

Abstract
Cancer is rapidly becoming the top killer in the world. Most of the FDA approved anticancer drugs are organic molecules, while metallodrugs are very scarce. The advent of first metal based therapeutic agent, cisplatin, launched a new era in the application of transition metal complexes for therapeutic design. Due to their unique and versatile biochemical properties, ruthenium-based compounds have emerged as promising anti-cancer agents that serve as alternatives to cisplatin and its derivertives. The ruthenium(III) complexes have successfully been used in clinical research and their mechanisms of anticancer action have been reported in large volumes over the past few decades. Ruthenium(II) complexes have also attracted significant attention as anticancer candidates; however, only few of them have been reported comprehensively. In this review, we discuss the development of ruthenium(II) complexes as anticancer candidates and biocatalysts, including arene ruthenium complexes, polypyridyl ruthenium complexes, and ruthenium nanomaterial complexes. This review focuses on the likely mechanisms of action of ruthenium(II)-based anticancer drugs and the relationship between their chemical structures and biological properties. This review also highlights the catalytic activity and the photoinduced activation of ruthenium(II) complexes, their targeted delivery, and their activity in nanomaterial systems.
Graphical abstract
This review covers ruthenium(II) complexes as anticancer drugs in single molecules and nanomaterials including targets, mechanisms, SAR, PDT and nano-systems.
1. Introduction
Due to a rapid increase in cancer cases worldwide, there is an indispensable need for the development and screening of potential anticancer agents. In this regard, metal complexes hold potential as novel anticancer agents against a wide majority of cancer types.1–7 Cisplatin or cis-diamminedichloroplatinum(II) is the most widely known metal-based anticancer drug. Cisplatin has been shown to have efficacy against lung, head, ovarian, neck, and esophageal cancers.8–10 Although cisplatin and its derivatives are efficacious against the vast majority of cancers, they also produce non-cancer cell toxicity, thereby causing severe adverse effects, including peripheral neuropathy, hair loss and myelotoxicity in patients.11–17 The resistance of tumors to platinum decreases the efficacy of platinum-based or even renders them ineffective, causing treatment failure.18–22 In the design of new anticancer drugs,23–29 the ruthenium complexes have raised great interest and have been tested against a number of cancer cell lines,30–36 and are regarded as promising candidates for alternative drugs to cisplatin and its derivatives.
Ruthenium is a transition metal in group 8, the same chemical group as iron. Ruthenium has two main oxidation states, Ru(II) and Ru(III). Ruthenium(IV) compounds are also possible, but they are generally unstable due to their higher oxidation states.37 The ruthenium ion is typically hexa- coordinated with octahedral coordination geometries. Generally, the thermodynamic and kinetic stability of Ru(III)complexes are lower than that of Ru(II) complexes, and the kinetics of the hydration of Ru(II/III) compounds depends significantly on the nature of their ligands and net charge.38 Many Ru(III) compounds contain exchangeable ligands and require activation by the tumor microenvironment.39 The antitumor properties of the Ru(III) complexes occur when they are reduced to their corresponding Ru(II) counterparts in vivo. Under biological circumstances of low oxygen concentration, acidic pH and high levels of glutathione, the Ru(II/III)redox potential can be altered, and thus, Ru(III) complexes can be readily reduced to Ru(II) complexes.40–42 As the first approved ruthenium complex in clinical trials, NAMI-A, [ImH][trans-RuCl4(DMSO)(Im)] (Im = imidazole, DMSO = imethylsulfoxide; Fig 1), has low potency in terms of direct cytotoxicity towards cancer cells in vitro; however, in vivo, it has significant efficacy in inhibiting tumor metastasis.43–48 The mechanism of action of NAMI-A remains to be elucidated. There are data suggesting that NAMI-A is capable of binding to DNA and RNA. It can bind to the histidine residues of serum albumin (has or bsA) under physiological conditions.49–51 However, the low therapeutic efficiency, the progressive disease in the clinical studies (phase I) and partial response (phase I/II) limited the further clinical use of NAMI-A and resulted in the failure of the clinical investigations.52 Alessio et al. believed that the main reason of the failure is more philosophical, but nevertheless fundamental.53 Subsequently, the KP1019 [trans-tetrachlorobis -(1H-indazole)ruthenate(III)] designed by the Keppler group entered clinical trial.54,55 But its low solubility limits its further development and its better soluble sodium salt KP1339 is currently undergoing clinical trials.56
Fig. 1.
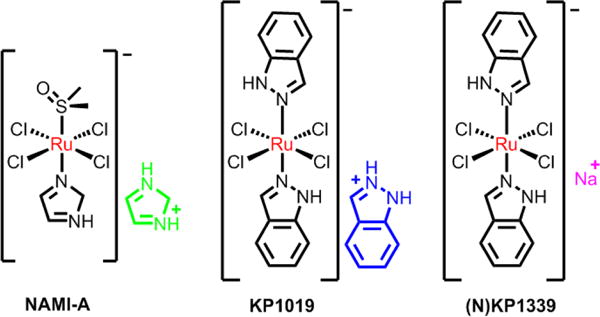
Three ruthenium(III) compounds in clinical trials.
Recently, many organometallic Ru(II), inorganic Ru(II) and nanomaterial Ru(II) complexes have been designed and developed into anticancer drugs, with potent therapeutic properties.57–61 With the development of new technology, such as photodynamic therapy (PDT) and nanomaterials,62–69 Ru(II) complexes can be photophysical and bioactive, improving the efficacy and selectivity of Ru(II) complexes as anticancer drugs, as well as allowing for the elucidation of their mechanism of action. The Ru(II)-polypyridyl compound (TLD-1433) recently entered phase IB clinical trials as PDT agent in patients with bladder cancer at 2015.70 Therefore, the direct study of Ru(II) complexes for cancer therapy contributes to the design of new metal-based drugs.
Generally speaking, the following options are viable in the design of ruthenium-based drugs: (i) constructing complexes with selective and specific targets; (ii) exploiting the potential targets and mechanisms; (iii) the evaluation of structure-activity relationships; (iv) exploiting prodrugs that can be activated by light; and (v) exploiting drug accumulation and activation at the tumour tissues with the nano drug-delivery system. This Review aims to present the reader with an impression of the latest progress of development of ruthenium complexes as anticancer agents as well as biocatalysts from single molecule compounds to nanomaterials. We present an overview of the field today, hoping that colleagues not only may taste a comprehensive development of ruthenium(II) complexes as metallodrugs, but that we can inspire more researchers to enter the charming field of metallodrugs.
2. The cellular uptake and potential targets of Ru(II) complexes
2.1 Cellular uptake
The uptake of ruthenium complexes by cancer cells or other cells is important for selective and effective cancer therapy. In order to move into living cells, molecules and atoms must cross or penetrate the cell membrane. The cell membrane contains diverse proteins and lipids, and it functions to regulate what substances enter into the cells. The general mechanisms of cellular uptake for small molecule drugs are shown in Fig. 2.71 Ru(II) complexes are known to enter cells through multiple mechanisms, such as passive diffusion, active transport, and endocytosis.71 However, it is noted that most nanostructured ruthenium complexes enter cells by endocytosis.72,73 Confocal laser scanning fluorescence microscopy, inductively coupled plasma mass spectrometry (ICP-MS), flow cytometry and transmission electron microscopy are often used to determine the intracellular accumulation of ruthenium complexes.74 As the changes in ligands and hydrophobicity can modulate cellular uptake and cellular localization, the intracellular distribution of ruthenium complexes in cells have been discussed with regard to: (a) the net ionic charge, which can undergo change from anionic to cationic; (b) the degree of lipophilicity based on the octanol/water partition coefficient; (c) the structures and sizes of the ligands.75
Fig. 2.
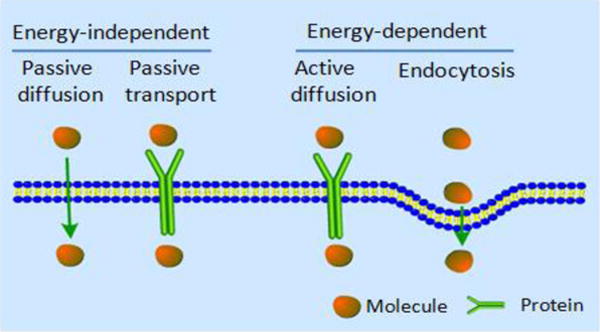
Conmmon cellular uptake mechanisms of drugs. Reproduced with permission from ref. 71. Copyright 2012, Royal Society of Chemistry.
2.1.1 Localization in nucleus
The cell nucleus is an enclosed membranous organelle that exists in eukaryotic cells, containing nucleic acids and proteins. Typically, the nucleus is regarded as one of the most important organelles in eukaryotic cells. Ruthenium complexes have been shown to interact with nucleic acids and proteins via multiple binding modes in the nucleus.73 The synthesis of Ru(II) complexes would represent an ideal scaffold for the optimization of therapeutic compounds targeted to the nucleus.76 In order to allow Ru(II) complexes to target the nucleus, a nuclear targeting peptide D-octaarginine (D-R8) was conjugated to a Ru(II) complex.77 The Ru-D-R8 significantly enhanced cellular uptake compare to the Ru(II) complex and was found to preferentially accumulate in the nucleus at high concentration (15–20 μM). The authors speculated that a high concentration of Ru-D-R8 could enter into the nucleus via a nonendocytic uptake mechanism. Similarly, the same research group found that Ru-RrRK, bearing a tetrapeptide, became localized in the nucleus at a high concentration (100 μM).78 It should be noted that a low concentration of the conjugated Ru(II) complexes had a less cellular uptake. The above studies mainly focused on the direct imaging of the nucleus and the compounds did not have significant cytotoxic efficacy. Subsequently, Tan et al. designed three Ru(II) complexes containing a β-carboline alkaloid. These complexes were shown to penetrate into the cell nucleus and to have significant cytotoxic efficacy.79 The most potent complex of this family had greater efficacy than cisplatin, and the mechanistic studies showed that reactive oxygen species (ROS), autophagy and increased sub-G1 phase arrest were involved in eliciting apoptosis. Recently, Chao et al. synthesized a new cycloruthenated [Ru(bpy)(phpy)(dppz)]+ (bpy=bipyridine, dppz = dipyridophenazine) by replacing the bpy ligand with the cycloruthenated ligand (Fig. 3), phpy (2-phenylpyridine) from the molecule [Ru(bpy)2(dppz)]2+. This compound readily entered into the nucleus and had IC50 values lower than cisplatin.80
Fig. 3.
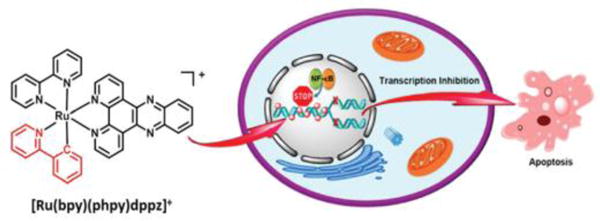
The schematic description of the anticancer effect of the nucleic targeting complex [Ru(bpy)(phpy)dppz]+. Reproduced with permission from ref. 80. Copyright 2015, American Chemical Society.
2.1.2 Localization in mitochondria
Although the nucleus is reported to be the key target for ruthenium complexes, many studies have demonstrated that the accumulation of some ruthenium complexes in the nucleus is far lower than that in other subcellular regions.74,81 Some nonnuclear targets, such as the cell surface, and especially mitochondria, have also been reported to be targets for the anticancer activity of some Ru(II) complexes. Mitochondria function plays a significant role in cellular metabolism and under certain cellular conditions, release molecules that can activate the extrinsic and intrinsic apoptotic pathways.82 Two key characteristics of mitochondria include mitochondrial DNA nucleoids anchored to the matrix side of the inner membrane, and the extremely negative membrane potential (−160 to −180 mV) caused by the proton gradient across its inner membrane.83–85 The negative potential of the inner membrane attracts lipophilic cations, including metal complexes such as the Ru(II) complexes. The lipophilicity of Ru(II) complexes can be modulated by adjusting the ligands and the valence of complexes, which partly affects the uptake and targeting of Ru(II) complexes. For examples, Gasser and co-workers found that [Ru(dppz)2(CppH)]2+ (1a, CppH = 2-(2′-pyridyl)pyrimidine-4-carboxylic acid) possesses two positive charges and accumulates in the mitochondria (Fig. 4). In addition, 1a had significant anticancer efficacy in A2780 cancer cells, with an IC50 value of 2.8 μM, which was similar to that of cisplatin (IC50: 2.9 μM).86, 87 Moreover, 1a was more efficacious in cisplatin-resistant A2780/CP70 cells than cisplatin and less cytotoxic than cisplatin in healthy MRC-5 cells.86 Dickerson and co-workers reported that the Ru(II) complex 1b, carrying an overall charge of +2, can localize to the mitochondria and induce rapid membrane depolarization and necrotic cell death.88 In contrast, its analogue Ru(II) complex (1c), carrying an overall charge of -4, does not localize to the mitochondria and lacks efficacy as it does not localize to the mitochondria.88 Chao et al. reported that the mitochondrial-targeting Ru(II) complexes, complexes 1d to 1f, induce cellular apoptosis via the mitochondrial pathway.89,90 In addition, the combination of mitochondrial-targeting and photodynamic therapy significantly increase the selectivity and anticancer efficacy of the Ru(II) complexes.91 Although many Ru(II) complexes target the mitochondria and induce cell apoptosis, most research has been directed towards DNA-targeting complexes. Therefore, there is potential for further study of mitochondrial-targeting Ru(II) complexes and the elucidation of their mechanisms for inducing cell apoptosis.
