

Abstract
Methylglyoxal (MGO) is a major glycating agent that reacts with basic residues of proteins and promotes the formation of advanced glycation end products (AGEs) which are believed to play key roles in a number of pathologies, such as diabetes, Alzheimer’s disease, and inflammation. Here, we examined the effects of MGO on immortalized mouse hippocampal HT22 nerve cells. The endpoints analyzed were MGO and thiol status, the glyoxalase system, comprising glyoxalase 1 and 2 (GLO1/2), and the cytosolic and mitochondrial Trx/TrxR systems, as well as nuclear Nrf2 and its target genes. We found that nuclear Nrf2 is induced by MGO treatment in HT22 cells, as corroborated by induction of the Nrf2-controlled target genes and proteins glutamate cysteine ligase and heme oxygenase 1. Nrf2 knockdown prevented MGO-dependent induction of glutamate cysteine ligase and heme oxygenase 1. The cystine/glutamate antiporter, system xc−, which is also controlled by Nrf2, was also induced. The increased cystine import (system xc−) activity and GCL expression promoted GSH synthesis, leading to increased levels of GSH. The data indicate that MGO can act as both a foe and a friend of the glyoxalase and the Trx/TrxR systems. At low concentrations of MGO (0.3 mM), GLO2 is strongly induced, but at high MGO (0.75 mM) concentrations, GLO1 is inhibited and GLO2 is downregulated. The cytosolic Trx/TrxR system is impaired by MGO, where Trx is downregulated yet TrxR is induced, but strong MGO-dependent glycation may explain the loss in TrxR activity. We propose that Nrf2 can be the unifying element to explain the observed upregulation of GSH, GCL, HO1, TrxR1, Trx2, TrxR2, and system xc− system activity.
Keywords: Methylglyoxal, Thioredoxin reductase, Nrf2, Glyoxalase, Glutathione, HT22 cells
1. Introduction
Methylglyoxal (MGO) is an endogenous by-product of the normal metabolism of carbohydrates, lipids, and proteins, inevitably produced spontaneously or enzymatically [1]. During glycolysis, MGO is readily formed under alkaline conditions from glyceraldehyde or dihydroxyacetone [2]. Intermediate metabolites such as acetoacetate from lipids, and succinylacetone and aminoacetone from threonine and glycine metabolism, are also endogenous sources of MGO [1]. Accumulation of MGO is highly deleterious, since it readily reacts in vivo with basic phospholipids and nucleotides, and with lysine and arginine residues of proteins, leading to advanced glycation end product (AGE) formation [3]. MGO can impair the antioxidant system by depleting GSH [1]. MGO has been considered as a possible causative agent in a number of pathologies, such as diabetes [4, 5], hyperalgesia and inflammation [6], aging disorders [7], Alzheimer’s disease [8], epilepsy [9], autism [10], and anxiety [11], among others.
MGO levels in human plasma are in the range of 0.1–0.6 μM [12–14], and about 10- to 20-fold higher in the cerebrospinal fluid [15]. These elevated levels of MGO can make nerve cells more prone to AGE formation, which is thought to be a relevant factor in the development of neurodegenerative diseases [8, 16, 17]. MGO can induce irreversible loss of protein function, including cross-linking, as well as contributing to oxidative stress [1, 8].
The glyoxalase system is the main enzymatic route for MGO elimination [3, 18]. Glyoxalase 1 (GLO1, EC 4.4.1.5) catalyzes the reduced glutathione (GSH)-dependent conversion of MGO to S-D-lactoylglutathione. The conversion of S-D-lactoylglutathione to D-lactate is catalyzed by glyoxalase 2 (GLO2, EC 3.1.2.6), thereby restoring the GSH consumed in the reaction catalyzed by GLO1. GLO1 activity is the rate limiting step in MGO degradation, which makes its regulation tightly associated with MGO toxicity [2, 19].
Experimental approaches to understanding MGO toxicity clearly show that thiols are important targets of MGO with GSH depletion being an almost ubiquitous finding [1]. Due to the high reactivity of MGO, spontaneous reaction with GSH may prevent it from reacting with more sensitive targets, avoiding irreversible enzyme inhibition and DNA modification [12]. In line with these findings, MGO-induced depletion of protein thiols has also been reported [20, 21]. Reactive thiols in proteins are particularly relevant targets for MGO toxicity. For instance, MGO-dependent posttranslational modification of cysteine residues in the TRPA1 pain receptor opens the channel, which has been proposed as a new mechanism for metabolic neuropathy [22]. In this regard, deleterious effects of MGO on thiolic proteins with reactive cysteines could be a general mechanism of MGO toxicity.
Thioredoxin (Trx), thioredoxin reductase (TrxR, EC 1.8.1.9), and NADPH constitute the Trx system and the main electron donor for enzymes such as ribonucleotide reductase (EC 1.17.4.1), methionine sulfoxide reductases (EC 1.8.4.11), peroxiredoxins (EC 1.11.1.15), and glutathione peroxidases (EC 1.11.1.9) [23, 24]. Similar to other proteins, Trx and TrxR have reactive cysteines that are among the most sensitive to oxidative stress [25, 26], and thus may be oxidized by reactive oxygen species or targeted by thiol-reacting molecules, such as MGO. There is some in vitro evidence that Trx [27] and TrxR [28] can be inhibited by MGO. Endothelial cells exposed to MGO present increased ROS production and typical signs of apoptosis. In this model, treatment with MGO downregulates Trx protein and mRNA copy number [29]. This information highlights the relevance of studying the Trx/TrxR system as a potential target of MGO in nerve cells.
Recently, it has been shown that inducers of the nuclear factor-erythroid 2 p45 related factor 2 (Nrf2) transcription factor can protect cells against MGO toxicity [30–33]. Nrf2 binds to the antioxidant response element (ARE), increasing the transcription of a number of protective genes. Glutathione S-transferase (EC 2.5.1.18), NAD(P)H: quinone oxidoreductase 1 (EC 1.6.5.2), glutamate-cysteine ligase (GCL, EC 6.3.2.2), and heme oxygenase 1 (HO1, EC 1.14.99.3) are representative enzymes whose expression is controlled by Nrf2 [34]. Along with other antioxidant genes, Trx and TrxR contain ARE sequences and therefore their transcription can also be controlled by Nrf2 [35]. Interestingly, one ARE sequence was recently found in the promoter region of the GLO1 gene, and Nrf2-dependent induction was also demonstrated [19].
The aim of the present study was to identify novel MGO targets in order to provide further insights into the mechanisms involved in neurodegenerative processes. For these studies, we used the immortalized mouse hippocampal HT22 nerve cell line, a well-established model for addressing neurodegenerative processes [36]. We report the contrasting effects of barely toxic (0.3 mM) and moderately toxic (0.75 mM) concentrations of MGO on thiol levels, system xc− activity, expression of Nrf2, GLO1, GLO2, and the mitochondrial and cytosolic Trx/TrxR pairs in the HT22 nerve cells. Collectively, the data show that MGO can modulate Nrf2 protein levels, which reflects on Nrf2-controlled genes (GCL and HO1). We also show that MGO either induces or impairs the glyoxalase system, depending on the concentration. The cytosolic and mitochondrial Trx/TrxR system is also affected by MGO in a concentration- and time-dependent manner, indicating a new possible route for MGO toxicity in nerve cells.
2. Materials and methods
2.1. Reagents
Tissue culture dishes were from NUNC; fetal calf serum (FCS) was obtained from Hyclone; high-glucose Dulbecco’s modified Eagle medium (DMEM) and TRIzol Reagent were from Invitrogen (Carlsbad, CA, USA). AMPD1 RT-Supermix (M-MuLV) was from Biopioneer (San Diego, CA, USA); RT-003 FastStart Universal SYBR Green Master was from Roche; methyl diaminobenzene-BODIPY (MBo) was synthetized as previously described [37]. Bovine serum albumin (BSA), N-(2-hydroxyethyl)-piperazine-N-(2-ethane-sulfonic acid) (Hepes), aminoguanidine, glutathione reductase (EC 1.8.1.7), GSH, 5,5-dithiobis(2-nitrobenzoic acid) (DTNB), homo-cysteate, 2-mercaptoethanol, NADPH, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), insulin, methylglyoxal, ethylenediaminetetraacetic acid (EDTA), ethylene glycol tetra-acetic acid, 1,4-dithio-D-threitol, tris(hydroxymethyl)amino-methane) (Tris), protease inhibitor cocktail, and DNase I were from Sigma–Aldrich. L-[3H]Glutamate was obtained from Perkin Elmer NEN. Primary antibodies, respective dilutions, and antigen were Nrf2 (sc-13032, 1/500, rabbit), TrxR1 (sc-31057, 1:500, goat), GLO1 (sc-67351, 1:3000, rabbit), GLO2 (sc-31057, 1:1000, goat), anti-GCLC (sc-22755, 1/1000, rabbit) polyclonal antibodies from Santa Cruz Biotechnology; Trx1 (C63C6, 1:3000, rabbit) monoclonal antibody from Cell Signaling; Trx2 (Ab71261, 1:1000, rabbit), TrxR2 (Ab58445, 1:500, rabbit) polyclonal antibodies from Abcam; HO1 (SPA-896, 1/5000) was from Stressgen (Victoria, BC, Canada); mouse anti-MGO (STA-011, 1/1000) was from Cell BioLabs (San Diego, CA USA). Nrf2 siRNA (sc-37049) was purchased from Santa Cruz Biotechnology, control siRNA was from Qiagen (1027280) and RNAiMAX reagent from Invitrogen.
