

Abstract
The predominant working hypothesis of Alzheimer's disease is that the proximate pathologic agents are oligomers of the amyloid β-protein (Aβ). “Oligomer” is an ill-defined term. Many different types of oligomers have been reported and they often exist in rapid equilibrium with monomers and higher-order assemblies. This has made formal structure-activity determinations difficult. Recently, Ono et al. (Ono, K. et al. (2009) Proc Natl Acad Sci USA 106, 14745–14750) used rapid, zero-length, in situ chemical cross-linking to stabilize the oligomer state, enabling the isolation and study of pure populations of oligomers of specific order (number of Aβ monomers per assembly). This approach was successful, but highly laborious and time-consuming, precluding general application of the method. To overcome these difficulties, we developed a “continuous flow reactor” with the capability of producing theoretically unlimited quantities of chemically stabilized Aβ oligomers. We show, in addition to its utility for Aβ, that this method can be applied to a wide range of other amyloid forming proteins.
For Table of Contents Use Only
Alzheimer’s disease (AD) is the most common age-related dementia, affecting over 5.3 million in the U.S. alone(1). This number is expected to triple by 2050(2). Increasing evidence suggests that oligomeric forms of the amyloid β-protein, Aβ, may be the proximate neurotoxins in AD(3). Oligomeric assemblies have been shown to exist in increased concentrations in vivo in AD patients and in AD animal models(4, 5). These oligomers are associated with significant neuronal dysfunction before any amyloid deposits are observed(6). In addition, rats injected with oligomeric Aβ assemblies display significant inhibition of hippocampal long-term potentiation, a measure of learning and memory(7). In vitro studies have shown that Aβ oligomers are more toxic than either monomers or amyloid fibrils(8). Similar findings exist with other diseases associated with amyloid forming proteins, including Parkinson’s disease(9) (α-synuclein: αSyn), type II diabetes(10, 11) (islet amyloid polypeptide: IAPP), familial amyloid polyneuropathy(12) (transthyretin: TTR), dialysis-related amyloidosis(13) (β2-microglobulin: β2M) and medullary carcinoma of the thyroid(14) (calcitonin: CT). These observations have led to a paradigm shift in target identification for drug discovery, namely away from the importance of amyloid deposits and fibrils per se toward the central role of protein oligomers.
A complex equilibrium exists among monomers and oligomers that involves both monomer conformation (secondary and tertiary structure) and order (quaternary structure)(3, 15, 16). Various oligomer structures have been described(3), but no consensus exists with respect either to oligomer structure or biological activity i.e., structure-activity (neurotoxicity) relationships (SAR). This fundamental problem exists because of the metastable and heterogeneous nature of oligomers(15). For this reason, a detailed structural and functional characterization of the oligomers, particularly of the more toxic 42 amino acid form of Aβ, Aβ42, has proven difficult(17).
To address the metastability problem, we used the method of photo-induced cross-linking of unmodified proteins (PICUP) to “freeze” the oligomer population, allowing quantitative determination of the oligomer size frequency distribution(18). When we originally published our application of the PICUP method in the Aβ system, we did extensive control experiments to convince ourselves that "physical reality" was being represented(19), which showed that the technique accurately reflected the oligomer frequency distribution in solution at the moment of cross-linking, for system sizes of <20 monomers per oligomer. This system size appears to be the one most likely to contribute to the pathogenesis of AD, as suggested by many (for a review, see(3)). The data were consistent with studies of assembly size done using dynamic light scattering(19) and with later discrete molecular dynamics simulations of Aβ oligomerization(20). The technique revealed that other amyloidogenic proteins yielded distinct oligomerization patterns whereas non-amyloidogenic, monomeric proteins yielded distributions consistent with concentration-dependent, diffusion limited cross-linking—namely ladders of bands, the nodes of which were determined by protein concentration, as would be expected from random collisioninduced cross-linking. These data provided further evidence that the cross-linking system does reflect what exists in solution.
Ono et al. recently showed that the PICUP technique yielded stable, low-order Aβ40 oligomers of constant quaternary structure and restricted conformational complexity, which allowed the performance of formal SAR studies for monomer through tetramer(17). Although successful, the studies of Ono et al. required the laborious and time-consuming preparation of hundreds of small batches of each oligomer that were pooled to provide sufficient material for study. Such an analytical scale process precludes production of the quantities of pure oligomers required for the broad range of studies necessary to fully characterize Aβ oligomer structure (e.g., NMR and x-ray crystallography), examine biological activity (in cell culture and animals), produce specific antibodies, or prepare oligomer immunogens for therapeutic purposes.