Fig. 4.
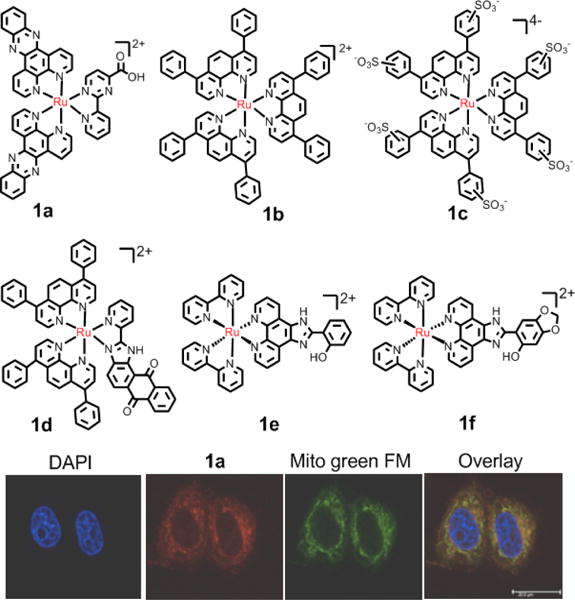
Representation of Ru(II) compounds that accumulate in mitochondria. Bottom image:fluorescence confocal microscopy images of HeLa cells incubated with 1a with commercial dyes. Reproduced with permission from ref. 86. Copyright 2015, American Chemical Society.
2.1.3 Localization in lysosomes
Lysosomes are spherical vesicles found in almost every eukaryotic cell and contain hydrolytic enzymes that degrade numerous biomolecules.92 Lysosomes play an essential role in many physiological processes and cell signaling pathways, including intracellular transport, protein degradation and recycling, endocytosis and apoptosis. Differing from two-membrane systems, such as cell membranes and mitochondria, lysosomes are single-membrane organelles. Lysosomes are an intracellular target and the final destination for numerous macromolecules, including drugs formulated as nanomaterials.93 The luminal environment pH is 4.6–5.0 in lysosomes, which helps optimize hydrolysis and digestion. Because the cell membrane is typically impermeable to complexes larger than 1 kDa, and given that most medical nanomaterials range from tens to hundreds of nanometers in diameter, nanomaterials generally enter cells through an endocytic process.93 In the endocytic process, nanomaterials are transported to the endosomes and then fuse with lysosomes, where the nanomaterials begin to degrade. Moreover, due to the acidic environment of lysosomes, some nanomaterials are designed to release ruthenium complexes or other drugs, preferentially in an acidic environment, to improve the stability and selectivity of the nanomaterial. Although lysosomes are regarded as the targets of nanomaterial drugs, some organometallics have been found to target organelles.94–97 The highly positively charged Ru(II) polypyridyl complex selectively localizes in the lysosomes, and can be used in photodynamic therapy as photosensitizers (PS).98 Moreover, lysosomes participate in an autophagic process that is linked to carcinogenesis and resistance to chemotherapy.99 Targeting these pathways could constitute a novel approach to cancer therapy.
2.2 The potential targets of ruthenium complex
There are some different molecules with special structures and distinct function, especially DNA and proteins, that have critical roles in determining cellular activity.76,100 The understanding of how ruthenium complexes interact with these two specific targets within cell is therefore important for exploring the anticancer mechanism and selecting the most potent ruthenium complex for selective and effective therapy.
2.2.1 DNA as a target
Cancer cells have a high rate of proliferation due to a loss of control of the normal restraints on cell cycle division. Also, cancer cell proliferation is regulated by DNA. Many ruthenium compounds are known to have high selectivity for binding to DNA.101–105 The electron-deficient metal atoms in these complexes might act as electron acceptors for electron-rich DNA nucleophiles by the hydrolysis of ligands. Furthermore, Ru(II) complexes can bind to DNA via interaction with aromatic ligands. There are two main categories of binding modes between DNA and Ru compounds: covalent and noncovalent binding.l The covalent binding is irreversible and forms adducts consisting of DNA and Ru(II) complexes. For example, the complex [(η6-arene)Ru(en)Cl]PF6] (arene= biphenyl, en= ethylenediamine) can bind to the N7 atom in guanosine.106 The covalent mode of binding in Ru–DNA distorts the DNA backbone, which impairs DNA replication and transcription. The non-covalent binding of Ru(ll) complexes is usually reversible and occurs as electrostatic binding, intercalation, and groove binding, amongst which intercalation has received the most attention. Intercalation occurs when planar aromatic compounds are inserted between adjacent base pairs in the DNA double helix.107 Many Ru(II) polypyridyl complexes can intercalate DNA with high affinity in vitro, and serve as DNA molecular probes. For example, the well known complex [Ru(bpy)2(dppz)]2+ was reported as a DNA light switch in vitro,108 but this complex did not readily cross the cell membrane. Gill et al. reported that the multi-intercalator,109 2a, can bind DNA with a binding constant of 2.5× 106 M−1. Moreover, it can bind cellular DNA in fixed and membrane-permeabilized HeLa cells (Fig. 5).109 Compound 2a immediately stalled the progression of the replication fork in HeLa cells, resulting in the activation of DNA damage checkpoints and blocking the cell cycle between the G1 and S stage. This complex induced death in HeLa cells, with an IC50 value of 38 μM.109 Önfelt et al. reported that the binuclear Ru(II) complex, Δ-Δ [μ-C4(cpdppz)2-(phen)4Ru2]4+ (C4(cpdppz)2 = N,N’ bis-(cpdppz)-1,4-diaminobut-ane; cpdppz = 12-cyano12,13-carbonyl-11H-cyclopenta[b]- dipyrido[3,2-h:2′3’-j]phen-azine-12-carbonyl; phen = 1,10-phenanthroline), binds with high affinity to DNA (Kb ∼108 M−1).110 The entry of this complex into the nucleus was facilitated by electroporation, and it induced the run apoptosis at 10−4 M. Thomas et al. showed that the dinuclear compound 2b (Fig. 5) rapidly targets the nuclei of MCF-7 cancer cells via a non-endocytic mechanism. However, its IC50 value, after 24 h of incubation with MCF-7 cells, was 138 μM.111,112
Fig. 5.
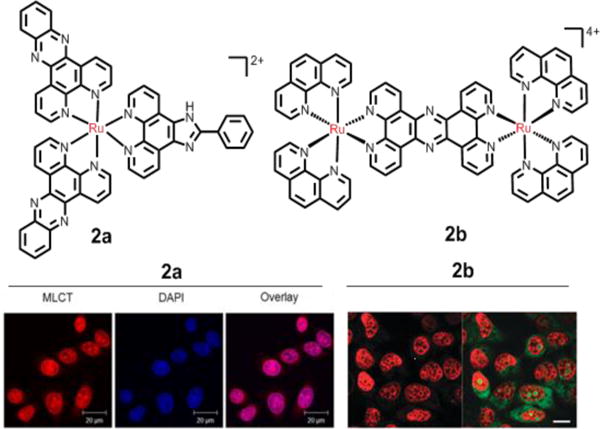
Representation of Ru(II) compounds that target DNA. Left-hand fluorescence image: the nuclear DNA staining of 2a in PFA-fxed HeLa cells, as evident by red luminescence, with co-staining by nuclear DNA dye DAPI. Right-hand fluorescence image: the nuclear DNA staining of the dinuclear 2b in MCF-7 cells, as evident by the red luminescence, with co-staining SYTO-9 (green). Reproduced with permission from ref. 109 and 111, respectively. Copyright 2015, Nature Publishing Group.
2.2.2 Proteins as targets
The primary cellular target of ruthenium complexes is DNA. However, data also indicates that certain proteins may be targets for ruthenium complexes, especially protein kinases.113–116 It is well known that certain enzymes play key roles in metabolic pathways associated with intercellular uptake, cancer cell proliferation, and cell death. The targeting of these biological processes with metal complexes has shown promising anticancer efficacy (Fig. 6).117 Ru(II) complexes have the potential to inhibit protein kinases due to their facile, three-dimensional structure and physicochemical properties. These Ru(II) complexes are highly stable, kinetically inert and stable in biological environments. The targeting capability and diverse functions of ruthenium complexes can be realized by modification of the ligands.118,119 In the early work of Dwyer and colleagues, the Ru(II) polypyridyl complex 3a and 3b were designed to act as acetylcholinesterase (AChE) inhibitors by a combination of electrostatic and hydrophobic interactions between the Ru(II) complexes and the peripheral anionic site of AChE.120 Generally, most of the Ru(II) complexes bind to the active site of enzymes through their biologically active ligands. The natural organic product staurosporine is a potent ATP-competitive inhibitor of protein kinase.121,122 Meggers and co-workers designed several ligands that retained the active core of staurosporine and coordinated this to ruthenium and this yielded compound 3c, 3d and 3e which were inhibitors of the protein kinases Pim1, MSK1, and GSK3R, respectively123,124 The kinetic inertness of the coordination bonds allowed these Ru(II) compounds to successfully mimic the organic product staurosporine, thereby inhibiting these protein kinases. The binding of ATP and the active core of staurosporine was illustrated by the co-crystal structures of the protein kinase Pim-1 with the Ru(II) complexes, which verified that the ruthenium ion only has a structural role and is not involved in binding with the active site of ATP.125–128 Similarly, Dyson et al. designed a compound, 3f, which contains a benzimidazole-phenoxazine derivative that significantly inhibited P-glycoprotein (P-gp). 3f was shown to inhibit P-gp activity and thus has the potential to attenuate multidrug resistance due to the overexpression of P-gp.129 Dyson et al. also designed 3g and its analogues that contained ethacrynic acid (EA) ligands that inhibited glutathione-S-transferase (GST) in A2780 and cisplatin-resistant A2780cisR cell lines.130
Fig. 6.
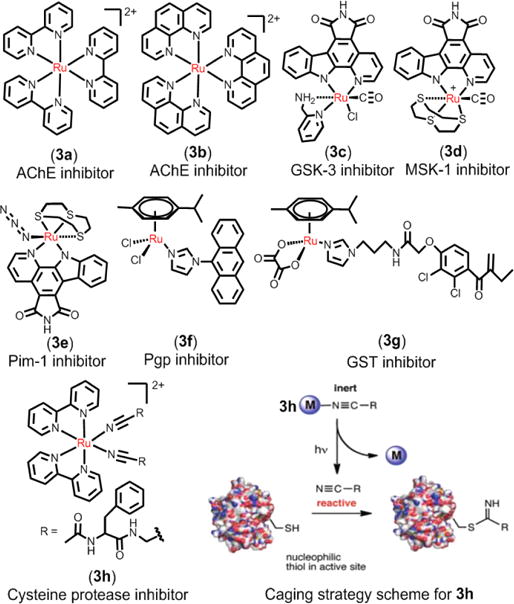
Representation of Ru(II) compounds that target proteins as enzymes inhibitors. Right-bottom image: caging strategy for comound 3h as photoinduced cysteine protease inhibitors. Reproduced with permission from ref. 131. Copyright 2015, American Chemical Society.
Respondek et al. developed a novel method to inhibit the activity of cysteine protease by using the photoactive 3h containing the cysteine protease inhibitor Ac-Phe-NHCH2CN.131 As a result of binding to the Ru(II) center, the nitriles of Ac-Phe-NHCH2CN were caged, and thus 3h could not bind to cysteine protease (Fig. 6). However, when 3h was irradiated, it released Ac-Phe-NHCH2CN, which can potently inhibit the cysteine proteases papain and cathepsin B. This method provides a novel approach to control the activity of enzymes. In addition, the identification of cellular target proteins is a major challenge in drug development. However, Hartinger et al. first identify 15 cancer-related proteins that associated with the observed antimetastatic ruthenium organometallic complex based on 1,3,5-triaza-7-phosphaadamantane (RAPTA) by chemical proteomic.116 As shown in Fig. 7, this methodology has broad applicability beyond RAPTA complexes and enables, for the first time, the direct identification of intracellular interactions of metallodrugs with proteins.
Fig. 7.