2.2. Cell culture and treatments
Immortalized mouse hippocampal HT22 cells were grown on tissue culture dishes in high-glucose DMEM that was supplemented with 10% FCS as previously published [38]. For viability assays, 5 × 103 HT22 cells were plated in 96-well plates and grown to semiconfluence.
Following treatment for the indicated times and concentrations of MGO, the medium was exchanged with fresh medium without MGO, and cell viability was quantified by the MTT assay [39]. Results obtained in the MTT viability assay correlate well with the extent of cell death as confirmed visually [40], and by the extent of protein lost. For other assays, 1.5 × 105 cells were plated in 60 mm plate dishes and the cells were grown to semiconfluence. Prior to harvesting the cells, the medium was aspirated and the cells rinsed twice with cold PBS.
For siRNA transfection, HT22 cells were plated in 60 mm dishes at 5 × 105 cells/dish and 166 pmol Nrf2 siRNA or control siRNA was used along with RNAiMAX according to the manufacturer’s instructions.
2.3. Fluorometric assay of methylglyoxal in culture media
After treatment of cells with MGO in 60 mm culture dishes, the appropriate amount of medium was sampled at the indicated time points. Due to interference with the assay, phenol red was not included in the media. The MGO present in the media was diluted to give 3 μM, based on the starting concentration, and incubated with 5 μM MBo, which was obtained from a 2 mM stock solution diluted in dimethyl sulfoxide. The reaction was allowed to occur in the dark at 37 °C for 1 h, and the fluorescent product was quantified in a 96-well plate reader fluorimeter using 485 nm for excitation and 528 nm for emission [37]. Background fluorescence was subtracted from the readings by using samples without the addition of MBo. The background fluorescence of MBo in PBS was also subtracted from the readings. Values were obtained based on a standard curve containing known amounts of MGO diluted in PBS. The specificity of the MBo reaction with MGO was checked in parallel aliquots of samples previously treated with 10 mM aminoguanidine for 2 h to quench MGO [41]. Aminoguanidine-treated samples produced marginal fluorescence, indicating that this assay specifically detects the fluorescent MBo-MGO reaction product (data not shown).
2.4. Quantification of total GSH and protein thiols (PSH)
HT22 cells (1.5 × 105) were seeded in 60 mm dishes, and after 24 h cells were treated as described under Cell culture and treatments. Cells were harvested in cold 0.5 M perchloric acid and centrifuged (15,000 g, 5 min). The supernatant was neutralized in 0.1 M potassium phosphate buffer, pH 7.4, containing 1 mM EDTA and assayed enzymatically for total GSH (GSH + GSSG) based on the reduction of DTNB with glutathione reductase and NADPH [42]. For protein thiol (PSH) measurements, a colorimetric test based on the reaction of DTNB with thiols was used [43]. The acid-precipitated pellet was washed with 0.5 M perchloric acid, and solubilized in 0.5 M Tris/HCl, pH 8.0, 1% SDS, 1 mM EDTA. A sample aliquot was incubated without (sample blank) or with 0.2 mM DTNB (sample). After 30 min at 37 °C under agitation, readings were performed at 412 nm in a 96-well plate reader. The sample blank and the DTNB-background absorbance were subtracted, and thiol content was obtained by using a standard curve with known amounts of GSH. Obtained data from both assays were normalized to protein content.
2.5. Measurement of system xc− activity
For measurement of system xc − activity, 2.5 × 104 HT22 were plated in 24-well dishes. After 24 h, the cells were washed three times with sodium-free Hank’s balanced salt solution (HBSS). System xc− activity was measured as sodium-insensitive, homocysteic acid-inhibitable uptake of [3H]glutamate (Perkin Elmer, Waltham, MA, USA) as described [44]. Briefly, cells in triplicate were incubated in 10 μM glutamate ([3H]glutamate/cold glutamate 1:1000) with or without 1 mM homocysteic acid adjusted to pH 7.4 in sodium-free HBSS. Cells were washed three times with ice-cold, sodium-free HBSS and lysed in 0.2 N NaOH. Radioactivity was measured by liquid scintillation counting and normalized to protein measured by the Bradford method (Pierce).
2.6. Enzyme assays
The GLO1 assay was based on the rate of formation of S-D-lactoylglutathione from the hemithioacetal formed nonenzymatically from the reaction of MGO and GSH [45]. Briefly, 2 mM GSH was allowed to react with 2 mM MGO in potassium phosphate buffer (50 mM, pH 6.6) for 10 min at 37 °C and immediately added to the sample. The linear appearance of S-D-lactoylglutathione was followed for 3 min, at 240 nm, in a 96-well plate reader. The activity was calculated based on the molar absorptivity of S-D-lactoylglutathione (2860 M−1 cm−1) and normalized to protein content.
TrxR catalyzes the reduction of DTNB at the expense of NADPH, whose decay was used to estimate TrxR activity [46]. Briefly, after harvesting cells in 0.1 M phosphate buffer, 1 mM EDTA, pH 7.4, samples were centrifuged (15 min, 15,000 g) and an aliquot was incubated with DTNB for 15 min, at room temperature, to allow reaction with thiols present in the sample. Thereafter, NADPH was added to start the reaction that was followed for 5 min by readings at 340 nm in a 96-well plate reader. Spontaneous reaction was subtracted from actual reading in a duplicate sample that was run without the addition of NADPH. NADPH molar absorptivity of 6.22 M−1 cm−1 was used and activity normalized to protein content.
Trx activity was determined by the endpoint method based on Trx-catalyzed insulin reduction [46]. After reaction with insulin, Trx is reduced to its thiol form by TrxR added to the assay, and the amount of thiols formed (reduced insulin) is measured by reaction with DTNB, following the manufacturer’s instructions (IMCO, Stockholm, Sweden). In brief, samples were incubated in Trx activation buffer containing 0.5 mM dithiothreitol, 75 mM Hepes, pH 7.4, 2 mM EDTA, 1.7 mg/ml BSA, for 30 min at 37 °C. After Trx activation, samples were assayed in 75 mM Hepes, pH 7.4, 8 mM EDTA, 20 mM Tris/HCl, 1.8 mg/ml insulin, 0.8 mM NADPH, 15 μg/ml TrxR. After 30 min at 37 °C, the reaction was stopped with a solution containing 0.8 mM DTNB, 6.4 M guanidinium chloride, and 0.16 M Tris/HCl, pH 8.0. A sample blank made without the addition of TrxR was run in parallel and subtracted from the readings. An additional blank without sample was run to subtract spontaneous color development. Activity was based on the total amount of 5-thionitrobenzoate formed, using the molar absorptivity of 14.1 M−1 cm−1, and normalized to protein content
Protein was determined by the bicinchoninic acid assay (Thermo Sci.), using bovine serum albumin as a standard.
2.7. Real-time quantitative PCR
RNA isolation was carried out using TRIzol Reagent, followed by DNase treatment. M-MuLV reverse transcriptase was used to convert 1 μg RNA to cDNA per the manufacturer’s protocol (Biopioneer, RT-003). The 300 nM gene-specific primers, in conjunction with FastStart Universal SYBR Green Master Mix (ROX) (Roche), were used to perform real-time qPCR reactions on an Applied Biosystems 7900HT Fast Real-Time PCR System located in Salk Institute’s Functional Genomics Core Facility using the following conditions: standard curve method, 96-well plates (10 μl reaction), SYBR Green (Fast/Regular) with melting curve standard: 50 °C for 2 min, 95 °C for 10 min, 95 ° for 15 s, 60 °C for 1 min: 40 cycles. Melting-point dissociation curves were used to determine reaction specificity with SDS 2.3 software while relative expression levels were normalized to β-actin using the 2−ΔΔCt method. Primer sequences are as follows: GCLC forward, CACCCCCGCTTCGGTACTCT; GCLC reverse, GA-CAGCAGTTGCCCATCCCG; HO1 forward, GACAGAAGAGGCTAA-GACCGC; HO-1 reverse, TGGAGGAGCGGTGTCTGG. Experimental data were normalized to control conditions, pooled, and analyzed.