We report here that preparative-scale amounts of cross-linked Aβ oligomers can be produced easily using a continuous flow reaction system. We show that other amyloid proteins also can be cross-linked successfully using this system. The ability to produce large quantities of stable oligomers now should enable the conduct of a variety of biophysical and biological experiments that heretofore have been precluded due to sample scarcity and oligomer instability.
Experimental Procedures
Aβ was prepared by the Biopolymer laboratory at UCLA. αSyn was a gift from the laboratory of David Eisenberg. Other proteins were purchased from Sigma (TTR), Anaspec (CT), Poly Peptide, San Diego, CA (IAPP) and Lee Biosolutions, St. MO (β2M). 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was obtained from TCI America, Portland, OR. Tris(2,2'-bipyridyl)dichlororuthenium(II) hexahydrate (Ru(II)) and ammonium persulfate (APS) were purchased from Sigma. Dithiothreitol (DTT) was purchased from Fisher.
Preparation of LMW Aβ
Lyophilized Aβ was dissolved in 60 mM NaOH, diluted into 1 v Milli-Q water, and then mixed with an equal volume of 22 mM sodium phosphate buffer, pH 7.4. The material was sonicated in an ultrasonic water bath (model 1510, Branson Ultrasonics Corp., Danbury, CT), for 1 min at 22 °C, transferred into centrifugal filters (30,000 molecular weight cut-off, Centricon YM-30, Millipore Corp. Billerica, MA), and centrifuged at 16,000 × g using a bench top microcentrifuge (Eppendorf model 5415C, Brinkmann Instruments) for 30 min. The eluate from the filtration was collected and Aβ was quantified by UV absorbance (ε280=1280 cm−1 M−1) using a 1 cm quartz cuvette (Hellma, Plainview, NY) and a Beckman DU-640 spectrophotometer (Beckman Instruments, Fullerton, CA). All measurements were performed at 22°C. This protocol results in uniform and reproducible material termed low molecular weight (LMW) Aβ(21), containing monomeric Aβ in equilibrium with low order, unstructured oligomers. LMW Aβ was cross-linked immediately upon preparation.
Preparation of αSyn and IAPP
To prepare aggregate-free αSyn and IAPP, the lyophilized peptide was dissolved with ice cold HFIP and then dried in silicon-coated low-adsorbent tubes, as described previously(22). These “protein films” were stored airtight at −20°C. Prior to use, the peptide film was re-solubilized, initially in 60 mM NaOH(22).
Photochemical cross-linking using PICUP
Immediately after preparation, peptide solutions were diluted to concentrations of 5–40 µM and then they were cross-linked using the PICUP method, as described previously(19). Briefly, 18 µL of protein solution was added to 2 µL of 2 mM Ru(II)bpy32+ and 2 µL of 40 mM APS. The mixture was irradiated for 1 s with a 150 W visible light source and the reaction was immediately quenched with 2 µL of 1 M DTT. For each reaction, the mixture was vortexed for 1 s before and after addition of the cross-linking reagents and immediately preceding irradiation. To determine the frequency distribution of oligomers(23), the material was fractionated on 4–20% gradient Tris-Glycine or 10–20% gradient Tricine polyacrylamide gels (Invitrogen, Eugene, OR), depending on the size of the protein of interest. Samples were mixed with an equal volume of 2× SDS sample buffer (Invitrogen) and boiled for 10 min prior to loading. After electrophoresis, the gels were stained with SilverXpress (Invitrogen, Eugene, OR), according to the manufacturer’s protocol.
Densitometry
The gels were scanned at 600 dpi and converted to 8-bit grayscale and analyzed densitometrically by ImageJ 1.43r. Densitometry profiles were measured for each lane. Normalized relative percentages of each band were determined by dividing baseline-corrected peak intensities by the sum of all intensities in a lane.