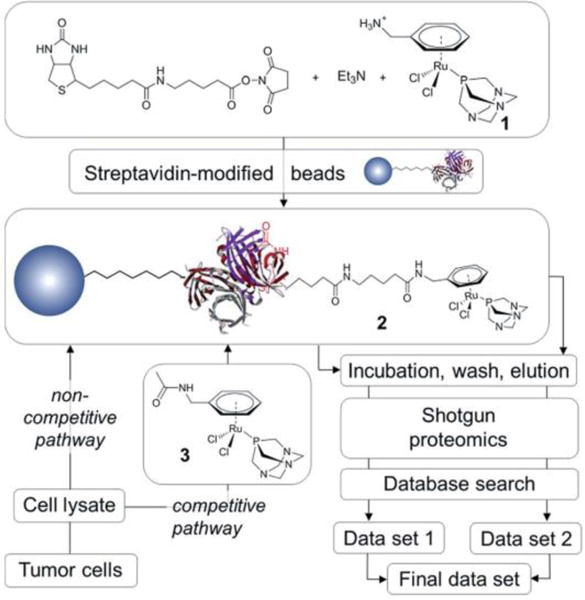
Schematic representation of the work-flow used in the metallodrug pull-down experiments performed by Hartinger et al. Reproduced with permission from ref. 116. Copyright 2012, Royal Society of Chemistry.
3. Cell death process
Cell death can occur via several mechanisms (Fig. 8).132 Apoptosis is primarily induced by the activation of two pathways in cells, the intrinsic and extrinsic (death receptor- mediated) pathways.132 The intrinsic pathway, also known as the mitochondria-mediated pathway, is activated by DNA damage, oxidative stress and endoplasmic reticulum (ER) stress.133 These stimuli can induce the mitochondrial release of the cytochrome c, which can activate the apoptotic protease activating factor 1, and regulate other proteins involved in apoptosis. Chen and Chao et al. conducted experiments to determine the effect of Ru(ll) complexes on the intrinsic pathway of apoptosis.89,90,134,135 Typically, non-apoptotic cell death occurs by necrosis and ER stress.132 The process of autophagy is regulated by autophagy-related (ATG) proteins and the role of autophagy in cancer is complex and is contextually dependent.136 For example, autophagy can inhibit or facilitate cell death. The process of necrosis differs from the apoptosis in a number of ways.137 Interestingly, certain stimuli that induce apoptosis can also cause necrosis, such as ROS. It has been shown that Ru(ll) complexes can induce autophagy and necrosis, as well as apoptosis.138–140
Fig. 8.
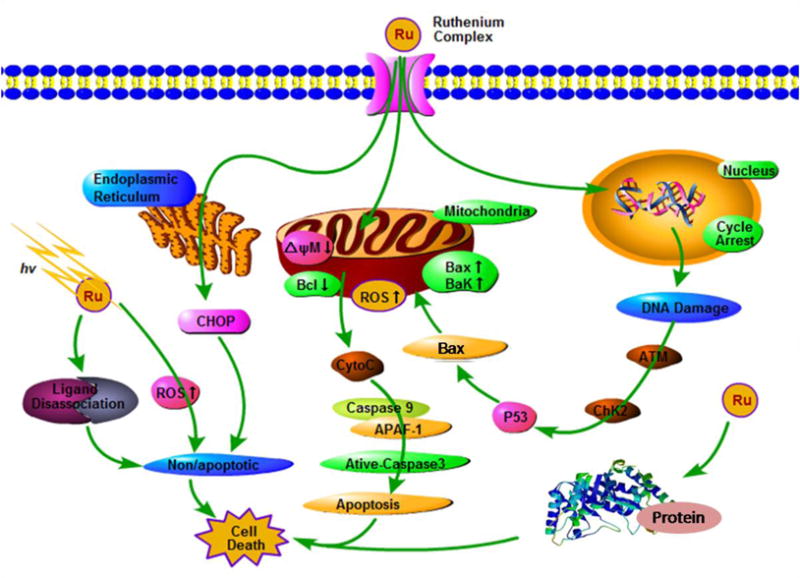
General representation of the main targets and proposed mechanisms of action of ruthenium compounds as anticancer drugs.
4. Arene ruthenium(II) complexes
Arene ruthenium(II) complexes are also called piano stool complexes.141 Sadler and Dyson are the pioneers in the field of anticancer arene ruthenium complexes.142,143 Arene Ru (II) complexes have the general formula [(η6-arene)Ru(X](Y)(Z)], as shown in Fig. 9. Common arene rings include benzene (ben), methylisopropyl benzene (cym), biphenyl (bip) and dihydroanthracene (dha). The ligands X and Y can be two monodentate ligands or one bidentate ligand, and Z is usually a leaving group, such as a halogen.144 The arene is regarded as the core component of arene Ru(II) complexes. Furthermore, the arene rings are hydrophobic, which facilitates the entry of Ru(II) complexes into cells. The arene rings determine the electron distribution of the Ru(II) complex, which affects the stability of the Ru(II) complexes. However, the hydrolysis of Ru-Z bonds is also affected by pH and the concentration of Z in the environment. The water solubility and volume of the chelating ligand and leaving group can also affect the anticancer efficacy of arene Ru (II) complexes.141
Fig. 9.
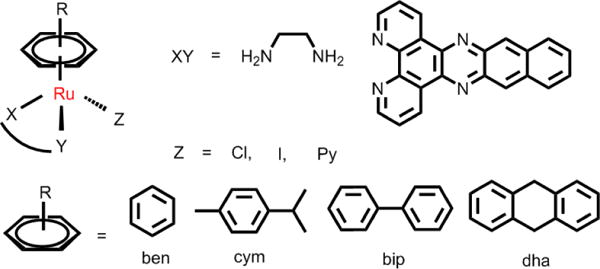
Common structures of [(η6-arene)Ru(X](Y)(Z)].
4.1 Arene Ru(II) complexes with N,N-chelating ligands
Common N,N-chelating ligands include aliphatic diamine, aromatic diamine and pyridine derivatives. The arene ruthenium complexes containing ethylenediamine (en) chelating ligands have been studied systematically by Sadler.58 Sadler’s group reported that variation in the leaving group, the N,N-chelating ligand and the arene ring, can have a significant effect on chemical and biological activity.58 The complex [(η6-C6H5)Ru(en)Cl]+ (4a, Fig. 10) had anticancer efficacy against A2870 cancer cells, with an IC50 value of 17 μM.58 Another compound, 4b, was synthesized by replacing the benzene with a more hydrophobic biphenyl group, and 4b had similar anticancer efficacy compared to carboplatin in A2780 cells.58 In order to gain insight into the mechanism of arene Ru(II) complexes as anticancer drugs, Chen and co-workers reported the recognition of nucleic acid derivatives with the compound [(η6-C6H5)Ru(en)X], where arene = Bip, Tha, Dha, Cym or Ben, X = Cl− or H2O.106 At neutral pH, pseudo-octahedral diamino arene Ru(II) complexes were found to discriminate between G and A nucleobases. In kinetic studies (pH 7.0, 298 K, 100 mM NaClO4), the rates of reactions of cGMP (3′, 5′-cyclic-GMP) with X (Cl− or H2O) decreased according to arene present as follows: Tha > Bip > Dha, Cym > Ben (Scheme 1). The cytotoxicity study showed of the family [(η6-C6H5)Ru(en)Cl]+ was distinctly correlated to the above reactions rates.103 These findings indicate that diamine NH2 groups, the hydrophobic arene, and the chlorine leaving group all have important roles in the interaction of nucleic acids with arene Ru(II) complexes. In addition, Romero-Caneló et al. determined the activity of iodido versus chlorido N,N-chelated arene Ru(II) complexes with imino-pyridine ligand.26 The subtle changes in the monodentate ligands (Cl, I) can lead to major changes in cellular metabolism and mechanisms of anticancer efficacy. The iodido complex, 4d, was more potent and selective than the chlorido analogue, 4c, towards cancer cell lines and was not cross-resistant to platinum-based drugs. Moreover, these two halido ligands were found to affect cellular uptake of the arene Ru(II) complexes.145 The chloride complex, 4c, was largely taken up through active transport, whereas the iodide complex 4d entered cells through passive transport. After 24 h of drug exposure, the ruthenium accumulation of 4d was 1.6 times greater than that of 4c in A2780 cells.145 Additional experiments showed that the ABC transporter P-gp contributed to the efflux of 4c, but had little effect on the cellular efflux of 4d, indicating that the passive transport may help drugs circumvent some resistance mechanisms. Montani et al. determined the antitumor activity of 4e in vivo and found that 4e effectively inhibited the growth of A17 triple negative breast cells transplanted into mice.146 4e was rapidly eliminated from the liver, kidney and bloodstream due to its high hydrosolubility, and it has with excellent therapeutic efficacy and minimal adverse effects. Immunohistological studies showed a significant reduction in the number of tumor-infiltrating regulatory T cells caused by 4e.
Fig. 10.
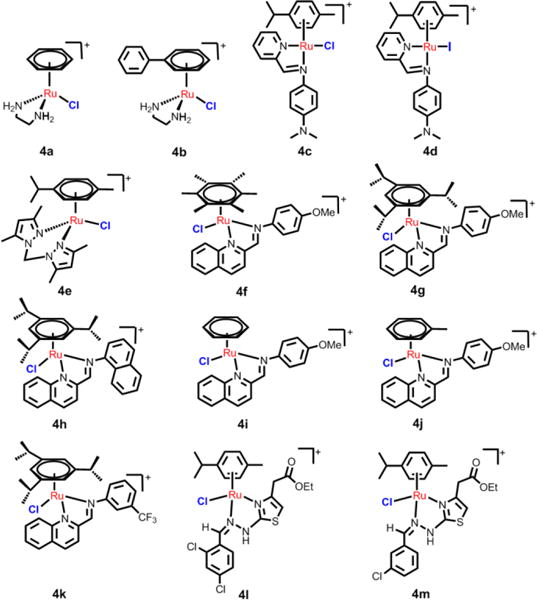
Common structures of arene Ru(II) compounds with N,N-chelating ligands.
Scheme 1.
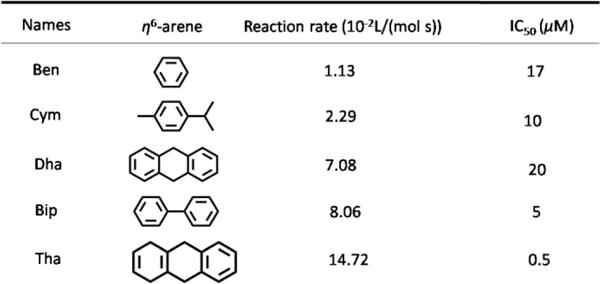
The reaction rate of [(η6-C6H5)Ru(en)Cl]+ family with cGMP and their cytotoxicity towards A2780 cells. Data originate from ref. 103 and 108.
Chow et al. synthesized a more effective arene Ru(II) compound, 4g, compared to cisplatin, by using high-throughout screening.147 Compound 4g displayed low micromolar IC50 values against A2780, A2780cisR, MCF7, HCT116 and SW480 cells. The authors also found that the water-soluble and stable half-sandwich arene Ru(II) Schiff-base (RAS) complexes, 4f and 4g, can induce non-apoptotic programmed cell death (PCD) through the ER stress pathway.148 The mechanism of action was significantly different between the two complexes, despite modest structural variations. 4g elicited ROS-mediated ER stress, while 4f’s efficacy was independent of ROS. These two complexes were more efficacious against apoptosis-resistant cells compared to clinical drugs such as oxaliplatin. This work provides the basis for targeting ER stress modulation using Ru(II) complexes to bypass apoptosis resistance. Recently, the same group study of the relationship between the structures and cytotoxicities of a series of arene Ru(II) Schiff-base (RAS) complexes was conducted.149 They reported that the RAS complexes with more hydrophobic ligands displayed higher intercellular accumulation. For example, 4g, with more hydrophobic π-donating arene ligands, accumulated 7.5-fold more than the least hydrophobic compound, 4i. 4g exhibited more than a 150-fold increase in cytotoxicity compared to 4i in HCT116 cells. The authors also found that 4h accumulated 3 times less than 4f, but had a higher cytotoxicity, which suggested that the higher cytotoxicity was partially correlated to the intercellular uptake of the RAS complexes. In addition, 4j and 4k, with more π-acidic 3-CF3 or 3-Cl substituent groups, were more likely to be hydrolyzed compared to 4g, which had a 4-OMe substituent group. Further experiments indicated that all of the RAS complexes induced p53-independent cytotoxicity. Therrien et al. designed hydrazinyl-thiazolo arene Ru(II) complexes and these compounds were more cytotoxic to HeLa, A2780 and A2780cisR cells compared to cisplatin and oxaliplatin.150 The representative complexes, 4l and 4m, were induced HeLa cell death by disrupting mitochondrial membranes and damaging the nucleus. The biological activity of the two compounds was first evaluated using microarray gene expression assay and ingenuity pathway analysis. 4I and 4m affected p53 signaling, which induced apoptosis. In addition, 4l and 4m activate the genes that correlate with overcoming cisplatin-resistance, such as PRMT2, FAS, ZMAT3, BBC3/PUMA, and PDCD4. It is hypothesized that 4l and 4m will surmount cisplatin resistance in ovarian cancer therapy.