2.8. Cell fractionation and Western blot
HT22 cells were plated at a density of 1.5 × 105 cells per 6 cm dish. After 24 h, the cells were treated as indicated, rinsed twice in ice-cold, Tris-buffered saline (TBS), and harvested in sample buffer (25 mM Tris, pH 8.0, 2% SDS, 25 mM 2-mercaptoethanol, 1 mM Na3VO4) for whole cell extracts. For cell fractionation, cells were rinsed twice in ice-cold, TBS scraped into an ice-cold nuclear fractionation buffer (10 mM Hepes, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM ethylene glycol tetraacetic acid, 1 mM 1,4-dithio-D-threitol, 1 mM Na3VO4, 1X protease inhibitor cocktail), and incubated on ice for 15 min. NP40 was then added at a final concentration of 0.6%, the cells were vortexed, and their nuclei pelleted by centrifugation. The supernatant was collected as the cytosolic/membrane fraction. Nuclear proteins were extracted from the pellet by sonication in the nuclear fractionation buffer and the extracts were cleared by additional centrifugation.
Samples were analyzed by SDS-PAGE using 10% Criterion XT Precast Bis-Tris Gels (Bio-Rad). Proteins were transferred to poly-vinylidene fluoride membranes and probed with the desired primary antibody as detailed elsewhere [40]. Immunodetection was performed by using Super Signal West Pico Substrate (Pierce) with the appropriated secondary antibody. For all antibodies, the same membrane was reprobed for actin to normalize the protein load. Nuclear extracts were probed with Nrf2 primary rabbit polyclonal antibodies, while other antibodies were tested in whole cell or cytosolic protein extracts. Horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) were diluted 1/5000 in 5% skim milk in TBS/0.1% Tween 20 prior use. Autoradiographs were scanned using a Bio-Rad GS800 scanner, and band density was measured using the Quantity One software. Protein expression was normalized to actin band intensity. Each Western blot was repeated at least twice with independent protein samples.
2.9. Immunoprecipitation
TrxR1 was immunoprecipitated using 5 μg of TrxR1 antibody (sc-31057, Santa Cruz Biotechnology). Complexes were collected on protein A-Sepharose, washed twice in 20 mM Hepes, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol, and then once in PBS. Bound proteins were eluted by boiling in SDS sample buffer, separated by SDS/PAGE, transferred to nitrocellulose, and probed with anti-TrxR1 or anti-MGO primary antibodies, as above.
2.10. Statistical analysis
Data are presented as mean ± SEM and the statistical significance assessed by one-way ANOVA with Tukey’s post test, and P values < 0.05 were considered significant.
3. Results
3.1. MGO and GSH metabolism
The cytotoxicity of MGO was examined using the MTT viability test as shown in Fig. 1. MGO induced a time- and concentration-dependent decrease in HT22 viability. For further experiments, we decided to use 0.3 mM MGO, a barely toxic concentration, causing only a 10% decrease in viability at 24 h, and full restoration at 48 h. A more toxic concentration of MGO (0.75 mM) was also selected for further studies. At this concentration, viability decreased by 40–60% after 24 h of exposure with no further mortality over an additional 24 h period.
Fig. 1.
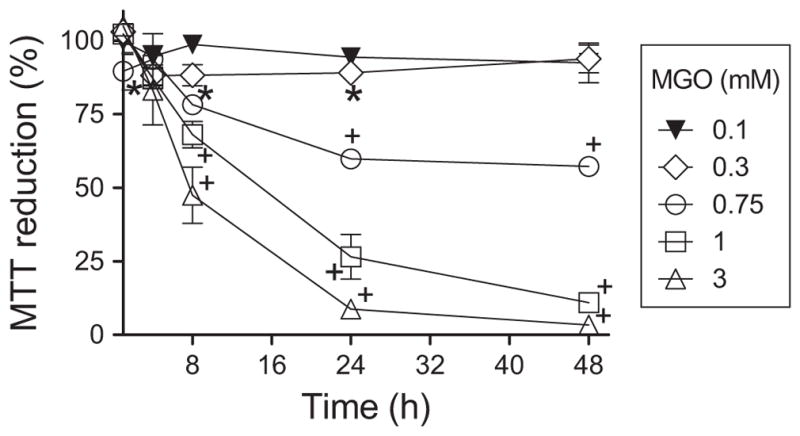
Concentration and time course viability of MGO-treated HT22 cells. Data are means + SEM of at least three independent experiments. Some error bars are enclosed by symbols. Statistical differences are represented by P<0.05 (*) or <0.01 (+).
In order to understand the time course of MGO consumption by the cells from the incubation media, we took advantage of a fluorescent probe for MGO named MBo that was recently developed [37]. MGO is stable in PBS for up to 48 h (data not shown), and in DMEM without the addition of FCS (Fig. 2A). In regular media, supplemented with 10% FCS, MGO is no longer stable (Fig. 2A), presenting a half-life of about 4.6 h. In the presence of cells, this half-life was observed to decrease to 40–50 min, a 5- to 7-fold decrease versus DMEM (Fig. 2A). This decrease in stability did not depend on the starting MGO concentration, indicating active MGO consumption by the HT22 cells.
Fig. 2.

Consumption of MGO in culture media. (A) MGO is stable in DMEM without FCS (open triangle), but not in DMEM media supplemented with 10% FCS (open circles); HT22 cells increase MGO decay in DMEM + 10% FCS at both 0.3 (squares) and 0.75 mM (diamonds). HT22-dependent consumption of MGO in HBSS media containing 0.3 (B) or 0.75 mM (C) MGO. MGO is stable in PBS and HBSS for at least 48 h (data not shown). Data points represent the average of three independent experiments made in duplicate (A) or one out of two experiments made in duplicate (B and C)
Since MGO presented a low stability in DMEM even in the absence of cells, we performed additional experiments to evaluate the cell-dependent MGO consumption at both 0.3 and 0.75 mM MGO (Fig. 2B and C) using HBSS media, in which MGO is stable (Fig. 2A). The rate of decay of MGO in HBSS fitted well to a one-phase decay, confirming the cell’s ability to handle excess MGO.
Next, we investigated whether MGO treatment leads to a perturbation in thiol homeostasis in the HT22 cells, as previously reviewed for other cell types [1]. One hour after MGO exposure, the HT22 cells showed an ~50% loss of GSH, regardless of the concentration used (0.3 or 0.75 mM) (Fig. 3A), consistent with the rapid rate of MGO depletion from the culture media. Thereafter, the pattern of GSH metabolism differed between the low and the high MGO concentrations. GSH remained low (−44%) in the 0.75 mM MGO group at 8 h, but HT22 cells exposed to 0.3 mM not only recovered GSH but also showed a 37% rebound effect. At 24 h, GSH was about 50% higher with both MGO concentrations, as compared to untreated controls.
Fig. 3.

Glutathione (GSH-t), protein thiol (PSH) levels, and system xc− activity in HT22 cells exposed to MGO. (A) Glutathione, (B) system xc− activity (glutamate uptake,) and (C) PSH levels were determined in HT22 cells exposed to MGO 0.3 or 0.75 mM for the indicated periods. Values were normalized as percentage of untreated cells and presented as means + SEM. Sample size were N=5–9 for GSH-t and PSH, and N=3–4 for system Xc−. P <0.05 (*), 0.01 (**), or 0.001 (***) are indicated relative to untreated control (dashed line).
The changes in GSH levels could be a consequence of a disturbance in cysteine availability whose transport depends on the glutamate/cystine antiporter, system xc− [47]. To test this hypothesis system xc− activity was assayed at 4, 8, or 24 h after MGO treatment (Fig. 3B). At 0.3 mM MGO, a progressive increase in system xc− activity, from 4 (133%) to 8 (200%) h, was observed, as compared to untreated cells. This effect was no longer present at 24 h. With the same pattern of change as for GSH, at 0.75 mM MGO, system xc− activity remained 25–50% lower than that of the control group for up to 8 h, followed by a 25% increase in activity by 24 h.
Protein thiols decreased only after 24 h with 0.75 mM MGO, but remained unaltered with the 0.3 mM MGO treatment (Fig. 3C).
Since MGO removal is mostly dependent on the glyoxalase system, we also determined if GLO1 and GLO2 expressions were affected by MGO (Fig. 4). An ~20% increase in GLO1 and GLO2 expression was observed at earlier time points, 0.5/2.5 h after treatment with 0.3 or 0.75 mM MGO, but no changes in GLO1 expression were observed after 8 or 24 h of treatment (Fig. 4A and C). In contrast to the GLO1 expression pattern, GLO2 content was strongly affected by MGO treatment at later time points (8/24 h, Fig. 4B and E). Despite the observation that MGO treatment increased GLO2 levels at earlier time points, by 8 h 0.3 mM MGO produced a 20% decrease in GLO2 protein content, while 0.75 mM MGO led to a 45% decrease. At this latter concentration GLO2 levels remained low at 24 h. In contrast, 0.3 mM MGO produced a 63% increase in GLO2 expression at 24 h (Fig. 4B and E).