Continuous flow PICUP reactor
Peptides were prepared as described above and then diluted to 5–40 µM and combined in the dark with Ru(II)bpy32+ and APS. A small aliquot of each sample was used as a control to be cross-linked in the traditional camera system. The continuous flow reactor was fabricated using opaque 1 mm I.D. PEEK tubing (GE life Sciences) coupled to a peristaltic pump (Bio-Rad). Tubing passed from the peristaltic pump into a light-tight chamber and then back out into a collection vessel. An illumination volume was created by splicing in ≈4 cm of clear tubing. The lamp (20 W tungsten halogen, Cedarberg Industries, Eagan, MN) was positioned approximately 5 cm above a 6 cm wide opening in the chamber, and turned on to equilibrate the temperature at the illumination point for 15 minutes prior to flow of the sample. The tubing was washed with >10 mL of 4 °C buffer immediately prior to each sample. The Aβ, Ru(II)bpy32+, APS mixture was kept on ice, shielded from light, and pumped through the illumination chamber into a collection tube containing 1M DTT.
Results
PICUP chemistry
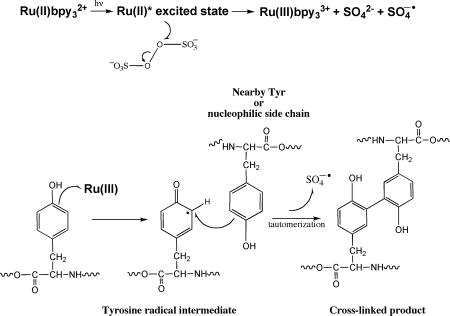
The PICUP chemistry is radical based (Fig. 1). The protein complex of interest is mixed with a water-soluble metal complex, such as tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate (Ru(II)bpy32+), and ammonium persulfate (APS). To initiate the chemistry, the ruthenium complex (λmax = 452 nm) is photolyzed with visible light. Photolysis of the Ru(II)bpy32+ complex produces an excited state that results in the donation of an electron to persulfate, causing cleavage of the persulfate O-O bond(24). The products are Ru(III), a potent one-electron oxidant(25–27), the sulfate radical, which is a good hydrogen atom abstraction agent, and sulfate anion. The activated Ru(III) metal complex can abstract an electron from an amino acid, particularly Tyr, Trp, His, or Met. These amino acids have higher propensities to form radicals due to their ability to stabilize the radical state through aromatic or neighboring-group effects. This produces a protein radical species that can then attack a variety of other groups, leading to covalent cross-linking among proteins within a complex. A hydrogen atom must be lost to quench the radicals and form stable products(28). The PICUP method is a rapid (<1 s), efficient (80–90%), zero-length, in situ chemical cross-linking method that works across a wide pH range(29). PICUP does not require pre facto structural modification of the peptide of interest, e.g., the introduction of a spacer or an unnatural photoreactive group into the protein, as do other chemical(30) or photoaffinity(31) cross-linking methods. The rapidity of the PICUP reaction is important, because it limits cross-linking between protein chains not already in oligomers. That is, the resulting covalently stabilized protein population reflects pre-existing complexes and not simply complexes formed by collisions of monomers or oligomers in the bulk solution phase(19).
Figure 1.

Photo-induced cross-linking of unmodified protein (PICUP). The chemistry is initiated by photolyzing Ru(II) (λmax = 452 nm) with visible light. Photolysis of this metal complex produces an excited state, which can donate an electron to persulfate, cleaving its O-O bond. The products are Ru(III), the sulfate radical, and the sulfate anion. The activated metal complex Ru(III) can abstract an electron from a nearby amino acid (Tyr, Trp, His, or Met are the most reactive). This produces a protein radical species that can then attack a wide variety of other groups on nearby proteins. This chemistry results in a covalently cross-linked protein complex without any prior modification to the protein of interest.