In addition, Betanzos-Lara et al. investigated the effect of arene Ru(II) complexes on DNA. Their results indicated that arene Ru(II) complexes [(p-cym)Ru(bpm)(py)][PF6]2 (where p-cym = para-cymene, bpm = 2,2′-bipyrimidine and py = pyridine) can selectively photodissociate a monodentate ligand (py) when excited by visible light.151 Betanzos-Lara et al. also studied the relationship between photoactivity and structure of the arene Ru(II) pyridine and pyridine-derivative complexes with N,N-chelating ligands. These complexes can activate the release of its monodentate ligands by photoirradiation to increase the activities of these photoactivatable arene ruthenium complexes.152 They found that increasing the electron-donating substituents in the Py ring moderately increased the extent of the photoinduced hydrolysis. In contrast, more electron-donating substituents on the arene ring increased both the extent and the rate of photoinduced hydrolysis, and increasing the aromatic N,N-chelating ligands decrease the extent of photoinduced hydrolysis. These complexes were only tested for cytotoxicity in the dark, with the IC50 values in the range of 9.0–60 μM in A2870 cells.152 However, Wang et al. reported that the ferrocenly pyridine-based arene Ru(II) complex, 5a, was an efficacious photosensitizer that killed cancer cells (Fig. 11).153 Interestingly, this complex could produce both hydroxyl radicals and 1O2 as well as photoinduced monodentate ligand dissociation upon visible light irradiation (> 400 nm). The complex produced DNA photodamage under light irradiation and had significant photoactivated anticancer efficacy, at 70 uM and irradiated for 30 min, decreased the viability of SKOV3 and A549 cells by 70% and 62%, respectively. In addition, the same research group also determined the effect of substituents on the photoactivity of the [(η6-p-cymene)Ru(dpb)(py-R)]2+ (Fig. 11).154 The complexes induced DNA photocleavage and DNA photobinding by photoinduced ligand dissociation and 1O2. The magnitude of 1O2 production by the complexes was: 5e > 5f > 5d > 5h ≈ 5c ≈ 5b, and the order of ligand dissociation rates were: 5h > 5f > 5e > 5d > 5c > 5b. 5e produced the most potent phototoxicity under irradiation in A549 cells. It was reported that the difference in the photoactivities may result from the influence of the substituent groups on the energy levels of 3MLCT and 3MC, and the energy gaps between 3MLCT, 3MC and 3IL.
Fig. 11.
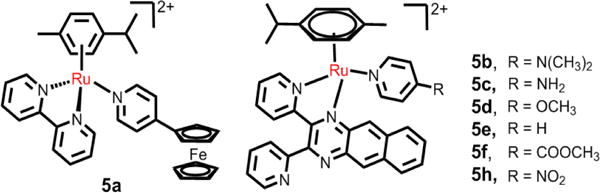
The N,N- ligands arene Ru(II) compounds with good photoactivity.
4.2 Arene Ru(II) complexes with N,O-, O,O- and C,N- ligands
N,O-chelating ligands, including tetrahydroisoquinoline, select amino acid ligands, and the O,O-ligands, are common β-diketonate and pyrone ligands.155 Chelopo et al. determined the anticancer efficacy of several arene Ru(II) complexes containing 1,2,3,4-tetrahydroisoquinoline amino alcohol ligands (6a-6d, Fig 12) in the human cancer cell lines MCF-7, A549, and MDAMB-231.156 These complexes were moderately efficacious against only MCF-7 cells, with the lowest IC50 value of 34 μM for complex 6d. These complexes displayed much lower activity (> 250 μM) in the normal MDBK cells.156 The results indicated that certain Ru(II) N,O-complexes are cytotoxic in certain cancer cell lines but do not elicit significant toxic effects in normal cells. Sadler et al. determined the structure-activity relationships in cytotoxic arene Ru(II) complexes containing N,O-, and O,O-chelating ligands (Fig. 12, Scheme 2).157 Their results indicated that the amino acidato (N,O-) complexes were inactive in A2780 cells, with IC50 values > 100 μM. However, the O,O-chelated arene Ru(II) complexes were efficacious in A2780 cells (Scheme 2). Most of the O,O-chelated arene Ru(II) complexes were poorly soluble and had a moderate rate of hydrolysis. Their efficacy was significantly dependent upon the substituent and the arene ring. The IC50 values of neutral Ru(II) acetylacetonate (acac) complexes in A2780 cells had the following differences in efficacy, depending upon the arene: p-cymene, biphenyl, dihydroanthracene < benzene, indan.157 The effect of substituents in the acac backbone has been investigated in p-cymene complexes, where the IC50 values vary with the acac substituents in the order: Ph, tert-butyl < Me, Me/naphthyl < CF3, Ph/phenyl.157 In general, acac-type complexes have low cytotoxicity, poor aqueous solubility and rapid hydrolysis compared to the ethylenediamine analogs, which is likely due to the protonation and irreversible displacement of the acac derivatives under some conditions.
Fig. 12.
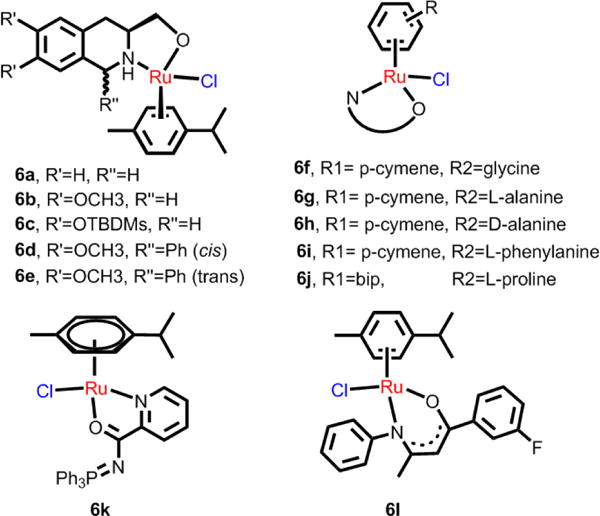
Ru(II) arene complexes bearing N,O- chelating ligands.
Scheme 2.
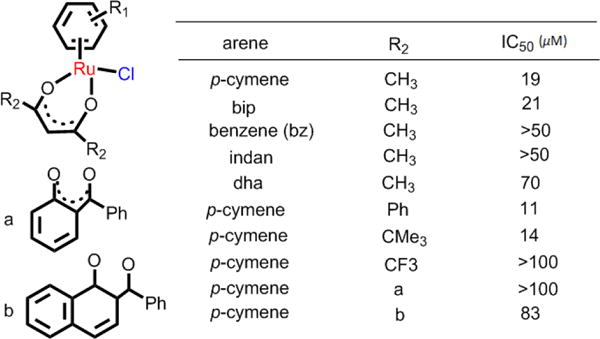
Ru(II) arene complexes bearing O,O-chelating ligands, and their cytotoxicity towards A2780 cells. Data originate from ref. 157.
A series of water-soluble iminophosphorane Ru(II) complexes were synthesized by Frik et al. Most of the complexes were found to be more cytotoxic than cisplatin in several human cancer cell lines.158 The most effective complex, 6k, produced a 56% decrease in tumor size in mice with xenografted breast carcinoma MDA-MB-231 cells, with low systemic toxicity after 28 days of treatment (14 doses of 5 mg/kg, every other day). Pharmacokinetic studies indicated that 6k appeared rapidly in plasma, with high uptake in the breast tumor tissues compared to the kidneys and liver. Mechanistic studies indicated that this complex did not interact with DNA or inhibit protease cathepsin B and induced cell death mainly through canonical or caspase-dependent apoptosis, independent of p53. Ru(II) complexes with N,O-ligands were synthesized by Lord et al.159 Most of the complexes were cytotoxic effect in HT-29 and MCF-7 cell lines, with significantly greater efficacy in A2780 and A2780cisR cells. In addition, the complexes were also cytotoxic to the hypoxic HT-29 cells. The complexes were found to inhibit the thioredoxin reductase (TrxR), with IC50 values in the nanomolar range, in combination with significant single-strand DNA breaks. The representative, 6l, was the most active against all cancer cell lines (IC50 = 1.9 μM for MCF-7 cells) and 6l was ∼3-fold more efficacious than cisplatin against A2780cisR cells.
Dyson et al. also designed another two arene Ru(II) complexes, 7a and 7b, with O,O-ligands (see Fig. 13).160 7b was more cytotoxic than 7a in A2780 cells (IC50 = 22.4 and 73 μM, respectively). These two complexes could selectively inhibit the migration of MDA-MB-231 tumor cells. Moreover, the compounds displayed more potent antivascular effects in vivo (chicken chorioallantoic membrane) model compared to RAPTA-C. The Turel group discussed the anticancer efficacies of a series of fluorinated O,O-ligands arene Ru(II) compounds with different monodentate ligands (Cl, pta, Fig. 13).161 They found that all complexes were efficacious against two cancer cell models (ovarian, osteosarcoma), but did not produce toxic effect nonmalignant keratinocytes. The analogues of pta Ru(II) complexes showed lower cellular Ru accumulation, but higher efficacy, especially in the osteosarcoma cells, compared to the chloride analogues. All chlorido complexes significantly induced ROS production, DNA damage, and apoptosis. Although the pta compounds did not significantly induce ROS production, they blocked cell cycle progression in the G0/G1 phase. 7c and 7c-I had the highest cytotoxicity in ovarian CH1 cells, with IC50 values of 17 μM and 8 μM, respectively. Pettinari et al. determined the anticancer efficacies of arene Ru(II) complexes (7i-7q, Fig. 13), with the 4-(biphenyl-4-carbonyl)-3-methyl-1-phenyl-5-pyrazolonate ligand and different monodentate ligands (Cl, CH3OH, pta).162 The nature of the monodentate ligands were critical in terms of the DNA binding affinities of the Ru(II) complexes, with a rank order of: pta analogues > CH3OH analogues > chloride analogues. They found that the three Ru(II) complexes with a hexamethyl benzenearene ring (hmb) had IC50 values of 9–34 μM in cancer cells, whereas the other Ru(II) complexes were much less active. The three hexamethylbenzene-Ru(II) complexes induced cell apoptosis by activating caspases. The active complexes produced DNA fragmentation, accumulation of pro-apoptotic proteins (i.e. p27, p53, p89 PARP fragments), and the down-regulation of the antiapoptotic protein Bcl-2.
Fig. 13.
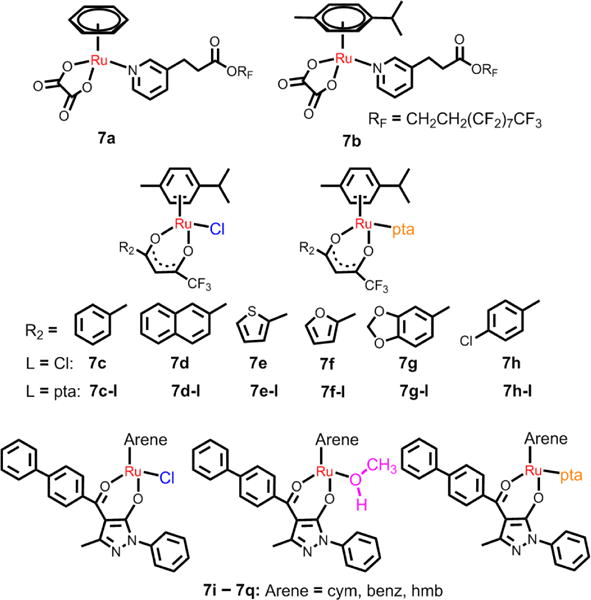
0,O- ligands arene Ru(II) complexes with different monodentate ligands.
Typically, arene Ru(II) complexes with C,N-cyclometalated ligands are more efficacious than cisplatin as anticancer compounds. Yellol and co-workers synthesized some neutral benzimidazole C,N-cyclometalated arene Ru(II) complexes (see Fig. 14), and these complexes were efficacious in HT29, T47D, A2780 and A2780cisR cancer cell lines.163 The IC50 value of 8a, a representative compound, was 2.18 μM in HT29 cancer cells, indicating that it is more efficacious than cisplatin (IC50: 9 μM). Further studies indicated that 8a induces a high rate of apoptosis, good accumulation, S-phase cell cycle arrest, strong binding to HSA at sites I and II, and weak binding to DNA in the minor groove. Subsequently, Yellol et al. determined the effects of varying substituents (H, Me, F, CF3, MeO, NO2, and Ph) in the R4 position of the phenyl ring of 2-phenylbenzimidazole chelating ligand on the anticancer efficacy of the complexes.164 The hydrolysis of the ruthenium−chlorido bond was relatively rapid for 8c, 8d, and 8h. The relative hydrophobicities, according to RP-UPLC-QTOF-MS studies, were: 8d < 8c < 8h. There was no distinct variation in the cytotoxicities for these complexes due to the substitutions, but the CF3 substitution was found to increase the efficacy of 8e in almost all of the cell lines.164 Most of these compounds were more efficacious than cisplatin in A427 and HT29 cell and were able to kill A2780cisR cells with IC50 values of 0.96–3.26 μM.