Fig. 4.

GLO1 and GLO2 expression and GLO1 activity in HT22 cells exposed to MGO. (A) GLO1 and (B) GLO2 expression and their respective quantification (C and E); (D) GLO1 activity was determined in HT22 cells exposed to 0.3 or 0.75 mM MGO for the indicated time periods. Densitometric data were normalized to actin content and expressed as a percentage of the untreated control. Vertical dashed lines represent non-adjacent lanes from the same run. Data on graphs are presented as means ± SEM (N=5–9). P<0.05 (*), 0.01(**), or 0.001 (***) indicate significant differences relative to untreated control (horizontally dashed lines).
A modest decrease in GLO1 activity (20%) was only observed after 24 h of exposure to 0.75 mM MGO (Fig. 4D).
3.2. MGO-dependent induction of GCL and HO-1 is under the control of Nrf2
The low concentration of MGO (0.3 mM) produced a rapid and sustained (1–8 h) increase in nuclear Nrf2 content (Fig. 5). This rapid response contrasts with a more delayed (24 h) increase in nuclear Nrf2 induction when HT22 cells were treated with a more toxic concentration of MGO (0.75 mM). No other significant changes in Nrf2 were observed at this concentration.
Fig. 5.

MGO modulates nuclear Nrf2 protein content. (A) Nrf2 expression in HT22 cells exposed to MGO, and the respective quantification. (B) Densitometric data were normalized to actin content and expressed as percentage of untreated control. Vertical lines separate lanes from different runs. Data on graphs are presented as means + SEM (N=4–9). P<0.05 (*) or 0.01 (**) indicate significant differences relative to untreated control (horizontally dashed lines).
There is a significant linear dependence of Nrf2 expression on the MGO concentration (0–0.3 mM) for each of the time points 1, 4, and 8 h (P<0.002), as evaluated by Pearson’s linear correlation (data not shown). However, as the time progresses, the slope decreases, indicating a weakening of the signal, which vanished by 24 h.
To confirm the increase in nuclear Nrf2, we investigated the expression of two target genes, the GCL catalytic subunit GCLC and HO1 (Fig. 6). Indeed, 0.3 mM MGO treatment induced an increase in GCLC (Fig. 6A and C) and HO1 (Fig. 6B and D) protein expression after 8 h of exposure. However, at higher levels of MGO (0.75 mM), GCLC and HO1 expressions were significantly increased only at 24 h (Fig. 6B and D). At 2.5 h posttreatment with 0.75 mM MGO, GCLC expression was significantly decreased by 15%.
Fig. 6.

MGO induces Nrf2-dependent genes. GCL catalytic subunit GCLC (A) and HO1 (B) expression in HT22 cells exposed to MGO, and their respective quantification (C and D). Densitometric data were normalized to actin content and expressed as percentage of untreated control. Vertical dashed lines separate lanes from the same run. Data on graphs are presented as means + SEM (N=4–9). P<0.05 (*), 0.01(**), or 0.001 (***) indicate significant differences relative to untreated control (horizontally dashed lines).
Corroborating the data on protein expression, GCLC and HO1 transcripts were increased by both MGO concentrations at 4 and 8 h posttreatment. At 4 h and 0.3 mM MGO, a modest effect was observed, presenting an average of 3.8 +/− 1.9 (N=3)- and 2.2 +/−0.5 (N=3)-fold increase in transcripts for GCLC and HO1, respectively. In contrast, higher than 26 (N=2)- and 17 (N=3)-fold induction in GCLC and HO1 transcripts were observed at 8 h and 0.75 mM MGO.
Cells transfected with siRNA to Nrf2 presented very low levels of Nrf2 protein, as compared to control cells (Fig. 7A). Transfection of Nrf2-si also prevented the induction of Nrf2 protein expression by fisetin, a known Nrf2 inducer [40], further showing the effectiveness of the siRNA transfection (Fig. 7A). The MGO-induced increase in GCLC (8 h, 0.3 mM and 24 h, 0.75) was completely abolished by Nrf2-si transfection (Fig. 7B–E). The MGO-dependent induction of HO-1 was not attenuated by Nrf2-si at 8 h and 0.3 mM MGO (Fig. 7B and D); however, the 2.7-fold induction of HO-1 at 24 h and 0.75 mM MGO was completely prevented by transfection with Nrf2-si (Fig. 7C and E).
Fig. 7.

siRNA targeting of Nrf2 prevents the MGO-induced increase in GCL and HO1 expression in HT22 cells. (A) Cells were transfected with control (Ct-si) or Nrf2 siRNA (Nrf2-si), and further treated for 4 h with fisetin (10 μM), a known Nrf2 inducer. Nuclear extracts were prepared and blotted for Nrf2. Note that basal Nrf2 expression was decreased by its siRNA, and the strong fisetin-dependent induction of Nrf2 was abolished in cells treated with siRNA to Nrf2. After transfection with control-si or siRNA to Nrf2, cells were exposed to 0.3 mM MGO for 8 h (B and D), or 0.75 mM for 24 h (C and E). Cytosolic extracts were probed with GCL or HO-1 antibodies, shown as representative images (B and C) and their respective quantification (D and E). Densitometric data were normalized to actin content and expressed as percentage of untreated control. Data on graphs are presented as means ± SEM (N=3–5). P<0.01 (**) or 0.001 (***) indicate significant differences relative to their respective control siRNA (Ct-si).
3.3. MGO targets the Trx/TrxR system
Since there are some data in the literature indicating that Trx and TrxR can be affected by MGO [27–29], we also examined the effect of MGO on the expression of the cytosolic Trx/TrxR couple in our model. Trx1 levels and activity were not significantly affected by 0.3 mM MGO (Fig. 8A, B, and C). In contrast, 0.75 mM MGO-treated cells presented a marked decrease in Trx1 expression (Fig. 8A and B) after 24 h, which was accompanied by an equivalent decrease in Trx activity (Fig. 8C). Although the Trx activity assay cannot discriminate between cytosolic and mitochondrial Trx isoforms, the use of a cytosolic preparation excludes the mitochondrial fraction (see Material and methods), so the activity assay should predominantly reflect the activity of the cytosolic isoform.
Fig. 8.

MGO targets the cytosolic Trx/TrxR system. Trx1 (A) and TrxR1 (D) expression in HT22 cells expose to MGO, and their respective quantification (B and E). Activities of Trx (C) and TrxR (F) are also presented. Densitometric data were normalized to actin content and expressed as percentage of untreated control. Enzymatic activity was quantified as described under Materials and methods and presented relative to untreated control (dashed line). Data on graphs are presented as means + SEM (N=4–9). P<0.01 (**) or < 0.001 (***) indicate significant differences relative to untreated control (dashed lines).
TrxR1 expression was decreased after 30 min of exposure to both MGO concentrations (Fig. 8D and E). The only other change in TrxR1 content was observed at 24 h, when 0.75 mM MGO produced an ~100% increase (Fig. 8D and E).
Similar to Trx, the use of the cytosolic fraction to assay TrxR activity should reflect the activity of the cytosolic isoform. Both MGO concentrations produced a clear decrease in TrxR activity at earlier time points (0.5 and 2.5 h) after MGO treatment (Fig. 8F). Despite being unaltered relative to control values at 24 h, the TrxR activity (Fig. 8F) was low when the 100% increase in protein content is taken into consideration (Fig. 8D and E).
The influence of MGO on the mitochondrial Trx/TrxR system has not been investigated previously. MGO treatment mostly presented an inducing pattern on Trx2/TrxR2 (Fig. 9). Both Trx2 (Fig. 9A and C) and TrxR2 (Fig. 9B and D) showed increased expression at early time points (0.5/2.5 h) with both MGO concentrations. Trx2 presented a 20–38% increase in expression, while TrxR2 presented a large 2.5-fold increase. Although 0.3 mM MGO had no effect on Trx2 and TrxR2 expression at later time points, 0.75 mM MGO had a biphasic effect on TrxR2 expression with a significant decrease (25%) at 8 h and a 100% increase at 24 h.
Fig. 9.

MGO-induced changes in the mitochondrial Trx/TrxR system. Trx2 (A) and TrxR2 (B) expression in HT22 cells exposed to MGO, and their respective quantification (C and D). Densitometric data were normalized to actin content and expressed as percentage of untreated control. Data on graphs are presented as mean + SEM (N=4–9). P<0.05 (*), 0.01 (**), or 0.001 (***) indicate significant differences relative to untreated control (dashed lines).
Immunoprecipitation of TrxR1 was performed followed by Western blots to detect glycated TrxR1 (Fig. 10). MGO-treated cells presented a significant amount of MGO-glycated TrxR1, which was not seen in the untreated control cells.
Fig. 10.