Concept and design of a continuous flow reactor (CFR) for large-scale oligomer production
The “continuous flow reactor” (CFR; Fig. 2) is a simple, versatile instrument in which each component of our prior analytical-scale single-reaction system(32) has been modified for continuous flow chemistry. The cross-linking chemistry proceeds at a negligible rate unless Ru(II)bpy32+ is photooxidized, thus the peptide, Ru(II)bpy32+ complex, and APS are pre-mixed in a light-tight reservoir kept on ice. Pre-mixing the reactants in this way eliminates the need for even a rudimentary on-line fluidic mixing device. The reactants are pumped through opaque (light tight) tubing into an exposure chamber using a peristaltic pump, which allows precise control of flow rates, and thus irradiation time. The exposure chamber is a black box (literally) through which the tubing passes. A small opening is created in the box to allow the entrance of light from a 20 W tungsten halogen lamp. Various lamps will work. The primary lamp design consideration was photon flux, which determines irradiation time (flow rate), although cost and maneuverability are also considerations. To create an irradiation volume, a section of clear tubing was “spliced” into the tubing within the exposure chamber. The volume of exposed sample was calculated to be 34.5 µL (4.4 cm of 1 mm i.d. tubing). This region of tubing was 6 cm from the light source. Quenching of the reaction is done by placing the outflow tube from the exposure chamber into a vessel containing 1M dithiothreitol in water.
Figure 2.

Continuous flow reactor. The peptide is mixed with APS and Ru(II)bpy32+ at 4°C and shielded from light. The reaction mixture is pumped through a variable flow rate pump into an exposure chamber in which a section of clear tubing is placed approximately 10 cm away from a light source. The fluid flows out of the exposure chamber to a collection vessel containing reaction quencher (e.g., 2-mercaptoethanol).
Because differences in irradiation time can affect the cross-linking efficiency, reproducibility requires that each element in the reaction volume be irradiated identically. To determine if this was so, we calculated the Reynolds number of the system. The Reynolds number(33), Re, is a dimensionless number that gives a measure of the ratio of inertial forces to viscous forces and thus can determine if fluid flow is laminar or turbulent. The instrument should display laminar flow so that the residence time of each molecule is constant. Using the equation where solvent density ρ = 1,000 kg·m−3, mean velocity V = 0.036545 m·s−1, cylinder diameter D = 0.001 m, and dynamic viscosity μ = 8.98 ×10−4 kg·m−1s−1, we determined the Reynolds number to be between 30 and 40, which is far below the upper limit (2300) of the laminar flow regime. Thus, the geometry and flow rates for the CFR system described above will produce laminar flow of the reactants, leading to homogeneous irradiation and consistent cross-linked products.
Establishment of operating parameters
Temperature control
We found that heat radiating from the lamp caused an increase in the temperature of the illuminated tubing over time, and as a result, increased fluid temperatures and altered reaction rates. The distance of the light source to the irradiation site is proportional to the heating effect. To ensure a constant reaction temperature, we placed an infrared filter between the lamp and the irradiated volume. This reduced the temperature increase (data not shown), although 4°C water immediately came to room temperature (22–23°C) upon flowing through the previously empty, room temperature, tubing of the CFR system. This temperature jump is likely due to the friction of the peristaltic pump, and heat transfer from the room temperature CFR tubing. We determined that the reactor could be equilibrated prior to use by pumping 4°C water through the tubing for 10 min at 1 ml/min. After this equilibration process, a constant reaction temperature could be maintained.
Irradiation time determination
The CFR design allows control of irradiation time by simple adjustment of the flow rate. When the PICUP technique originally was adapted to the Aβ system, we observed that short irradiation times resulted in inefficient cross-linking but long irradiation times resulted in peptide damage(19). To establish optimal irradiation times for the CFR, we performed reactions at flow rates from 0.02–6.0 ml/min. We evaluated cross-linking quality by performing SDS-PAGE and silver staining on samples from each flow rate. "Quality" was determined by comparison of results from the CFR with those obtained using our camera-based method (Fig. 3). At the fastest flow rate, 6.0 ml/min, the monomer is much more abundant than it is using the camera-based PICUP method, and fewer high-order oligomers are observed. This reflects lower cross-linking efficiency. There is a gradual decrease in the amount of monomer and a concomitant increase in the amounts of higher-order oligomers as the flow rate is decreased. At the slowest flow rates (longest exposure times), little or no monomer is observed and the oligomer distribution "collapses" into one comprising primarily dimer and trimer. In addition, the oligomer bands become more nebulous, likely due to formation of multiple species (intra- and intermolecularly cross-linked) with similar molecular masses but slightly different mobilities in SDS-PAGE. Bitan et al. carefully described the effect of extended irradiation times on the relative abundance of Aβ oligomers when initially characterizing the application of PICUP to Aβ, concluding that irradiation time had little effect on the relative abundances of low-order oligomers(19). Optimal flow rates were found to be between 0.2–1.0 ml/min. At these rates, cross-linking occurred efficiently without peptide damage. The calculated exposure time for a flow rate of 1 ml/min is 2 s. For the analytical scale system, the typical exposure is 1 s. The longer exposure time for the CFR likely results from a combination of the use of a weaker light source, 20 W, as compared to 150 W, and the difference in system geometry.