Fig. 14.
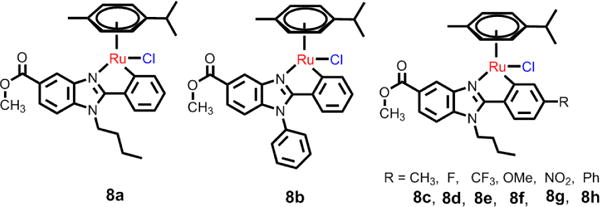
C,N-cyclometalated (η6-p-cymene) Ru(II) complexes.
4.3 Arene Ru(II) complexes with RAPTA ligands
RAPTA compounds are characterized by a monodentate phosphane ligand pta (1,3,5-triaza-7-phosphaadamantane) and a η6-arene ligand bound facially to the metal center, with the general formula [(η6-arene)Ru(X)(Y)(pta)], where X and Y are most commonly chlorine.141 The hydrophilic pta ligand had good aqueous solubility and is preferentially protonated in a low pH environment. RAPTA derivatives containing two chloride ligands were susceptible to hydrolysis in a low chloride environment.165 RAPTA-C is the prototype of this class of organometallic, half-sandwich compounds and had properties similar to the toluene derivative RAPTA-T. In vitro, RAPTA-C and RAPTA-T lack significant cytotoxicity, but they inhibited lung metastasis in CBA mice bearing the MCa mammary carcinoma, while having only mild effects on the primary tumor.166,167 Weiss et al. recently reported that RAPTA-C can reduce the growth of primary tumors in preclinical models for ovarian and colorectal carcinomas.156 When administered daily, at a relatively low dose (0.2 mg/kg), RAPTA-C significantly reduced the growth of A2780 cells transplanted onto the chicken chorioallantoic membrane (see Fig. 15). RAPTA-C had similar efficacy in LS174T colorectal carcinoma in athymic mice, albeit at a higher dose. This complex restricted microvessel density and was cleared from the organs and the bloodstream.168
Fig. 15.
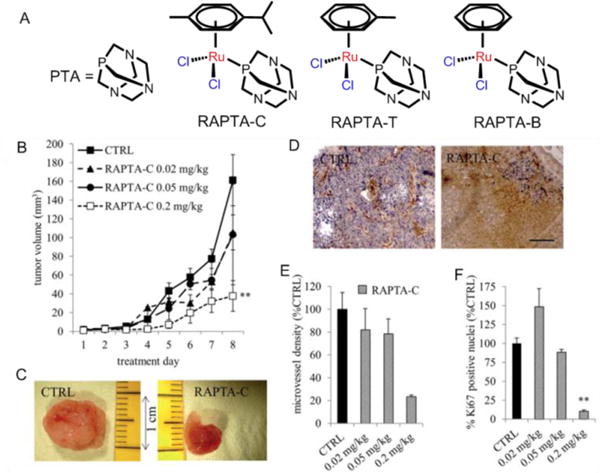
(A) The structures of PTA, RAPTA-C, RAPTA-T and RAPTA-B. (B) Growth curve of A2780 tumors with respect to RAPTA-C treatment. (C) Images show representative tumors from the vehicle treated (CTRL) and RAPTA-C (0.2 mg kg-1) treated CAMs. (D) Representative images of the immunohistochemical staining of the endothelial cell marker CD31 (in brown) showing reduced microvessel density per mm2 in tumors treated with RAPTA-C normalized to the tumor surface area and provided as a % of the control (E) and Ki-67 positive nuclei (in blue) (D) and quantification of the percentage of the tumor surface area staining positive for Ki-67 (as a % of CTRL) (F). Black bar in the right image of (D) represents 500 mm and is valid for both images. Reproduced with permission from ref. 168. Copyright 2012, Royal Society of Chemistry.
RAPTA must undergo hydrolysis to be active in vivo, and the extent of hydrolysis depends upon the pH and the amount of chloride present in solution. The chloride ligands of RAPTA-C were replaced with bidentate oxalate to create RAPTA derivatives (9a-9f, Fig. 16) that were more inert towards hydration, and some of these complexes displayed efficacy similar to RAPTA-C in vitro.169 However, the RAPTA derivatives were highly cytotoxic in A2780 and A2780cisR cell lines (IC50 0.14–1.15 and 0.27–1.18 μM, respectively) and all the complexes with curcuminoid ligands were more efficacious than cisplatin.169
Fig. 16.
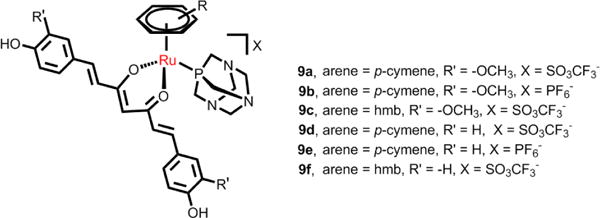
Curcuminate complexes derived from the RAPTA structure.
It was first hypothesized that DNA was the primary target of the RAPTA derivatives.170 Allardyce et al. reported that RAPTA-C exhibited pH-dependent DNA damage; the pH where damage was greatest was significantly correlated with the pH environment of the cancer cells.170 As shown in Fig. 17, Davey et al. found that the RAED-C complex preferentially targets the DNA of chromatin with cytotoxic effects.171 However, the relatively non-cytotoxic antimetastasis RAPTA-C was found to interact with the proteins in A2780 cells and formed protein-Ru(II) complex adducts. About 85% of the adducts resulting from the total intracellular ruthenium content were bound to histone proteins, and the authors hypothesized that histone lesions may contribute to the efficacy of this compound.171 Recently, Dyson and Davey et al. firstly found that the RAPTA-T can combine with auranofin to yield a synergistic activity in killing cancer cells via the allosteric cross-talk in chromatin.172 In addition, the RAPTA derivatives inhibited the activity of glutathione transferase, lysozyme, cathepsin B (Cat B) and TrxR.173 Thus, RAPTA complexes can readily react with proteins and inhibit enzymes, but there is no significant correlation between this reactivity and toxicity in cancer cells. Dyson et al. postulated that the RAPTA derivatives induced cell death via multiple modes of action.173
Fig. 17.
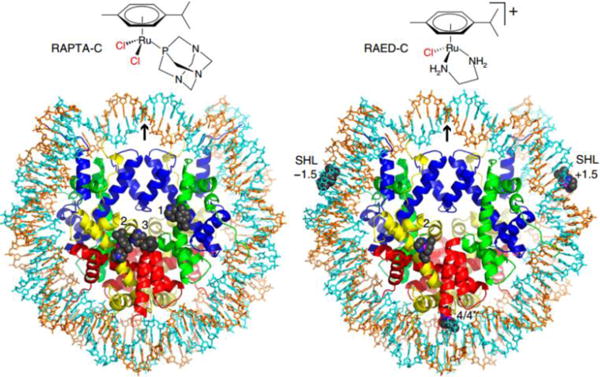
Chemical structures and nucleosomal adducts of RAPTA-C and RAED-C by X-ray. Reproduced with permission from ref. 171. Copyright 2015, Nature Publishing Group.
4.4 Multinuclear arene Ru(II) complexes
The introduction of ligands with multiple ligand-binding sites enables arene Ru(II) compounds to occur in multinuclear complexes.174,175 The lipophilicities and water solubilities of the new multinuclear ruthenium complexes with large positive charges are distinct from the mononuclear arene ruthenium complexes. l Multinuclear ruthenium compounds mainly contain dinuclear, trinuclear, tetranuclear, hexanuclear, octanuclear, or supermolecular derivatives.176 Among the multinuclear arene ruthenium complexes, dinuclear complexes have a variety of significant biological effects. Keppler et al. designed a series of dinuclear arene Ru(II) complexes and investigated the relationships related to spacer length, lipophilicity, modes of action and cell toxicity (Fig. 18A).177,178 Increasing the chain length of the dinuclear arene Ru(II) complexes from 2 to 6 and 12 CH2 groups, increased the lipophilicity and the in vitro efficacy, respectively. The most potent dinuclear compound, 10f, had an IC50 of 0.29 μM in SW480 cancer cells and there was no cross-resistance between oxoplatin and 10f in three oxoplatin-resistant cell lines (5637-oxo, SISO-oxo, and KYSE70-oxo).177 The mechanism study indicated that DNA molecules were the main targets for the dinuclear arene Ru(II) compounds. The dinuclear arene Ru(II) complexes were rapidly hydrolyzed to predominantly form diaqua species that interacted with transferrin, indicating proteins were potential targets. Brabec and co-workers reported that the dinuclear arene Ru(II) complexes with long linkers bind DNA by forming intrastrand and interstrand cross-links in one DNA molecule in the absence of proteins and that they can crosslink two DNA duplexes, as well as proteins, to DNA-a feature not observed for other antitumor ruthenium complexes.179 Gras et al. designed some thiophenolato-bridged dinuclear arene Ru(II) complexes (Fig. 18B) and found the complexes to be highly cytotoxic in A2780 and A2780cisR cells, with IC50 values in the submicromolar range.180
Fig. 18.
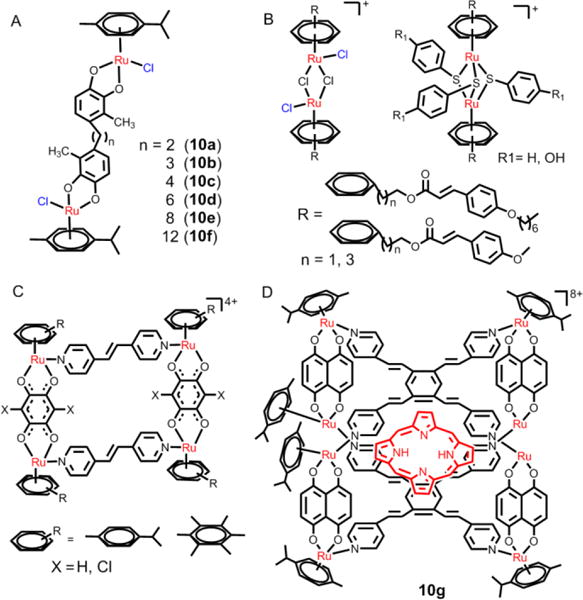
(A), (B), (C) and (D) The structures of selective dinuclear and tetranuclear arene Ru(II) complexes.
Another feature of multinuclear arene Ru(II) compounds is supramolecular self-assembly: the spontaneous association of two or more moieties under equilibrium conditions into stable, structurally well-defined aggregates through either covalent or non-covalent interactions.181 These compounds can form both two-dimensional and three-dimensional structures. Generally, these supramolecular arene ruthenium compounds have large charges, good solubility in water and a large, hollow space capable of encapsulating guest molecules.182 These multinuclear arene ruthenium compounds have antitumor efficacy in certain cancer cells. The Therrien and Barea research groups reported that some p-cymene ruthenium- based metalla-cycles (see Fig. 18C) have significant biological activity and the metalla-cycles had IC50 values in the micromolar range in A2780 cancer cells.174,183 In addition, the three-dimensional cages on these multinuclear compounds can serve as drug delivery vectors to control the release of the guest molecule, and may become a new platform for future development of efficacious anticancer drugs.184–186 Schmitt et al. used the water-soluble Ru(II) metalla-cage, 10g, to deliver hydrophobic porphyrin molecules to cancer cells (Fig. 18D).182 After photoactivation, there was a significant increase in cellular cytotoxicity.
5. Ruthenium(II) polypyridyl complexes
Recently, many studies have reported that ruthenium(II) polypyridyl complexes have a number of significant biological properties.187–190 l Most Ru(II) polypyridyl complexes have excellent reactivity, imaging capability, binding ability, and redox chemistry, giving them potential as diagnostic and therapeutic drugs for cancer. These ruthenium complexes frequently contain N,N-chelating ligands with octahedral structures, and they are kinetically inert. Ru(II) polypyridyl complexes can reversibly interact as probes or inhibitors with important biological molecules including DNA, proteins and RNA. The interaction of ruthenium complexes with biological molecules often leads to damage or toxicity to the biological targets. In addition, many Ru(II) polypyridyl complexes have photophysical and photothermal properties, including a large stoke shift, long luminescent lifetime, significant two-photon absorbing and photostability, which endows Ru(II) polypyridyl complexes with the properties of photosensitization for use in cancer photodynamic therapy (PDT).68,191 PDT is a non-invasive and effective method for localized tumor treatment.l PDT, as a new multi-modality treatment platform, requires both a non-toxic photosensitizer and a harmless light source which matches the absorption spectrum of the photosensitizer. PDT can cause a direct effect on cancer cells, inducing cell death by necrosis or apoptosis.