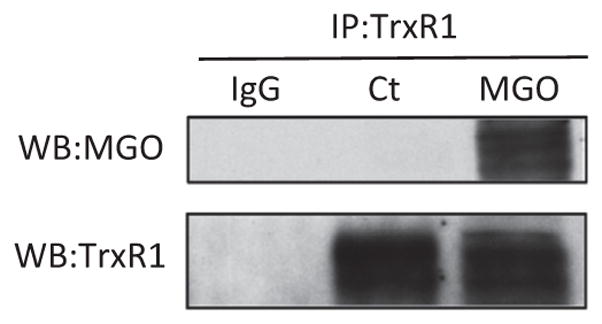
Glycation of TrxR1 induced by MGO treatment. HT22 cells were treated with or without 0.75 mM MGO for 24 h. After MGO treatment TrxR1 was immunoprecipitated (lower panel), and probed with an anti-MGO antibody (upper panel), showing a strong signal indicating the presence of glycated TrxR1. Note that MGO-dependent glycation of TrxR1 could not be detected in control cells (Ct), in opposition to the strong band intensity in MGO-treated cells. IgG indicates a control where normal rabbit IgG was used instead of the TrxR1 antibody for the immunoprecipitation.
4. Discussion
Cells follow divergent regulatory pathways depending on the nature and intensity of a stress. Often a specific stress can elicit diametrically opposed consequences triggering either adaptation or death pathways. As recently reviewed, HT22 nerve cells provide a very useful model for studying how various types of stress can contribute to neurodegenerative diseases [36]. In the current study, we focused on MGO because its accumulation is implicated in multiple neurodegenerative diseases [8,47–49]. Although a high neuronal susceptibility to MGO toxicity has been previously described [50–53], in this study we were interested in the spectrum of stress responses to MGO treatment. Thus, we took advantage of stress intensity to follow for 24 h some adaptive responses of surviving nerve-derived HT22 cells challenged with a barely toxic (0.3 mM), or a more toxic (0.75 mM) concentration of MGO. The toxic MGO concentration led to an ~50% decrease in cell viability, leading to a proportional decrease in viable cells or in protein content (data not shown), but no further mortality was found for an additional period of 24 h.
4.1. Methylglyoxal metabolism
The data on MGO levels allow the following conclusions: (a) MGO reacts rapidly with FCS components present in DMEM, mostly probably the proteinaceous components [41], given that MGO is stable in DMEM without addition of FCS; (b) cells rapidly consume MGO (t1/2 ~50 min), probably as a consequence of the hemithioacetal formed from the nonenzymatic reaction between MGO and GSH [18]. However, a reaction with other cellular components is obviously not excluded; (c) MGO consumes GSH to a large extent, as reviewed elsewhere [1]; (d) most MGO is consumed within ~4 h of treatment, suggesting that responses after this period are secondary to direct MGO exposure.
Limitations of our study relate to the pharmacologic concentrations of MGO used, and that cell culture models are not ideal for studying chronic exposure due to methodological limitations. This scenario contrasts with the delayed progression of neurodegenerative diseases, which are generally associated with a progressive imbalance in one or more physiological processes. Nevertheless, the concentrations of MGO used here are within those usually found in the literature [50–53]. Due to its high reactivity, as exemplified by the rapid metabolism found in HT22 cells, it is estimated that more than 90% of cellular MGO is bound to macromolecules. It has been estimated that actual levels of MGO can reach up to 300 μM if bound MGO is taken in consideration [54,55]. Also, considering that cerebrospinal fluid has an overall concentration of 10–20 μM of free MGO [15], it is possible that at a given time and site of production, MGO levels can be significantly higher [1].
4.2. Glutathione metabolism
The GSH response to MGO was biphasic. At 0.3 mM, MGO produced a decrease in GSH (1 h), followed by a rebound effect by 8 and 24 h. A similar response was observed with 0.75 mM MGO, although GSH depletion persisted for a longer period (1–8 h) and increased levels of GSH could only be seen later (24 h). Our studies identified three factors that could be responsible for the changes in GSH levels: (a) the rate limiting step in GSH synthesis that is catalyzed by GCL [56]; (b) cysteine availability that is controlled by the amount of cystine (oxidized form of cysteine) in the media [57–59], and (c) the activity of system xc− which can control cystine uptake by the cell [47].
Initially GSH is depleted by MGO, as expected [1, 18], which triggered regulatory cellular responses. Our results suggest that the increased levels of GSH at 8 h and 0.3 mM MGO can be explained by the induction of GCL, and the higher activity of system xc−. Conversely, the longer depletion of GSH (8 h) at 0.75 mM MGO can be explained by the lower activity of system xc− and the lack of GCL induction. This scenario is reversed at 24 h, given that 0.75 mM MGO induced GCL, which resulted in elevated levels of GSH due to increased GSH synthesis. At this time point, the activity of system xc− was increased by 25%, as compared to control levels, also contributing to GSH buildup.
Not only did MGO (0.3 mM) treatment result in a sustained increase in GSH levels by 8–24 h, and 0.75 mM at 24 h, but it also increased the expression of GCL and HO1, classical markers of Nrf2 activation [34]. System xc− is also under Nrf2 control [47–60]. Together these data are in line with the idea that the observed coordinated cellular response to MGO, leading to an increase in GSH, is governed by Nrf2 translocation to the nucleus.
4.3. Nrf2 is induced by methylglyoxal
The results presented here clearly show that MGO treatment of HT22 cells induces an increase in nuclear Nrf2. This effect was observed earlier (1–8 h) for 0.3 mM MGO, as compared to 0.75 mM MGO (24 h). MGO induced GCLC and HO1 proteins and transcript levels, markers of Nrf2 induction [34–61]. Protein induction was observed at 8 h for 0.3 mM, while at 0.75 mM MGO it was observed at 24 h. An increase in the GCL and HO1 transcript levels support data on protein expression, indicating an MGO-dependent activation of Nrf2. Our findings suggest caution when interpreting data where Nrf2 inducers are used along with MGO treatment, given that MGO is also an Nrf2 inducer.
To confirm that Nrf2 is actually controlling GCL and HO-1 induction, cells were transfected with siRNA to Nrf2 and thereafter treated with MGO. Transfection with Nrf2-si was effective in preventing GCL and HO-1 induction in MGO-treated cells, in line with the idea that expression of these proteins is under Nrf2 control.
4.4. The glyoxalase system
The early (0.5/2.5 h) increase observed for GLO1 and GLO2 expression at both MGO concentrations, and the 60% increase in GLO2 expression induced by 0.3 mM MGO at 24 h could stimulate MGO elimination but the more toxic dose of MGO led to decreased GLO1 activity (24 h), and GLO2 expression at 8 and 24 h (−40% 0.75 mM). The obvious consequence of an impairment in the glyoxalase system would be an increased susceptibility to MGO and oxidative stress [3,18,50,53]. This is consistent with the increased toxicity of 0.75 mM MGO and suggests that, despite the increases in GSH, at this time point and dose of MGO the impairment of the glyoxalase system plays a key role in decreasing cellular viability.
There is strong evidence for the importance of GLO1 in preventing AGE formation and being a critical enzyme in MGO metabolism [3, 18, 32]. Nevertheless, there are uncertainties regarding the importance of GLO2, the enzyme most affected by MGO treatment. For instance, the functionality of GLO2 activity, as the second step in MGO detoxification, has been questioned [2]. On the other hand, in a model of fructose feeding, MGO was inversely related to adipose tissue GLO2 activity [62]. This same correlation was not found for GLO1, suggesting that GLO2 is an important determinant of MGO detoxification in this tissue. GLO2 overexpression is anti-apoptotic, and a deficiency is proapoptotic [63]. An interesting study [64] showed that transduced recombinant Tat-GLO1, Tat-GLO2, or the combination of both can protect HT22 cells from MGO and H2O2 toxicity. Protection has also been shown in vivo in mouse models of diabetes for Tat-GLO1 [65] or ischemic damage for Tat-GLO1 and Tat-GLO2 [64]. Interestingly, a substantial increase in intracellular Tat-GLO2 protected the HT22 cells, albeit to a lesser extent as compared to Tat-GLO1. One possible conclusion from this work is that any increment in either GLO1 or GLO2 activity will increase MGO elimination, favoring the GLO1/GLO2 equilibrium toward MGO elimination, in line with the protective effects against oxidative stress (H2O2) or MGO burden [64]. The converse would say that any decrease in GLO1 or GLO2 could turn cells more sensitive to MGO or oxidative stress. Additionally, the lower Kcat and higher Km of GLO2, as compared to GLO1 [66], suggests that GLO2 is a less efficient enzyme and would be rate limiting if the activity ratio GLO1/GLO2 is low. If this premise were correct, then MGO should increase the MGO burden by limiting its own elimination, consistent with our observation of a 40% decrease in GLO2 expression and a 15% decrease in GLO1 activity in HT22 cells treated with 0.75 mM MGO. MGO-induced inhibition of GLO1 has been shown in nerve cells, associated to GSH depletion, impairment in glutathione peroxidase activity, and increased oxidative stress [53]. These combined effects should increase MGO toxicity due to decreased clearance, and may be relevant to environments with lowered GLO activity and/or increased MGO production, such as during diabetes, ageing, and Alzheimer’s disease [4, 15, 49].