Figure 3.

Continuous flow rate optimization for Aβ40 (top) and Aβ42 (bottom) PICUP chemistry. 40 µM Aβ was and combined in the dark with Ru(II) and APS at a final concentration ratio of 0.72 : 1 : 20. The volume of the exposed sample in the apparatus is ≈35 µL and the exposure time is ≈2 seconds (at 1 ml/min). Negative control; non-cross-linked (XL−); positive control; cross-linked using the original camera system (XL+); 1.0*, the final lane examined the stability of Aβ pre-mixed with Ru(II) and APS, but not irradiated until 2 hours later at 1 ml/min.
Comparison of CFR and analytical scale cross-linking methods
To validate the CFR system relative to our analytical scale camera system, we compared the Aβ40 and Aβ42 oligomer distributions (Fig. 4). For each peptide, the two techniques produced similar distributions (XL−, XL+ and 0.2 lanes from Fig. 3, shown side by side in Fig. 4A). The CFR was marginally less efficient, as measured by monomer level. To compare the distributions quantitatively, we performed densitometry analyses using Image J software (Fig. 4B). The quantification showed that the CFR produced more higher-order oligomers for Aβ40, though this is potentially influenced by the smearing effect seen at the slowest flow rates. Quantification showed that dimer was the most abundant oligomer species for both Aβ40 and Aβ42. We observed monomer through hexamer oligomer states for Aβ40, and monomer through heptamer states for Aβ42. As expected(19), the pentamer and hexamer states in the Aβ42 distribution constituted a node, though this node was slightly less pronounced with the CFR (17% vs. 14% for pentamer, and 14% vs. 11.6% for hexamer).
Figure 4.

A) Analytical vs. continuous flow cross-linking of Aβ40 and Aβ42. The leftmost lanes are non-cross-linked protein (XL−), the middle lanes are analytical scale cross-linking (A), and the rightmost lanes are 0.2 ml/min continuous flow cross-linking (CFR), these data from Fig. 3 are shown here side-by-side for clarity of comparison. The number of monomers per oligomer is shown on the right. B) Densitometry analysis of oligomer abundance for analytical vs. continuous flow cross-linking. Oligomer order is plotted vs. oligomer frequency. Black bars show abundance from the analytical scale system. White bars represent the abundance from the continuous flow system.
General utility of the CFR
The Aβ system is not the only one in which oligomers exist or are thought to be important in disease causation. Proteins or peptides have been implicated in Parkinson's disease (α-synuclein; αSyn)(9), senile systemic amyloidosis and familial amyloid polyneuropathy (transthyretin; TTR)(12), dialysis-related amyloidosis (β2-microglobulin; β2M)(13), and medullary carcinoma of the thyroid (calcitonin; CT)(14), among others(13). To determine whether the CFR might be of utility for preparation of large quantities of chemically stabilized oligomers of these other proteins, we cross-linked αSyn, IAPP, TTR, β2M, and CT and compared the resulting oligomer distributions with those obtained after cross-linking 5–10 µg of each protein using the analytical scale PICUP method. In each system, PICUP was performed immediately upon protein dissolution (Fig. 5A).
Figure 5.

A) Silver stained SDS-PAGE analysis of analytical vs. continuous flow cross-linking of αSyn, IAPP, TTR, β2M and CT oligomers. For each peptide, the leftmost lanes are non-cross-linked protein (XL−), the middle lanes are analytical scale cross-linking (A), and the rightmost lanes are continuous flow cross-linking (CFR). The oligomer order is shown beside each distribution. B) Densitometry analysis of oligomer abundance for analytical vs. continuous flow cross-linking. Oligomer order is plotted vs. oligomer frequency. Black bars show abundance from the analytical scale system. White bars represent the abundance from the continuous flow system.