5.1 Ru(II) tris(polypyridyl) complexes
The Ru(II) polypyridyl complexes frequently contain chelating ligands such as polypyridine, 1,10-phenanthroline and their derivatives.190 These coordinated, saturated Ru(II) tris(bidenatate) complexes are lipophilic and cationic, and are strictly octahedral in their geometry. The unique construction of their three-dimensional and molecular geometries contribute to a variety of biological properties of the ruthenium complex.192 It has been more than 65 years since the biological activity of Ru(II) polypyridyl complexes was first reported by Dwyer et al.120 Dwyer and co-workers demonstrated that different types of enantiomeric Ru(bpy)32+ and Ru(phen)32+ have different biological activity. Ji et al. designed a number of Ru(II) tris(polypyridyl) complexes as potential anticancer drugs and systematically investigated the interactions between Ru(II) complexes and DNA molecules.190 As a source of positive charge and stable molecule construction, most Ru(II) tris(polypyridyl) complexes interact electrostatically with various biomolecules. The intercalating action of the Ru(II) polypyridyl complexes with DNA is a classical and frequent mechanism of anticancer activity.193
Barton et al. reported that Δ-[Ru(bpy)2dppz]2+ could bind to both mismatched and well-matched sites in the oligonucleotide 5′-(dCGGAAATTACCG)2-3′.194 Furthermore, the complex [Ru(bpy)2dppz]2+ was unable to permeate cells due to its poor lipophilicity. Strategies using either conjugation to cell-penetrating peptides, lipophilic ancillary ligands, such as dip or biochemical compounds (carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone, pentachlorophenol and tolfenamic acid) have been used to increase the membrane permeability of the Ru(II) complexes.74,77,195 Barton et al. systematically explored the cellular uptake of dppz Ru(II) polypyridyl complexes and reported that the complex [Ru(dip)2(dppz)]2+ entered the cell by passive diffusion.76,196 The cellular uptake of the complex appeared to be facilitated by the lipophilic dip ligand. Overall, the Ru(II) complexes with the greatest lipophilicity exhibited the highest uptake and [Ru(dip)2(dppz)]2+ mainly accumulated in the mitochondria and ER.76,196
The modification of ligands also affected the uptake and intracellular localization of ruthenium complexes. Gill et al. changed the ligands of the ruthenium complexes, and found that the modification of lipophilicity and cellular uptake by modulating ligands has the great effect on the cytotoxicity and intercellular targets of Ru(II) complexes.197 Compound 1e targeted the cell nucleus in the MCF-7 cells, with an IC50 value of 138 μM. However, its analogue, with more hydrophobic ligands, targeted the cellular membrane structures in MCF-7 cells, with an IC50 value of 7.0 μM.197 Recently, Zeng et al. reported that the chiral structures of Ru(II) complexes can also affect the intercellular localization of the complexes (Fig. 19). Their study showed that the complex Λ-11, mainly located in the cell nucleus, slightly inhibited the growth of MDAMB-231 cancer cells.198 However, Δ-11 was mainly enriched in the cytoplasm and produced no significant cytotoxicity in MDAMB-231 cells. These results demonstrated that the ligands can affect the anticancer efficacy of Ru(II) complexes by affecting their uptake and intracellular localization.
Fig. 19.
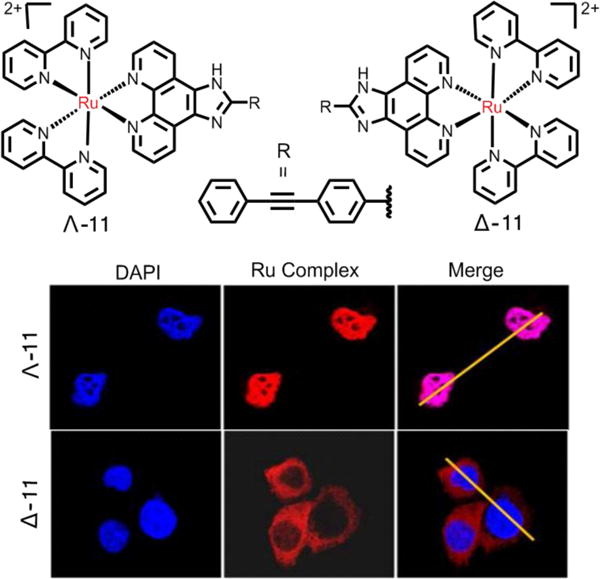
Cellular localization of Λ- and Δ-11 in MDA-MB-231 cells. Reproduced with permission from ref. 198. Copyright 2015, American Chemical Society.
In addition, both the Liu and Chen group designed a series of mitochondria-targeted, Ru(II) complex 12a-12e (see Fig. 20), based on a 2-phenylimidazo[4,5-f][1,10]phenanthroline (PIP) Ru(II) polypyridyl complex. Their results indicated that these complexes induced apoptosis by a mitochondrial pathway.199–204 Moreover, the Chen group found that 12f triggered cell apoptosis via the extrinsic pathway by inducing the activation of caspase-9.205 This complex firstly accumulated on cell membranes by a transferrin receptor-mediated endocytosis, promoting access to the cell cytoplasm and ultimately, the nucleus. Notably, the complex possessed more prolonged circulation time in the blood, comparable antitumor efficacy and significant lower toxicity in vivo experiments in mice, compared to cisplatin.
Fig. 20.
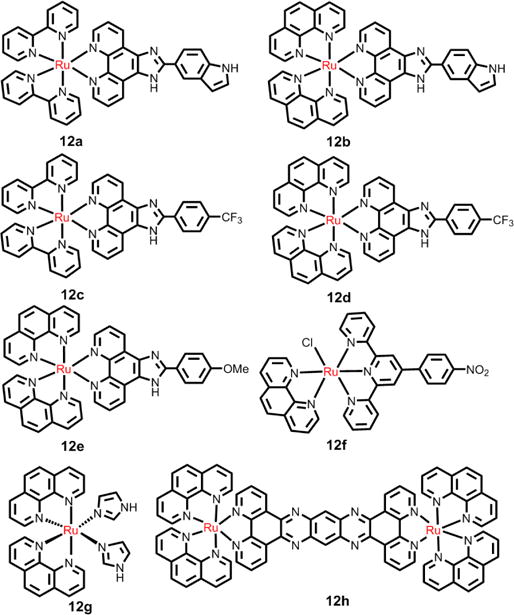
The structures of selective Ru(II) polypyridyl compounds as anticancer drugs.
The luminescent complex, 12g, was used as a theranostic compound by Cardoso et al.206 This complex was taken up in high amounts by HCT116 cells. In addition, it was cytotoxic in HCT116/p53+/+ and HCT116/p53−/−, with IC50 values of 0.1 and 0.7 μM, respectively. The cytostatic mechanism indicated that the complex induced cell cycle arrest in the G1 phase in both cell lines, and it activated proapoptotic PARP in p53−/−, but not in p53+/+ cells. Chao et al. reported that mixed ligand Ru(II) complexes had potent activity against a variety of tumor cell lines.89 The Ru(II) complex 1d accumulated preferentially in the mitochondria of HeLa cells. Furthermore, 1d induced apoptosis via the mitochondrial pathway, with an IC50 value of 7.9 μM after 48 h of incubation, which involved ROS generation, mitochondrial membrane potential depolarization and activation of Bcl-2 and caspase family members.89 l Recently, MacDonnell et al. determined the cytotoxicity of 12h in H358, HCC2998, HOP-62, Hs766t cells, in vitro, under normoxia (18% O2) and hypoxia (1.1% O2).207 The cytotoxicity of 12h was increased in Hs766t and HOP-62 cells under hypoxia compared to normoxia; however, neither H358 and HCC2998 cells were significantly affected by 12h. The authors hypothesized that a mechanistic model where single-strand cleavage of the DNA-bound complex was due to redox-cycling mediated by the concentration of GSH and O2 in a low O2 environment.
However, other mechanisms are assumed to be operative and Ru(II) complexes also have anticancer efficacy by inhibiting topoisomerases,208–210 G-quadruplex stabilizers,211–213 telomerase,214–216 histone deacetylase, and thioredoxin reductase,217,218 as well as stabilizing G-quadruplexes in DNA.
5.2 Cyclometallated Ru(II) complexes
Generally, the cyclometallated ligands are N- and C- donors.219 The metal-to-ligand bond distances of Ru-C are significantly shorter than that of Ru-N, and the binding of cyclometallated ligands with Ru is very stable.220,221 In addition, the cyclometalation can decrease the valence charge of the Ru(II) complexes, which contributes to an increase in the lipophilicity and cellular uptake of Ru(II) complexes. Therefore, the cycloruthenated complexes may be more stable in biological systems and exhibit distinct biological activities compared to non-cycloruthenated complexes (see Fig. 21, Table 1).222 Loeffler et al. evaluated the biological effects of cycloruthenated complexes in 2005 and reported that some cycloruthenated complexes display significant efficacy as antitumor compounds in vitro, compared to the anticancer efficacy of cisplatin.223 Subsequently, Meng et al. reported that the cycloruthenated complex, 13b inhibited the growth of various tumors implanted in mice more efficaciously than cisplatin.224 Further experiments indicated that this complex induced cell death through at least two pathways: the DNA damage/p53 and the ER stress/CHOP pathways.224 Recently, Licona et al. reported that 13b could interact with histones to induce cell death. Three histones (H3.1, H2A, H2B) were regarded as possible targets for 13b.225
Fig. 21.
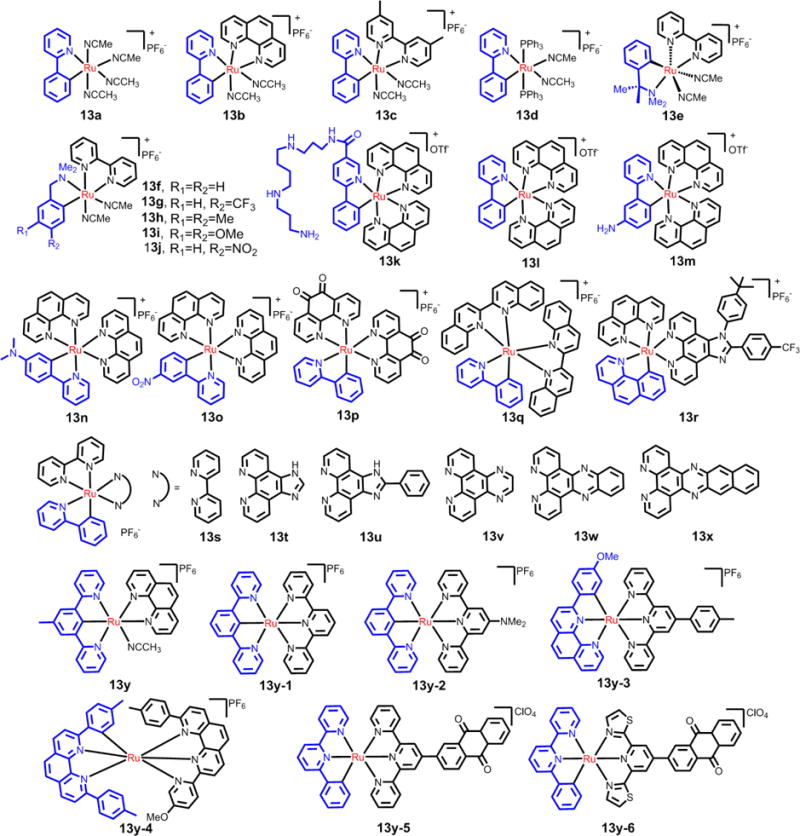
The structures of the selective cyclothenated compounds used in anticancer durgs.
Table 1.
