, N66001-14-2-4051), and the Welch Regents Chair to EVA (F-0046).
Footnotes
Electronic Supplementary Information (ESI) available: [HRMS, Analytical HPLC ]. See DOI: 10.1039/x0xx00000x
References
- 1.Swaminathan J, Boulgakov AA, Marcotte EM. PLoS Comp Bio. 2015;11(2):1–17. doi: 10.1371/journal.pcbi.1004080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yao Y, Docter M, Ginkel JV, Joo C. Phys Biol. 2015;12:1–6. doi: 10.1088/1478-3975/12/5/055003. [DOI] [PubMed] [Google Scholar]
- 3.Julka, Regnier F. J Proteome Res. 2004;3:350–363. doi: 10.1021/pr0340734. [DOI] [PubMed] [Google Scholar]
- 4.Cockrill SL, Foster KL, Wildsmith J, Goodrich AR, Dapron JG, Hassel TC, Kappel WK, Scott GBI. Biotech. 2005;38:301–304. doi: 10.2144/05382PT02. [DOI] [PubMed] [Google Scholar]
- 5.Frey BL, Ladror DT, Sondalle SB, Krusemark CJ, Jue AL, Coon JJ, Smith LM. J Am Mass Spectrom. 2013;24:1710–1721. doi: 10.1007/s13361-013-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krusemark CJ, Frey BL, Smith LM, Belshaw PJ. In: Gel-Free Proteomics, Methods in Molecular Biology. Gevaert K, Vandekerckhove J, editors. Vol. 753. Humana Press; New York: 2011. pp. 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horton HR, Koshland DE. J Am Chem Soc. 1965;87:1126–1132. doi: 10.1021/ja01083a033. [DOI] [PubMed] [Google Scholar]
- 8.Scoffone E, Fontana A, Rocchi R. Biochem. 1968;7:971–979. doi: 10.1021/bi00843a014. [DOI] [PubMed] [Google Scholar]
- 9.Kuyama H, Watanabe M, Toda C, Ando E, Tanaka K, Nishimura O. Rapid Commun Mass Spectrom. 2003;17:1642–1650. doi: 10.1002/rcm.1100. [DOI] [PubMed] [Google Scholar]
- 10.Macdonald JI, Munch HK, Moore T, Francis MB. Nat Chem Bio. 2015;11:326–334. doi: 10.1038/nchembio.1792. [DOI] [PubMed] [Google Scholar]
- 11.Bantscheff M, Bantscheff M, Sweetman G, Rick J, Kuster B. Anal Bioanal Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 12.Spicer CD, Davis BG. Nat Comm. 2014;5:1–14. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- 13.Krall N, da Cruz F, Boutureira O, Bernardes GJL. Nat Chem. 2015;8:1–11. doi: 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- 14.Chalker JM, Bernardes GJL, Lin YA, Davis BG. Chem Asian J. 2009;4:630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- 15.Isidro-Llobet A, Alvarez M, Albericio F. Chem Rev. 2009;109:2455–2504. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- 16.Ko BJ, Brodbelt JS. J Am Soc Mass Spectrom. 2012;23:1991–2000. doi: 10.1007/s13361-012-0458-z. [DOI] [PubMed] [Google Scholar]
- 17.Moffet JR, Namboodiri MA. Immunol Cell Biol. 2003;81:247–265. doi: 10.1046/j.1440-1711.2003.t01-1-01177.x. [DOI] [PubMed] [Google Scholar]
- 18.Scoffone E, Fontana A, Rocchi R. Biochem Biophys Res Commun. 1966;25:170–174. doi: 10.1016/0006-291x(66)90575-4. [DOI] [PubMed] [Google Scholar]
- 19.Wittman V, Seeberger S. Angew Chem Int Ed. 2000;39:4348–4352. doi: 10.1002/1521-3773(20001201)39:23<4348::AID-ANIE4348>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 20.Tulla-Puch J, Albericio F. The (classic concept of) solid support. In: Tulla-Pucha J, Albericio F, editors. The power of functional resins in organic synthesis. Wiley; Weinheim: 2008. pp. 3–14. [Google Scholar]
- 21.Millington CR, Quarell R, Lowe G. Tett Lett. 1998;39:7201–7204. [Google Scholar]
- 22.Rosenbaum C, Waldmann H. Tett Lett. 2001;42:5677–5680. [Google Scholar]
- 23.Keough T, Lacey MP, Yongquist RS. Rapid Commun Mass Spectrom. 2000;14:2348–2356. doi: 10.1002/1097-0231(20001230)14:24<2348::AID-RCM175>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 24.Tang W, Becker ML. Chem Soc Rev. 2014;43:7013–7059. doi: 10.1039/c4cs00139g. [DOI] [PubMed] [Google Scholar]
- 25.Zervas L, Borovas D, Gazis E. J Am Chem Soc. 1963;85:3660–3666. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.