We found, as in the Aβ40 and Aβ42 cases, that the oligomer populations produced using each method were qualitatively similar or identical, within experimental error (cf. “A” and “CFR” for each sample). Densitometry analyses were consistent with this conclusion, but also suggested, in the cases of αSyn, β2M, and CT, that the CFR method was more efficient (Fig. 5B). This could be inferred by relative decreases in monomer frequency and increases in the frequency of higher-order oligomers when comparing the CFR results to those from the analytical-scale method.
We note that the most abundant oligomer species for αSyn and β2M was dimer. IAPP exhibited abundant monomer, followed by a sharp decrease in oligomer abundance as oligomer order increased. Interestingly, the oligomer distribution for IAPP resembled that predicted for high-efficiency cross-linking of proteins undergoing random diffusion-limited collisions(19), suggesting that substantial oligomerization had not occurred in the ≈4 min required to dissolve and cross-link IAPP. Cross-linked TTR displayed only tetramer and a very nebulous dimer band, whereas CT showed a Gaussian-like distribution of oligomers centered at trimer and tetramer. A three-dimensional representation of the oligomer abundance profiles for each protein illustrates these similarities and differences (Fig. 6).
Figure 6.

Oligomer frequency distributions of CFR products. For each protein, oligomer frequency (%) is plotted against oligomer order. The % abundance axis is shown at 60% for clarity, though TTR is 100% tetramer.
Discussion
One major obstacle to a detailed understanding of Aβ oligomer structure and activity is the diversity and complexity of oligomer preparation methods, and the widespread use of poorly defined materials derived there from in studies of the effects of oligomers. A critical aspect of the CFR method is that it reliably produces consistent starting material with stability far superior to non-cross-linked oligomer preparations and in quantities sufficient for examination by myriad in vitro, ex vivo and in vivo methods. These include studies of the effects of oligomers on cellular pathways (inflammation, signaling), animal models of disease, structural studies (CD, TEM, AFM, NMR, and crystallography), and effects on gene expression. Importantly, the CFR is a simple yet powerful device that can easily be fabricated. To assemble a CFR, a laboratory only needs to have: (1) a peristaltic pump; (2) a light source (as basic as a flashlight(28)); and (3) tubing.
Oligomer structure cannot be studied formally, at present, in the absence of oligomer stabilization. In fact, the difficulty in studying unstabilized oligomers has been a significant barrier to progress in the field. Even if one begins an experiment with an oligomer “enriched” fraction, the results thus obtained represent "population average" data because the oligomer fraction exists in an equilibrium among many different species. One cannot determine the structure of a particular oligomer, and how that oligomer contributes to this population average for any particular biological effect. Structure-activity relationships of Aβ oligomers thus have not been established so far without stabilization (see(17)).
The inability to determine the structures of unstabilized oligomers precludes a comparision of their structures with those of chemically stabilized oligomers. However, there are reasons to suggest that they may be very similar. The oligomers differ in primary structure only through the presence/absence of the carbon-carbon bond in the zero-length cross-linking procedure. No alterations in the solvent milieu of the peptides is made during the cross-linking. In addition, we have found that Aβ40 and Aβ42 monomers do exist in a partially folded state(34). This means, prior to cross-linking, that the conformational space of the oligomer population is relatively restricted and thus if that state is stabilized properly, it would be representative of the population of conformers at the time of cross-linking.
The benefit of the CFR could be far reaching in the general field of oligomer study. The reactor enables the study of the properties of chemically-stabilized oligomers not only of Aβ, but of αSyn, β2M, IAPP and CT. The CFR method thus is not restricted to the study of any one protein or disease, but can be applied broadly to many diseases in which oligomers may be involved.
Finally, a very significant practical advantage is provided by the CFR, namely time-effort efficiency (Table 1). Analytical-scale reactions each typically involve 20 µL of 80 µM Aβ (10 µg). In the continuous flow reactor, the quantity of sample is only limited by the amount of protein available. For example, to produce 1 mg of cross-linked protein using the analytical method would require 100 individual reactions, 400 individual pipetting steps, and turning the light source on and off (actuating the shutter release) 100 times to irradiate the samples–a total of ≈200 min of investigator time. In contrast, using the CFR system, the entire 1 mg is produced in one step, at 1 mL/min, with only 4 pipetting steps, with the lamp on for 1 min, finishing the reaction in ≈4 min, a 50-fold time savings. This becomes more dramatic when considering the production of larger volumes. It would take ≈200,000 min of investigator time to prepare 1 g of sample with the analytical method. If this time were expended during an 8 h work day, the entire process then would take 14 months. In contrast, using the CFR system, preparing 1 g would take just 2,000 min (33 hours) total time, with only 2 min of investigator time input, a 300,000-fold savings in investigator time and a 300-fold elapsed time savings.