The cytotoxicity of the selective cycloruthenated compounds used in anticancer drugs
| Compounds | Cell lines | IC50 (μM) | Ref. |
|---|---|---|---|
| 13a | A172 | > 50 | 223 |
| 13b | A172 | 1.9 | 223 |
| 13c | A172 | 12 | 223 |
| 13d | A172 | 15 | 227 |
| 13e | A172 | > 50 | 227 |
| 13f | A172 | 12 | 227 |
| 13g | A172 | 6.0 | 227 |
| 13h | A172 | 9.0 | 227 |
| 13i | A172 | > 50 | 227 |
| 13j | A172 | 15 | 227 |
| 13k | HCT116 | > 50 | 228 |
| 13l | HCT116 | 0.1 | 228 |
| 13m | HCT116 | 5.0 | 228 |
| 13n | HCT116 | 0.2 | 228 |
| 13o | HCT116 | 0.5 | 228 |
| 13p | HeLa | 57 | 230 |
| 13q | HeLa | 51 | 230 |
| 13r | HeLa | 2.4 | 233 |
| 13s | HeLa | 34 | 230 |
| 13t | HeLa | 6.1 | 231 |
| 13u | HeLa | 3.4 | 231 |
| 13v | HeLa | 3.7 | 80 |
| 13w | HeLa | 2.0 | 213 |
| 13x | HeLa | 7.0 | 230 |
| 13y | HCT116 | 1.4 | 228 |
| 13y-1 | HCT116 | 0.7 | 228 |
| 13y-2 | HCT116 | 2.5 | 228 |
| 13y-3 | HCT116 | 1.0 | 228 |
| 13y-4 | HCT116 | 0.3 | 228 |
| 13y-5 | HeLa | 0.51 | 232 |
| 13y-6 | HeLa | 1.05 | 232 |
Pfeffer and group reported the synthesis and biological applications of cycloruthenated compounds. The synthesized cycloruthenated complexes produced significant cytotoxic effects in mammalian tumor cells that were similar to cisplatin and were less likely to produce neurotoxicity compared to cisplatin.226,227 Fetzer et al. synthesized a series of cycloruthenated compounds with N-C-N and N-N-C pincer derivatives (Fig. 21).228 Most of the compounds were tested for their in vitro antitumor efficacy, which ranged from good to excellent. Several of these compounds had IC50 values in the nanomolar range.228 The efficacy of the aforementioned cycloruthenated compounds was tentatively correlated to their RuIII/II redox potential and lipophilicity (log P).228
In addition, Peña et al. reported some cycloruthenated compounds were cytotoxic, and their cytotoxic efficacy in the dark was similar to that of cisplatin.229 The compound 13x, was 6 times more than cisplatin, and it disrupted the mitochondria membrane potential. Although Peña et al. reported that 13p is not an optimal compound for PDT due to its high cytotoxicity in the dark and its lack of photochemical sensitivity.230 They found a 7-fold increase in the cytotoxicity of cycloruthenated complex 13p upon irradiation with light at 639 nm.
Chao et al. reported the efficacy of novel cycloruthenated compounds in different cancer cell lines. Interestingly, the Ru(II) complexes 13s to 13x had lower IC50 values against HeLa cells, resulting from an increase of the volume of the chelating ligands, which is partly due to the larger chelating ligands which produces higher cellular uptake and stronger interactions with DNA.80,229,231 Recently, the same group also designed some cycloruthenated compounds with significant potency in hypoxic and cisplatin-resistant cancer cell lines.232,233 Complex 13y-5 (see Fig. 21) was efficacious against hypoxic HeLa cancer cells (IC50 = 0.53 μM).232 This complex was 46-times more potent than cisplatin (IC50= 24.62 μM) in HeLa cells.215 The hydrophobicity and cellular uptake of the complexes were consistent with their cytotoxicity. They also postulated that the cycloruthenated compounds have the potential to surmount drug resistance.232
The mechanism of action and subcellular localization of the cycloruthenated compounds, 13k and 13l, in cancer cells and normal cells was reported by Klainer et al. (see Fig. 21).234 They first tested and compared the intrinsic luminescence of cycloruthenated compounds with an anionic cyclometalated 2-phenylpyridine chelate or neutral aromatic chelating ligands, using a special charge-coupled camera instrument.234 Both 13k and 13l had nonselective interactions with DNA, RNA and BSA in vitro. 13k had reduced cellular uptake and cytotoxicity due to its improved water solubility compared to 13l. Further studies showed that 13l preferentially accumulated in the ER and induced H2AX phosphorylation, as well as expression of CHOP/UPR and SATB2 (signaling pathways). 13l entered into cells via passive transport (at high concentration, 10 μM) and active/facilitated transport (at low concentration, 5 μM). 13l, at 5 μM, accumulated to a greater level in glial A172 cancer cells compared to normal cells (neurons or glial cells), which may have resulted from the above mentioned transport modes. 13l had some selectivity for glial cancer cells compared to healthy glial cells, with IC50 values of 0.25 μM and 1.2 μM, respectively.234 Mechanistic studies indicated that multi-mode ER stress mechanisms and mitochondrial stress responses were involved in producing 13l’s cytotoxic efficacy in the cancer cells. This work provides novel and critical information on the molecular mechanisms and direct targets of organoruthenium compounds.
5.3 Ru(II) polypyridyl complexes for PDT
Recently, a variety of Ru(II)polypyridyl complexes have been reported to have excellent photoactive properties, with certain compounds having enhanced cytotoxicity following light irradiation, providing a platform for relatively selective and improved tumor therapy (see Fig. 22, Table 2).235–237 The mechanism of action for most photoactive Ru(II) compounds involves: ligand dissociation and ROS/1O2 (reactive singlet oxygen).63,238 The ligand dissociation property is regarded as an O2-independent mechanism, and the Ru(II) dyes can undergo photoinduced ligand dissociation, which allows the Ru(II) dyes to bind DNA, typically resulting in DNA damage.238 For the ROS/1O2 mechanism, the Ru(II) complexes enter excited states under light irradiation, giving them the potential for producing ROS or 1O2 by electron/energy transfer. Moreover, ROS or 1O2 can produce direct damage to cancer cells.63
Fig. 22.
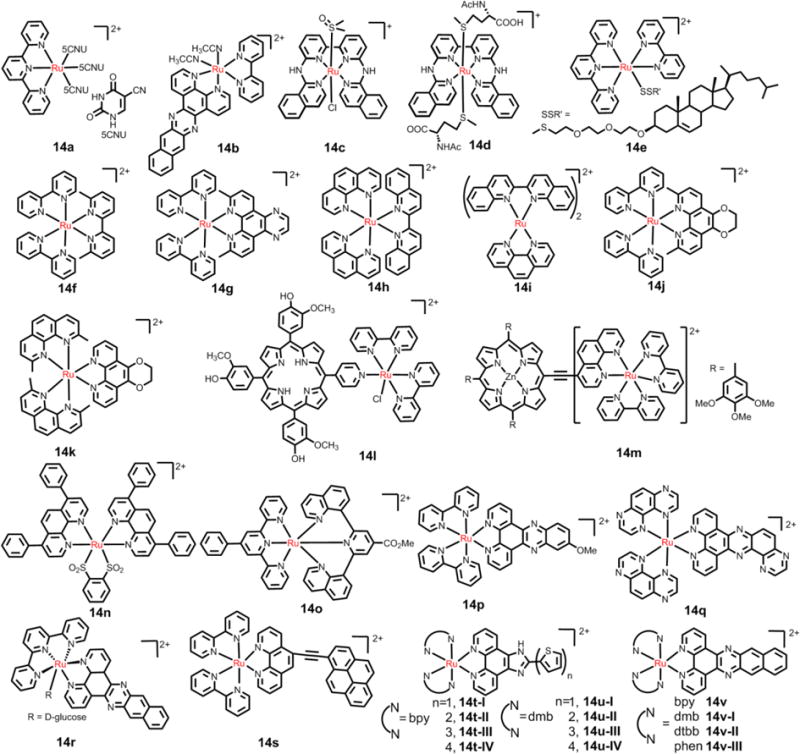
The structures of the selective Ru(II) compounds used in PDT
Table 2.
The cytotoxicity of the selective polypyridyl Ru(II)complexes used in PDT
| Compounds | Cell lines | Dark IC50 (μM) |
light IC50 (μM) |
PIa | Irradiation Wavelength (nm) |
Mechanismb | Ref. |
|---|---|---|---|---|---|---|---|
| 14b | HeLa | 334 | 0.47 | 1110 | 466 | A+B | 243 |
| 14c | A431 | 13 | 0.6 | 22 | 520 | A | 244 |
| 14d | A431 | 38 | 7.8 | 4.9 | 520 | A | 244 |
| 14f | HL60 | > 300 | 1.6 | > 188 | VLd (> 450) | A | 246 |
| 14g | HL60 | 108 | 2.6 | 42 | VL (> 450) | A | 246 |
| 14h | HL60 | 52.5 | 1.2 | 43.8 | Blue light | A | 247 |
| 14i | HL60 | 47.3 | 2.4 | 19.7 | Blue light | A | 247 |
| 14j | HL60 | 34 | 0.65 | 52 | VL (400) | A | 248 |
| 14k | HL60 | > 300 | 0.16 | 1880 | VL (400) | A | 248 |
| 14n | HeLa | 49.7 | 0.62 | 80 | 420 | B | 253 |
| 14o | HeLa | > 100 | 25.3 | > 4 | 420 | B | 254 |
| 14p | HeLa | > 100 | 20 | 5 | UVe | B | 254 |
| 14q | HeLa | 70 | 8.8 | 8 | VL | B | 255 |
| 14r | A549 | 19 | 0.72 | 26 | 454 | A+B | 256 |
| 14s | HL60 | 262 | 0.15 | 1747 | White light | B | 258 |
| 14u-I | HL60 | > 300 | > 300 | Nc | VL | B | 1 |
| 14u-III | HL60 | > 300 | 16 | > 20 | VL | B | 1 |
| 14v | HL60 | 135 | 34 | 4 | 625 | B | 261 |
| 14X | HL60 | > 300 | 0.741 | > 410 | VL | B | 262 |
| 15a | HeLa | > 100 | 1.9 | > 52 | 800 | B | 264 |
| 15b | HeLa | > 500 | 5.71 | 19 | 810 | B | 98 |
| 15c | HeLa | > 100 | 1.9 | 250 | 800 | B | 265 |
PI means IC50(dark)/IC50(light).
the ways of producing phototoxicity; A means ligand dissociation, B means ROS/1O2.
N means not.
VL means visible light.
UV means ultraviolet.
5.3.1 Ligand dissociation
The research groups of Turro and Sadler reported that Ru(II) complexes can lose the ligands following light irradiation, allowing them to covalently bind DNA to produce phototoxicity.239,240 They hypothesized that this class of complexes may provide valuable leads for new photoactivatable, antitumor Ru(II) complexes. l Turro et al. recently reported that irradiation of the cycloruthenated complex, 13b, at 690 nm, resulted in an IC50 value of 70 nM, representing a 14-fold increase in toxicity relative to the IC50 obtained in the dark.241 Furthermore, glutathione (GSH) facilitated ligand exchange of the cycloruthenated complex 13b, with solvent DMSO-d6 molecules. Intracellularly, in the absence of DMSO, a ligand exchange may result in the covalent binding of the complex to DNA and other biomolecules to produce phototoxicity. In order to increase the toxicity of the Ru(II) complex, Turro et al. used a dual-action therapeutic compound, 5-cyanouracil (5CNU), as a photoreleased ligand to combine with Ru obtain a photodynamic compound, 14a.242 14a binds to DNA following light (≥ 395 nm) irradiation and release of the 5CNU ligand, damaging HeLa cancer cells. The above mentioned research group also designed a new tris-heteroleptic, 14b, and this complex was more photocytotoxic due to both 1O2 production and ligand exchange upon irradiation.243 This dual-action is useful for increasing the efficacy of photochemotherapy drugs.
Bonnet et al. first obtained two Ru(II) complex prodrugs by using a tetrapyridyl biqbpy ligand and two trans monodentate ligands.244 The complexes 14c and 14d had trans geometries and the trans ligands were found to be photosubstituted by water under green light irradiation. The complexes were well taken up and had mild cytotoxicity in A431 and A549 cells in the dark. In contrast, when 14c and 14d were activated by green light irradiation, they produced significant cytotoxic, with EC50 (EC = effective concentration) values below 1 μM and a PI of up to 22 for complex 14c. Recently, the same group designed a photosensitive Ru(II) complex 14e.245 The complex was found to have similar cytotoxicity as cisplatin in the dark and increased photocytoxicity after 24 h incubation with blue light activation. Interestingly, the complex had monomer characteristics at a low concentration (< 3.5 μM) and it released the monodentate ligand under blue light activation. However, the complex formed supramolecular aggregates by self-aggregation at high concentrations (> 3–5 μM), which could induce non-apoptotic cell death by permeabilizing cell membranes and extracting membrane proteins and cell lipids.