Studies in vitro and with animal models show that classical inducers of Nrf2 increase protection against MGO toxicity, lowering AGE formation [30–33]. These protective mechanisms strongly suggest that the glyoxalase system is activated by Nrf2, including increases in GLO1 activity and expression [30,32,67]. Recently, it has been demonstrated that there is one ARE sequence in the promoter region of the GLO1 gene that can be activated by Nrf2 [19]. However, resveratrol-induced increases in GLO1 were not prevented by Nrf2 interfering RNA in HepG2 cells [30]. This observation is corroborated by a lack of GLO1 induction in MGO-treated rats [67], which is in line with a similar lack of response presented here for HT22 cells.
The control of GLO2 expression is less studied and our findings that MGO is able either to strongly increase or to decrease GLO2 expression in a dose-dependent manner are highly novel observations. To the best of our knowledge, there are no data in the literature regarding the transcriptional regulation of GLO2. Since we showed Nrf2 activation, the increased GLO2 expression with 0.3 mM MGO could be under Nrf2 control. Furthermore, a search in the oPOSSUM data base [68] for putative promotor regions in the mouse GLO2 gene revealed one ARE sequence (−140 bp), which is a putative Nrf2 binding site. However, transfection with siRNA to Nrf2 does not prevent the GLO2 induction 24 h after treatment with 0.3 mM MGO (data not shown). Furthermore, the observation that 0.75 mM MGO increased Nrf2 expression at 24 h but markedly decreased GLO2 expression at the same point indicates a more complex picture. An in-depth investigation is warranted to clarify which mechanisms are responsible for the observed changes in GLO2 expression in HT22 cells treated with MGO.
4.5. The cytosolic and mitochondrial thioredoxin/thioredoxin reductase systems
The literature shows clearly that cytosolic Trx1/TrxR1 are under Nrf2 control [35]. Indeed, 0.75 mM MGO produced a 100% increase in TrxR1 expression at a time point (24 h) when Nrf2 is also activated. However, the opposite effect was observed for Trx1, with 0.75 mM MGO producing a marked decrease in cytosolic Trx1 activity and expression, which is considered highly deleterious to the cells [24]. Lower Trx activity would impair peroxiredoxin activity, thereby limiting peroxide elimination, impair redox balance, and decrease cell survival [69–71].
TrxR activity was also lower than expected when the cells were treated with 0.75 mM MGO. At 8 h, TrxR1 expression remained at basal levels, but activity was ~40% lower, and at 24 h, expression of TrxR1 presented a 2-fold increase, as compared to the control group, but activity remained at basal levels. The mitochondrial isoforms of Trx and TrxR presented the same expression pattern; however, they are underrepresented in the TrxR activity assay. The inhibition of purified Trx1 and TrxR1 by MGO has been described [27, 28] and this also seems to be the case in the HT22 cells treated with MGO. This hypothesis was further corroborated by TrxR1 immunoprecipitation studies, showing that TrxR1 is heavily glycated after MGO treatment, as compared to control cells where it presents no signs of glycation. Together these findings highlight the importance of Trx/TrxR as possible cellular targets of MGO toxicity.
Despite Trx1 being under Nrf2 control, the opposite effect (lower activity and expression) from what would be expected was observed when Nrf2 was induced (0.75 mM MGO at 24 h). This suggests either that the Nrf2-dependent induction of Trx1 is impaired by MGO or that Trx1 is directly modified by MGO and targeted for degradation. In this regard, there is evidence that Trx1 can be inhibited, and expression of Trx1 decreased by MGO [27, 28], which seems to be controlled at the transcriptional level [29]. These observations combined with our data indicate that Trx1 is downregulated by MGO treatment in HT22 cells, a phenomenon not previously described for nerve cells.
Regarding the mitochondrial Trx2/TrxR2 system, there is only indirect evidence showing an upregulation of Trx2/TrxR2 via Nrf2 [72,73]. Also, mitochondrial Trx/TrxR upregulation is not always observed when Nrf2 is activated [74], which may depend on the specific model and conditions being studied. Given that expression of Nrf2-responsive genes only starts at 8–24 h following MGO treatment, Nrf2 is probably not responsible for the rapid (0.5/2.5 h) increase in Trx2 and TrxR2 content, which deserves further investigation.
5. Concluding remarks
There is strong evidence suggesting that the glyoxalase system is impaired in Alzheimer’s disease [49] with the brains of Alzheimer’s disease patients presenting increased levels of AGEs and oxidative stress, which are strictly correlated with MGO levels [8]. It has recently been shown that DJ-1 displays glyoxalase activity, and the absence of this activity has been suggested as the underlying cause of a rare autosomal-recessive genetic form of Parkinson’s disease [75,76]. Moreover, normal ageing is associated with a decline in GLO1 expression in rodents and human subjects [49]. These examples indicate the relevance of deciphering new mechanisms of MGO toxicity in nerve cells.
The major finding of the present work identified MGO as a foe and a friend of the glyoxalase and the Trx/TrxR systems. MGO detoxification is affected in two ways by MGO; at low MGO (0.3 mM) concentrations GLO2 is strongly induced, but at high MGO concentrations (0.75 mM) GLO1 is inhibited and GLO2 downregulated, and while the cytosolic Trx/TrxR system is impaired, the mitochondrial system is upregulated. The Trx1 and GLO2 downregulation by 0.75 mM MGO can be postulated as a possible MGO-induced disturbance in regulatory mechanisms, such as interferences in protein turnover or in Nrf2-dependent activation of these genes. On the other hand, the Nrf2-dependent expression of TrxR1/TrxR2 seems to be preserved, which would be considered a response to counteract the MGO-inhibitory effect on TrxR activity. As shown, TrxR1 is heavily glycated, which is compatible with a direct effect of MGO on TrxR1 resulting in inactivation.
The observed deleterious effects of MGO include GSH, PSH, GLO2, and Trx1 depletion, and decreased system xc−, Trx, and TrxR activity. To counteract the GSH depletion, the HT22 cells increased both cystine import (system xc− activity) and GCL expression, leading to increased GSH levels due to de novo synthesis. In addition, the cells also strongly induced TrxR1 expression, which may be associated with the depletion of Trx1 and TrxR inhibition. The mitochondrial Trx2/TrxR2 was also strongly induced by MGO.
Further studies are warranted to clarify the relevance of the novel MGO targets presented here. The modulation of cytosolic Trx/TrxR system by MGO is barely shown in the literature, and the modulation of mitochondrial Trx/TrxR induced by MGO has not been demonstrated. While the MGO-induced modulation of GLO1 is already known to have a major role in neurodegenerative diseases, the importance of MGO-dependent modulation of GLO2 is unknown.
We showed, by transfection with siRNA to Nrf2, that MGO-dependent induction of GCL and HO-1 is dependent on Nrf2. Given the demonstrated MGO-dependent modulation of Nrf2, synergistic effects with other Nrf2-inducing drugs, such as flavonoids may have clinical relevance in the treatment of neurodegenerative diseases.
Acknowledgments
A.L.D. is a research fellow of and awarded a travel grant from CNPq (National Counsel of Technological and Scientific Development), Brazil. This work was supported by NIH (R21 AG043816) to P.M.