Table 1.
Comparison of time and effort for analytical vs. continuous flow cross-linking. Production capacity and its related metrics are shown for each method. The continuous flow apparatus allows for production of theoretically limitless quantities, while at the same time reducing investigator time by four orders of magnitude (for 1 g preparation). Time and effort are shown for a flow rate of 0.5 mg/ml.
| Protein quantity |
Effort & Time |
Analytical PICUP | Continuous Flow PICUP |
Analytical /CF Ratio |
|---|---|---|---|---|
|
| ||||
| 1 µg | Effort | 1 individual reaction: 4 pipetting steps, 1 exposure. | 1 reaction: 4 pipetting steps, 1 exposure of <1 min | 1 |
|
|
||||
| Total Time | 2 min | 2 min | 1 | |
|
| ||||
| 1 mg | Effort | 100 individual reactions: 400 pipetting steps, 100 exposures. | 1 reaction: 4 pipetting steps, 1 exposure of 2 min | 100 |
|
|
||||
| Total Time | 200 min | 4 min | 50 | |
|
| ||||
| 1 g | Effort | 100,000 individual reactions: 400,000 pipetting steps, 100,000 exposures. 600,000 min investigator time* | 1 reaction: 4 pipetting steps, 1 exposure of 2000 min 2 min investigator time | 300,000 |
|
|
||||
| Total Time | ≈ 14 months | 1.4 days elapsed time | 300 | |
600,000 min total elapsed investigator time assuming an 8 hour work day (14 months).
Acknowledgments
We gratefully acknowledge the support of grants NS038328 and 5T32NS07449 from the National Institutes of Health. We also thank Drs. Gal Bitan, Robin Roychaudhuri, Mingfeng Yang and Dahabada Lopes for helpful comments and discussion. We thank Margaret Condron for synthesis of Aβ.
Funding Sources
This work was supported by grants from the National Institutes of Health (NF038328 and 5T32NS07449).
ABBREVIATIONS
- AD
Alzheimer's disease
- Aβ
amyloid β-protein
- αSyn
α-synuclein
- IAPP
islet amyloid polypeptide
- TTR
transthyretin
- β2M
β2-microglobulin
- CT
calcitonin
- SAR
structure-activity relationship
- PICUP
photo-induced cross-linking of unmodified proteins
- Ru(II)bpy32+
tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate
- APS
ammonium persulfate
- CFR
continuous flow reactor
- XL−
non-cross-linked
- XL+
cross-linked
Footnotes
Author Contributions
E.Y.H. and D.B.T. designed the experiments. E.Y.H. performed the experiments. E.Y.H. and D.B.T. wrote the manuscript.
References
- 1.World Alzheimer's Report. Alzheimer's Disease International; 2010. [Google Scholar]
- 2.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 3.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid β-protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Meyer G, Shapiro F, Vanderstichele H, Vanmechelen E, Engelborghs S, De Deyn PP, Coart E, Hansson O, Minthon L, Zetterberg H, Blennow K, Shaw L, Trojanowski JQ. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 7.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 8.Walsh DM, Selkoe DJ. Aβ oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 9.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O'Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010;176:861–869. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sorgjerd K, Klingstedt T, Lindgren M, Kagedal K, Hammarstrom P. Prefibrillar transthyretin oligomers and cold stored native tetrameric transthyretin are cytotoxic in cell culture. Biochem Biophys Res Commun. 2008;377:1072–1078. doi: 10.1016/j.bbrc.2008.10.121. [DOI] [PubMed] [Google Scholar]
- 13.Kad NM, Myers SL, Smith DP, Alastair Smith D, Radford SE, Thomson NH. Hierarchical Assembly of β2-Microglobulin Amyloid In Vitro Revealed by Atomic Force Microscopy. J. Mol. Biol. 2003;330:785–797. doi: 10.1016/s0022-2836(03)00583-7. [DOI] [PubMed] [Google Scholar]
- 14.Arvinte T, Cudd A, Drake AF. The structure and mechanism of formation of human calcitonin fibrils. J Biol Chem. 1993;268:6415–6422. [PubMed] [Google Scholar]
- 15.Teplow DB. Preparation of amyloid β-protein for structural and functional studies. Methods Enzymol. 2006;413:20–33. doi: 10.1016/S0076-6879(06)13002-5. [DOI] [PubMed] [Google Scholar]