Abbreviations
- AGE
advanced glycation end products
- ARE
antioxidant response element
- BSA
bovine serum albumin
- DMEM
high-glucose Dulbecco’s modified Eagle medium
- DTNB
5,5-dithiobis(2-nitrobenzoic acid)
- EDTA
ethylenediamine-tetraacetic acid
- FCS
fetal calf serum
- GCL
glutamate cysteine ligase
- GLO
glyoxalase
- GSH
glutathione
- HBSS
Hank’s balanced salt solution
- Hepes
N-(2-hydroxyethyl)-piperazine-N-(2-ethanesulfonic acid)
- HO1
heme oxygenase 1
- MBo
methyl diaminobenzene-BODIPY
- MGO
methylglyoxal
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Nrf2
nuclear factor-erythroid 2 p45 related factor 2
- PBS
phosphate-buffered saline
- PSH
protein thiols
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TBS
Tris-buffered saline
- Tris
tris(hydroxymethyl)aminomethane)
- Trx
thioredoxin
- TrxR
thioredoxin reductase
References
- 1.Kalapos MP. The tandem of free radicals and methylglyoxal. Chem Biol Interact. 2008;171:251–271. doi: 10.1016/j.cbi.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Sousa Silva M, Gomes RA, Ferreira AEN, Ponces Freire A, Cordeiro C. The glyoxalase pathway: the first hundred years… and beyond. Biochem J. 2013;453:1–15. doi: 10.1042/BJ20121743. [DOI] [PubMed] [Google Scholar]
- 3.Thornalley PJ, Rabbani N. Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev Biol. 2011;22:318–325. doi: 10.1016/j.semcdb.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Matafome P, Sena C, Seiça R. Methylglyoxal, obesity, and diabetes. Endocrine. 2013;43:472–484. doi: 10.1007/s12020-012-9795-8. [DOI] [PubMed] [Google Scholar]
- 5.Tian C, Alomar F, Moore CJ, Shao CH, Kutty S, Singh J, Bidasee KR. Reactive carbonyl species and their roles in sarcoplasmic reticulum Ca2+ cycling defect in the diabetic heart. Heart Fail Rev. 2014;19:101–112. doi: 10.1007/s10741-013-9384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koivisto A, Chapman H, Jalava N, Korjamo T, Saarnilehto M, Lindstedt K, Pertovaara A. TRPA1: a transducer and amplifier of pain and inflammation. Basic Clin Pharmacol Toxicol. 2014;114:50–55. doi: 10.1111/bcpt.12138. [DOI] [PubMed] [Google Scholar]
- 7.Desai KM, Chang T, Wang H, Banigesh A, Dhar A, Liu J, Untereiner A, Wu L. Oxidative stress and aging: is methylglyoxal the hidden enemy? Can J Physiol Pharmacol. 2010;88:273–284. doi: 10.1139/Y10-001. [DOI] [PubMed] [Google Scholar]
- 8.Angeloni C, Zambonin L, Hrelia S. Role of methylglyoxal in Alzheimer’s disease. BioMed Res Int. 2014;2014:e238485. doi: 10.1155/2014/238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albrecht P, Henke N, Tien MLT, Issberner A, Bouchachia I, Maher P, Lewerenz J, Methner A. Extracellular cyclic GMP and its derivatives GMP and guanosine protect from oxidative glutamate toxicity. Neurochem Int. 2013;62:610–619. doi: 10.1016/j.neuint.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 10.Maher P. Methylglyoxal, advanced glycation end products and autism: is there a connection? Med Hypotheses. 2012;78:548–552. doi: 10.1016/j.mehy.2012.01.032. [DOI] [PubMed] [Google Scholar]
- 11.Hambsch B. Altered glyoxalase 1 expression in psychiatric disorders: cause or consequence? Semin Cell Dev Biol. 2011;22:302–308. doi: 10.1016/j.semcdb.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Rabbani N, Thornalley PJ. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat Protoc. 2014;9:1969–1979. doi: 10.1038/nprot.2014.129. [DOI] [PubMed] [Google Scholar]
- 13.Kalapos MP. Where does plasma methylglyoxal originate from? Diabetes Res Clin Pract. 2013;99:260–271. doi: 10.1016/j.diabres.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Odani H, Shinzato T, Matsumoto Y, Usami J, Maeda K. Increase in three α,β-dicarbonyl compound levels in human uremic plasma: specific in vivo determination of intermediates in advanced maillard reaction. Biochem Biophys Res Commun. 1999;256:89–93. doi: 10.1006/bbrc.1999.0221. [DOI] [PubMed] [Google Scholar]
- 15.Kuhla B, Lüth HJ, Haferburg D, Boeck K, Arendt T, Münch G. Methylglyoxal, glyoxal, and their detoxification in Alzheimer’s disease. Ann N Y Acad Sci. 2005;1043:211–216. doi: 10.1196/annals.1333.026. [DOI] [PubMed] [Google Scholar]
- 16.Currais A, Maher P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid Redox Signal. 2012;19:813–822. doi: 10.1089/ars.2012.4996. [DOI] [PubMed] [Google Scholar]
- 17.Krautwald M, Münch G. Advanced glycation end products as biomarkers and gerontotoxins—a basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp Gerontol. 2010;45:744–751. doi: 10.1016/j.exger.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Thornalley PJ. Glyoxalase I—structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. 2003;31:1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 19.Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, Park BK, Souma T, Moriguchi T, Yamamoto M, Thornalley PJ. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem J. 2012;443:213–222. doi: 10.1042/BJ20111648. [DOI] [PubMed] [Google Scholar]
- 20.Morgan PE, Sheahan PJ, Davies MJ. Perturbation of human coronary artery endothelial cell redox state and NADPH generation by methylglyoxal. PLoS One. 2014;9:e86564. doi: 10.1371/journal.pone.0086564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okouchi M, Okayama N, Aw T. Hyperglycemia potentiates carbonyl stress-induced apoptosis in naive PC-12 cells: relationship to cellular redox and activator protease factor-1 expression. Curr Neurovasc Res. 2005;2:375–386. doi: 10.2174/156720205774962665. [DOI] [PubMed] [Google Scholar]
- 22.Eberhardt MJ, Filipovic MR, Leffler A, Roche J, de la Kistner K, Fischer MJ, Fleming T, Zimmermann K, Ivanovic-Burmazovic I, Nawroth PP, Bierhaus A, Reeh PW, Sauer SK. Methylglyoxal activates nociceptors through transient receptor ootential channel A1 (TRPA1). A possible mechanism of metabolic neuropathies. J Biol Chem. 2012;287:28291–28306. doi: 10.1074/jbc.M111.328674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.García-Giménez JL, Seco-Cervera M, Aguado C, Romá-Mateo C, Dasí F, Priego S, Markovic J, Knecht E, Sanz P, Pallardó FV. Lafora disease fibroblasts exemplify the molecular interdependence between thioredoxin 1 and the proteasome in mammalian cells. Free Radic Biol Med. 2013;65:347–359. doi: 10.1016/j.freeradbiomed.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal. 2012;17:1738–1747. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Z, Zhang J, Ballou DP, Williams CH. Reactivity of thioredoxin as a protein thiol-disulfide oxidoreductase. Chem Rev. 2011;111:5768–5783. doi: 10.1021/cr100006x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García-Santamarina S, Boronat S, Hidalgo E. Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry (Mosc) 2014;53:2560–2580. doi: 10.1021/bi401700f. [DOI] [PubMed] [Google Scholar]
- 27.Wang XL, Lau WB, Yuan YX, Wang YJ, Yi W, Christopher TA, Lopez BL, Liu HR, Ma XL. Methylglyoxal increases cardiomyocyte ischemia-reperfusion injury via glycative inhibition of thioredoxin activity. Am J Physiol Endocrinol Metab. 2010;299:E207–E214. doi: 10.1152/ajpendo.00215.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatsunami R, Oba T, Takahashi K, Tampo Y. Methylglyoxal causes dysfunction of thioredoxin and thioredoxin reductase in endothelial cells. J Pharmacol Sci. 2009;111:426–432. doi: 10.1254/jphs.09131fp. [DOI] [PubMed] [Google Scholar]
- 29.Oba T, Tatsunami R, Sato K, Takahashi K, Hao Z, Tampo Y. Methylglyoxal has deleterious effects on thioredoxin in human aortic endothelial cells. Environ Toxicol Pharmacol. 2012;34:117–126. doi: 10.1016/j.etap.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Cheng AS, Cheng YH, Chiou CH, Chang TL. Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in Hep G2 cells. J Agric Food Chem. 2012;60:9180–9187. doi: 10.1021/jf302831d. [DOI] [PubMed] [Google Scholar]
- 31.Hsu WH, Lee BH, Li CH, Hsu YW, Pan TM. Monascin and AITC attenuate methylglyoxal-induced PPARγ phosphorylation and degradation through inhibition of the oxidative stress/PKC pathway depending on Nrf2 activation. J Agric Food Chem. 2013;61:5996–6006. doi: 10.1021/jf401197r. [DOI] [PubMed] [Google Scholar]
- 32.Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One. 2011;6:e21226. doi: 10.1371/journal.pone.0021226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, Ohyama C, Itoh K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34:3421–3434. doi: 10.1128/MCB.00221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefanson AL, Bakovic M. Dietary regulation of Keap1/Nrf2/ARE pathway: focus on plant-derived compounds and trace minerals. Nutrients. 2014;6:3777–3801. doi: 10.3390/nu6093777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawkes HJK, Karlenius TC, Tonissen KF. Regulation of the human thioredoxin gene promoter and its key substrates: a study of functional and putative regulatory elements. Biochim Biophys Acta BBA Gen Subj. 2014;1840:303–314. doi: 10.1016/j.bbagen.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Prior M, Chiruta C, Currais A, Goldberg J, Ramsey J, Dargusch R, Maher PA, Schubert D. Back to the future with phenotypic screening. ACS Chem Neurosci. 2014;5:503–513. doi: 10.1021/cn500051h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang T, Douglass EF, Fitzgerald KJ, Spiegel DA. A “turn-on” fluorescent sensor for methylglyoxal. J Am Chem Soc. 2013;135:12429–12433. doi: 10.1021/ja406077j. [DOI] [PubMed] [Google Scholar]