- 16.Hayden EY, Teplow DB. Biophysical Characterization of Aβ assembly. In: Derreumaux P, editor. Alzheimer's Disease: Insights into low molecular weight and cytotoxic aggregates from in vitro and computer experiments: Molecular Basis of Amyloid-β Protein Aggregation and Fibril Formation. Imperial College Press; 2011. [Google Scholar]
- 17.Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc Natl Acad Sci U S A. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bitan G, Teplow DB. Rapid photochemical cross-linking–a new tool for studies of metastable, amyloidogenic protein assemblies. Acc Chem Res. 2004;37:357–364. doi: 10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 19.Bitan G, Lomakin A, Teplow DB. Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem. 2001;276:35176–35184. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 20.Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In silico study of amyloid β-protein folding and oligomerization. Proc Natl Acad Sci U S A. 2004;101:17345–17350. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 22.Rahimi F, Maiti P, Bitan G. Photo-induced cross-linking of unmodified proteins (PICUP) applied to amyloidogenic peptides. J Vis Exp. 2009 doi: 10.3791/1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, Baumketner A, Shea JE, Urbanc B, Cruz L, Borreguero J, Stanley HE. Elucidating amyloid β-protein folding and assembly: A multidisciplinary approach. Acc Chem Res. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- 24.Nickel U, Chen Y-H, Schneider S, Silva MI, Burrows HD, Formosinho SJ. Mechanism and Kinetics of the Photocatalyzed Oxidation of p-Phenylenediamines by Peroxydisulfate in the Presence of Tri-2,2'-bipyridylylruthenium(II) J. Phys. Chem. 1994;98:2883–2888. [Google Scholar]
- 25.Gray HB, Winkler JR. Electron transfer in proteins. Annu Rev Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 26.Yocom KM, Shelton JB, Shelton JR, Schroeder WA, Worosila G, Isied SS, Bordignon E, Gray HB. Preparation and characterization of a pentaammineruthenium(III) derivative of horse heart ferricytochrome c. Proc Natl Acad Sci U S A. 1982;79:7052–7055. doi: 10.1073/pnas.79.22.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berglund J, Pascher T, Winkler JR, Gray HB. Photoinduced Oxidation of Horseradish Peroxidase. J. Am. Chem. Soc. 1997;119:2464–2469. [Google Scholar]
- 28.Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci U S A. 1999;96:6020–6024. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fancy DA, Denison C, Kim K, Xie Y, Holdeman T, Amini F, Kodadek T. Scope, limitations and mechanistic aspects of the photo-induced cross-linking of proteins by water-soluble metal complexes. Chem Biol. 2000;7:697–708. doi: 10.1016/s1074-5521(00)00020-x. [DOI] [PubMed] [Google Scholar]
- 30.Das M, Fox CF. Chemical cross-linking in biology. Annu Rev Biophys Bioeng. 1979;8:165–193. doi: 10.1146/annurev.bb.08.060179.001121. [DOI] [PubMed] [Google Scholar]
- 31.Knorre DG, Godovikova TS. Photoaffinity labeling as an approach to study supramolecular nucleoprotein complexes. FEBS Lett. 1998;433:9–14. doi: 10.1016/s0014-5793(98)00860-6. [DOI] [PubMed] [Google Scholar]
- 32.Vollers SS, Teplow DB, Bitan G. Determination of Peptide oligomerization state using rapid photochemical crosslinking. Methods Mol Biol. 2005;299:11–18. doi: 10.1385/1-59259-874-9:011. [DOI] [PubMed] [Google Scholar]
- 33.Reynolds O. An Experimental Investigation of the Circumstances Which Determine Whether the Motion of Water Shall Be Direct or Sinuous, and of the Law of Resistance in Parallel Channels. Phil. Trans. R. Soc. Lond. 1883;174:935–982. [Google Scholar]
- 34.Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, Rigby AC, Teplow DB. Familial Alzheimer's disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc Natl Acad Sci U S A. 2007;104:16522–16527. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]