- 38.Lewerenz J, Albrecht P, Tien MLT, Henke N, Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P, Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- 39.Maher P, Lewerenz J, Lozano C, Torres JL. A novel approach to enhancing cellular glutathione levels. J Neurochem. 2008;107:690–700. doi: 10.1111/j.1471-4159.2008.05620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehren JL, Maher P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem Pharmacol. 2013;85:1816–1826. doi: 10.1016/j.bcp.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 41.Selwood T, Thornalley PJ. Binding of methylglyoxal to albumin and formation of fluorescent adducts. Inhibition by arginine, N-alpha-acetylarginine and aminoguanidine. Biochem Soc Trans. 1993;21:170S. doi: 10.1042/bst021170s. [DOI] [PubMed] [Google Scholar]
- 42.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 43.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 44.Lewerenz J, Maher P. Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem. 2009;284:1106–1115. doi: 10.1074/jbc.M807325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen RE, Lo TWC, Thornalley PJ. Purification and characterisation of glyoxalase II from human red blood cells. Eur J Biochem. 1993;213:1261–1267. doi: 10.1111/j.1432-1033.1993.tb17877.x. [DOI] [PubMed] [Google Scholar]
- 46.Arnér ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 47.Lewerenz J, Maher P, Methner A. Regulation of xCT expression and system xc− function in neuronal cells. Amino Acids. 2012;42:171–179. doi: 10.1007/s00726-011-0862-x. [DOI] [PubMed] [Google Scholar]
- 48.Distler MG, Gorfinkle N, Papale LA, Wuenschell GE, Termini J, Escayg A, Winawer MR, Palmer AA. Glyoxalase 1 and its substrate methylglyoxal are novel regulators of seizure susceptibility. Epilepsia. 2013;54:649–657. doi: 10.1111/epi.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xue M, Rabbani N, Thornalley PJ. Glyoxalase in ageing. Semin Cell Dev Biol. 2011;22:293–301. doi: 10.1016/j.semcdb.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 50.Bélanger M, Yang J, Petit JM, Laroche T, Magistretti PJ, Allaman I. Role of the glyoxalase gystem in astrocyte-mediated neuroprotection. J Neurosci. 2011;31:18338–18352. doi: 10.1523/JNEUROSCI.1249-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen YJ, Huang XB, Li ZX, Yin LL, Chen WQ, Li L. Tenuigenin protects cultured hippocampal neurons against methylglyoxal-induced neurotoxicity. Eur J Pharmacol. 2010;645:1–8. doi: 10.1016/j.ejphar.2010.06.034. [DOI] [PubMed] [Google Scholar]
- 52.Kikuchi S, Shinpo K, Moriwaka F, Makita Z, Miyata T, Tashiro K. Neuro-toxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J Neurosci Res. 1999;57:280–289. doi: 10.1002/(SICI)1097-4547(19990715)57:2<280::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 53.Di Loreto S, Zimmitti V, Sebastiani P, Cervelli C, Falone S, Amicarelli F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int J Biochem Cell Biol. 2008;40:245–257. doi: 10.1016/j.biocel.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 54.Chaplen FWR, Fahl WE, Cameron DC. Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal Biochem. 1996;238:171–178. doi: 10.1006/abio.1996.0271. [DOI] [PubMed] [Google Scholar]
- 55.Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification-A role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol Vasc Syst. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 56.Lu SC. Glutathione synthesis. Biochim Biophys Acta BBA Gen Subj. 2013;1830:3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frade J, Pope S, Schmidt M, Dringen R, Barbosa R, Pocock J, Laranjinha J, Heales S. Glutamate induces release of glutathione from cultured rat astrocytes—a possible neuroprotective mechanism? J Neurochem. 2008;105:1144–1152. doi: 10.1111/j.1471-4159.2008.05216.x. [DOI] [PubMed] [Google Scholar]
- 58.Lewerenz J, Sato H, Albrecht P, Henke N, Noack R, Methner A, Maher P. Mutation of ATF4 mediates resistance of neuronal cell lines against oxidative stress by inducing xCT expression. Cell Death Differ. 2012;19:847–858. doi: 10.1038/cdd.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sagara J, Makino N, Bannai S. Glutathione efflux from cultured astrocytes. J Neurochem. 1996;66:1876–1881. doi: 10.1046/j.1471-4159.1996.66051876.x. [DOI] [PubMed] [Google Scholar]
- 60.Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 61.Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masterjohn C, Park Y, Lee J, Noh SK, Koo SI, Bruno RS. Dietary fructose feeding increases adipose methylglyoxal accumulation in rats in association with low expression and activity of glyoxalase-2. Nutrients. 2013;5:3311–3328. doi: 10.3390/nu5083311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu Y, Chen X. Glyoxalase II, a detoxifying enzyme of glycolysis byproduct methylglyoxal and a target of p63 and p73, is a pro-survival factor of the p53 family. J Biol Chem. 2006;281:26702–26713. doi: 10.1074/jbc.M604758200. [DOI] [PubMed] [Google Scholar]
- 64.Shin MJ, Kim DW, Lee YP, Ahn EH, Jo HS, Kim DS, Kwon OS, Kang TC, Cho YJ, Park J, Eum WS, Choi SY. Tat-glyoxalase protein inhibits against ischemic neuronal cell damage and ameliorates ischemic injury. Free Radic Biol Med. 2014;67:195–210. doi: 10.1016/j.freeradbiomed.2013.10.815. [DOI] [PubMed] [Google Scholar]
- 65.Kim MJ, Kim DW, Lee BR, Shin MJ, Kim YN, Eom SA, Park BJ, Cho YS, Han KH, Park J, Hwang HS, Eum WS, Choi SY. Transduced Tat-glyoxalase protein attenuates streptozotocin-induced diabetes in a mouse model. Biochem Biophys Res Commun. 2013;430:294–300. doi: 10.1016/j.bbrc.2012.10.134. [DOI] [PubMed] [Google Scholar]
- 66.Creighton DJ, Migliorini M, Pourmotabbed T, Guha MK. Optimization of efficiency in the glyoxalase pathway. Biochemistry (Mosc) 1988;27:7376–7384. doi: 10.1021/bi00419a031. [DOI] [PubMed] [Google Scholar]
- 67.Hsu WH, Lee BH, Chang YY, Hsu YW, Pan TM. A novel natural Nrf2 activator with PPARγ-agonist (monascin) attenuates the toxicity of methylglyoxal and hyperglycemia. Toxicol Appl Pharmacol. 2013;272:842–851. doi: 10.1016/j.taap.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 68.Kwon AT, Arenillas DJ, Worsley Hunt R, Wasserman WW. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda) 2012;2:987–1002. doi: 10.1534/g3.112.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Floen MJ, Forred BJ, Bloom EJ, Vitiello PF. Thioredoxin-1 redox signaling regulates cell survival in response to hyperoxia. Free Radic Biol Med. 2014;75:167–177. doi: 10.1016/j.freeradbiomed.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 71.Mitozo PA, de Souza LF, Loch-Neckel G, Flesch S, Maris AF, Figueiredo CP, dos Santos ARS, Farina M, Dafre AL. A study of the relative importance of the peroxiredoxin-, catalase-, and glutathione-dependent systems in neural peroxide metabolism. Free Radic Biol Med. 2011;51:69–77. doi: 10.1016/j.freeradbiomed.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 72.Harris C, Hansen JM. Nrf2-mediated resistance to oxidant-induced redox disruption in embryos. Birth Defects Res B Dev Reprod Toxicol. 2012;95:213–218. doi: 10.1002/bdrb.21005. [DOI] [PubMed] [Google Scholar]
- 73.Mukherjee S, Gangopadhyay H, Das DK. Broccoli: a unique vegetable that protects mammalian hearts through the redox cycling of the thioredoxin superfamily. J Agric Food Chem. 2008;56:609–617. doi: 10.1021/jf0728146. [DOI] [PubMed] [Google Scholar]
- 74.Arredondo F, Echeverry C, Abin-Carriquiry JA, Blasina F, Antúnez K, Jones DP, Go YM, Liang YL, Dajas F. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free Radic Biol Med. 2010;49:738–747. doi: 10.1016/j.freeradbiomed.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 75.Bonifati V, Rizzu P, Baren MJ, van Schaap O, Breedveld GJ, Krieger E, Dekker MCJ, Squitieri F, Ibanez P, Joosse M, Dongen JW, van Vanacore N, Swieten JC, van Brice A, Meco G, Duijn CM, van Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset Parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 76.Lee J, Song J, Kwon K, Jang S, Kim C, Baek K, Kim J, Park C. Human DJ-1 and its homologs are novel glyoxalases. Hum Mol Genet. 2012;21:3215–3225. doi: 10.1093/hmg/dds155. [DOI] [PubMed] [Google Scholar]