Two photoactive [Ru(bpy)2(L)]2+ complexes (14f, 14g), with sterically clashing ligands, were designed by Glazer et al.246 The complexes were inert until activated by visible light, which induces ligand loss and covalent modification of DNA.246 Mechanistically, 14f lost the 6,6′-dimethyl-2,2′-bpy ligand and 4g lost the 2,9-dimethyl-dpq (dpq: dipyrido[3,2-f:2′,3′-h]-quinoxaline) under light irradiation. The photo-ejection kinetics was 30-fold faster for 14f compared to 14g. A greater than 100-fold increase in cytotoxicity of these two complexes occurred with light activation in HL60 and A549 cancer cells. Glazer et al. also found that Ru(II) polypyridyl complexes (14h, 14i), with biquinoline ligands, could photobind DNA following the loss of the biquinline ligands in the presence of visible and near infrared light.247 These two complexes were cytotoxic in HL60 cancer cells after irradiation with near infrared light, with phototoxicity indices (PI) of 3.32 and 9.2, for 14h and 14i, respectively. Recently, the same group designed two novel, strained Ru(II) polypyridyl complexes (14j, 14k), containing a 2,3-dihydro-1,4-dioxino[2,3-f]-1,10-phenanthroline ligand (dop), and these two complexes selectively lost a methylated ligand when irradiated with light > 400 nm.248 The compound 14k was 1880-fold more cytotoxic in HL60 cancer cells upon activation by light and was 19-fold more potent than cisplatin.248 Glazer et al. hypothesized that this O2-independent mechanism and ligand dissociation in photoactive metal complexes can simultaneously provide effective and selective phototherapy and overcome diminished efficacy due to hypoxia in the core of the tumor.65,249
5.3.2 ROS/1O2
As most Ru(II) complexes have high photostability and long fluorescence lifetimes, they can produce ROS or 1O2 when irradiated by light. The Ru(II) polypyridyl complex elevates ROS production, resulting in the efficient cleavage of supercoiled DNA and damage to certain biomarkers.63 Swavey et al. designed several Ru(II) polypyridyl complexes with porphyrin derivative ligands.250,251 The Ru(II) porphyryl complex, 14i, efficiently cleaved supercoiled DNA and induced apoptosis in melanoma cells after irradiation with light, while normal cells were unaffected. Ke et al. also designed a mitochondria-specific, porphyrin-Ru(II) conjugate (14m), with high a luminescence and a high singlet oxygen quantum yield.252 The porphyrin-Ru(II) complex held a large two-photon absorption cross section σ value of 1104 GM (GM = 10−50 cm s4 photon−1 molecule−1) upon light irradiation 800 nm. This complex was used to sensitize the formation of 1O2, with high 1O2 quantum yields (0.93) upon irradiation at 424 nm. This complex could kill 80% of HK-1 cells at a concentration of 1 μM and a light dose of 3 J/cm2. After a 5 minute flash excitation by an 850 nm laser on cells loaded with 5 μM of this complex, 90% of the HeLa cells were deformed or had lost their integrity.
Gasser’s group recently inactivated 1a by attaching it to a photo-labile protecting group (PLPG).87 Upon UV-A exposure, 1a was released from the PLPG and had effective phototoxicity. In addition, two other light sensitive Ru(II) polypyridyl complexes (14n and 14o) were tested for photosensitization efficacy.253 The two complexes had significant PDT activity in HeLa cancer cells, with IC50 values of 25.3 μM for 14o and 0.62 μM for 14n under light irradiation (420 nm, 6.95 J/cm2). Moreover, both complexes showed significant antimicrobial PDT activity against the Staphylococcus aureus and the Escherichia coli.253 Recently, the same group reported that 14o localized in the nucleus of various cancerous and normal cells, but only produced cytotoxicity upon irradiation.254 This complex induced the production of ROS, causing DNA damage, cell cycle arrest and cell death. Importantly, the complex had a 3.6-fold increase in its phototoxicity against mitotic cells. This dual mode of cell death upon photo irradiation of the complex may open new avenues in PDT. Cloonan et al. designed a new Ru(II) PDT candidate, 14q, that efficiently entered cells by incorporation of 1,4,5,8-tetraazaphenanthrene ancillary ligands and the lipophilic aromatic pdppz ([2,3-h]dipyrido[3,2-a:2′,3′-c]phenazine) ligand.255 The complex entered into cells via an energy-dependent mechanism and was localized in mitochondria, lysosomes and ER. The complex was nontoxic to HeLa cells, with an IC50 value 70 μM in the dark, and cellularclearance occurred within 96 h. However, it had significant photoactivity against HeLa cells, with an IC50 value 8.8 μM upon visible light activation. Further studies showed that the complex could induce DNA photocleavage and caspase-dependent and ROS-dependent apoptosis upon light activation. Recently, Bonnet et al. obtained two highly photosensitive Ru(II)-based anticancer prodrugs. The complexes contained either a D- or L-glucose.256 The D-glucose, 14r, was present in the mitochondria and had significant photocytotoxicity in A549 cells, with an IC50 value of 0.72 μM due to both 1O2 production and ligand exchange upon irradiation at 454 nm.
McFarland et al. designed and tested the PDT efficiency of Ru(II) polypyridyl complexes that had the lowest-lying 3IL-based excited states, with remarkably long lifetimes (from 22 to 270 μs), by linking pyrenylethynylene derivatives ligands,257–259 which made the complexes hold higher 1O2 quantum yields compared to [Ru(bpy)3]2+. These complexes interacted with DNA via intercalation and produced photocleavage of DNA in vitro at submicromolar concentrations when irradiated with visible light. These photosensitizers had highly potent photocytotoxicities in melanoma, HL60 and bacterial cells at very low concentrations. Compounds 14s induced cell death in a HL60 cells, and the LC50 (LC = lethal concentration) went from 262 μM in the dark to less than 0.15 μM upon visible light irradiation.258 Another class of Ru(II) polypyridyl complexes, with polythiophene chains of variable lengths, were obtained and studied by the same group.1,260 These compounds gave access to a low-lying, 3IL excited state and a strong interaction with plasmid DNA. These complexes were also found to possess high 1O2 quantum yields that increase with polythiophene chain length, and the complex, 14t-III, was found to targeted DNA in HL60 cells, inducing = DNA photodamage.1 The cytotoxicity of this family in the dark was very low, with an EC50 > 300 μM in the dark. However, there was a 200-fold increase cytotoxicity after light exposure compared to dark conditions as n was increased (n = 1 to 4) in polythiophene chain. Furthermore, animal survival was significantly increased after the administration of 14t-III and 14u-III. The complex 14u-III is currently undergoing human phase IB clinical trials.1,70 Interestingly, McFarland’s group found that the family of 14v, containing the π-expansive dppn (benzo[i]dipyrido[3, 2-a: 2′, 3′-c]phenazine) ligand, also shared the properties of low-energy and long-lived 3IL excited states, and had efficient photodynamic activity in HL60 cancer cells.261 In addition, a photoactive cycloruthenated complex [Ru(bpy)2(pbpn)]+, 14x, was developed by replacing dppn with the π-expansive cyclometalating ligand pbpn (4,9,16-triazadibenzo (a,c-napthacene).262 This complex displayed intense green ligand-centered fluorescence from inside the cell. This complex had very weak to no room temperature phosphorescence, extremely short phosphorescence state lifetimes (< 10 ns), low singlet oxygen quantum yields (0.5–8%) and lack of cytotoxicity in the dark (EC50 > 300 μM). However, it had significant phototoxicity in cancer cells, with an IC50 in the nanomolar range.262 The authors postulated that 3IL excited states affected the phototoxicity of 14x. Moreover, three cycloruthenated compounds of N-C-N pincer were designed by Tabrizi et al. The compounds showed high uptake and effective photocytotoxicity in HeLa cells under light irradiation (350 nm) with the formation of 1O2 and hydroxyl radicals.263
Recently, Chao et al. developed an efficacious cancer therapy by combining PDT with targeting.68 They found that 15a, (Fig. 23) with a triphenylphosphine (TPP) group, highly targeted mitochondria and was activated by 2 photons.264 15a had an IC50 value as low as 9.6 μM in one-photon PDT (λirr = 450 nm, 12 J cm−2) and an IC50 of 1.9 μM in two-photon PDT (λirr = 830 nm, 800 J cm−2) towards 3D HeLa, with multicellular spheroidal tumors (MCTSs). In addition, several highly charged Ru(II) complexes (15c-15e), were designed as two-photon photosensitizers by the same group.98 The three complexes were found to localize in the lysosomes via an endocytotic uptake pathway. 15c produced significant phototoxicity upon irradiation (800 nm, 10 J/cm2) in 3D HeLa MCTSs, with an IC50 value as low as 1.9 μM. Thus 15c has the potential to be used for two-photon photodynamic therapy. Moreover, Chao’s group utilized a GSH-activatable Ru(II)-azo photosensitizer, (15b), for two-photon PDT.265 15b did not show luminescence but exhibited a strong luminescence after incubation in HeLa cells due to the activation of intracellular GSH. 15b accumulated to a significant extent in mitochondria and could produce ROS in HeLa cells upon two-photon irradiation (810 nm, 100 mW). In dark conditions, 15b lacked significant cytotoxicity in 3D HeLa MCTSs (IC50 > 100 μM). In contrast, 15b was significantly cytotoxic in MCTSs, with an IC50 value as low as 5.71 μM upon two-photon irradiation.265 The Chao et al. study demonstrated that the combination of organelle-targeting and two-photon activation provides a valuable paradigm for developing Ru(II) complexes for PDT applications.
Fig. 23.
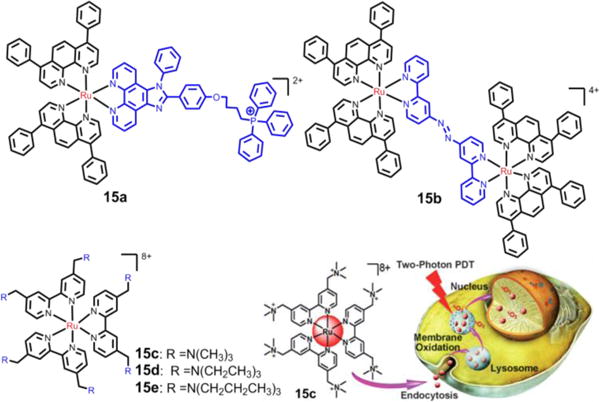
The Ru(II) compounds used for two-photon-PDT in Chao’s group, and the schematic description of 15c used for two-photon-PDT. Reproduced with permission from ref. 98. Copyright 2015, Wiley-VCH.
6. Ruthenium(II)-based nanomaterial systems
Over the years, numerous strategies have been used to deliver Ru(II)-based metal complexes into cancer cells. However, their particle size and specificity have always been major obstacles for effective tumor targeting. Nanomaterials represent a wide range of nanoscaled, hybrid components that are linked together by covalent or non-covalent interactions. l Recently, nanomaterials have been developed for a variety of biomedical applications for the treatment and imaging of diseases.266 Drug encapsulation into nanomaterials can provide significant improvements in pharmacokinetics, solubility, toxicity and biodistribution when compared to freely administered molecules.267 Importantly, once nanomaterial drugs are injected into the bloodstream, they preferentially accumulate in solid tumors due to the enhanced permeation and retention (EPR) effect.268–290 Past studies have shown that encapsulating Ru(II) metal complexes in a nanomaterial system improves their targeting and delivery into tumor cells. Ru(II) complexes in nanomaterial systems play four main roles: (a) to control the release of payloads (drugs) with better efficiency; (b) to work as drugs or catalysts in the nanomaterial systems; (c) to improve the photothermal efficacy and the stability of nanomaterials and (d) to act as theranostic tools to track the import of nanomaterials through luminescence imaging.
6.1 Ru(II)-selenium nanoparticles
Due to its biocompatibility, rapid degradation, low toxicity, facile synthesis, selenium has evolved as one of the most commonly used nanosystems to deliver drugs.271 Moreover, selenium shows promising chemopreventative efficacy when used as a nanosystem to deliver chemotherapeutic drugs such as 5-fluorouracil and doxorubicin.272,273 Several selenium nanoparticle systems have been utilized to deliver Ru(II) complexes as anti-cancer compounds. The Ru(II) complex, RuPOP, produced significantly greater anticancer efficacy and lower toxicity compared to cisplatin, but its use was limited due to poor aqueous solubility.274 Liu et al. loaded RuPOP in selenium-based nanoparticles (Fig. 24A) and determined its anti-cancer efficacy.274 They developed pluronics (a group of block copolymers that are amphiphilic) based on the ruthenium complex antagonism of the folate receptor and formulated a selenium-based nanosystem. This formulation (FA-SeNPs) entered cells via endocytosis. Furthermore, 20 μM of FA-SeNPs had significant cellular uptake after 8h of incubation in doxorubicin-resistant R-HepG2 cells. The intracellular translocation was achieved in 1 h and the pluronic-bound ruthenium was released in acidic conditions. In vitro, FA-SeNPs had significant cytotoxicity in the parental and drug-resistant hepatocellular carcinoma cell lines, with IC50 values of 0.33 and 0.24 μM, respectively.274 The mechanistic experiments indicated that FA-SeNPs induced cell apoptosis by up-regulating the level of ROS in cells to activate the MAPK and AKT pathways. Moreover, 0.48 μM of FA-SeNPs decreased the expression level of major drug efflux transporters, namely, ABCB1, ABCC1, and ABCG2 and induced apoptosis by stimulating the sub-G1 phase of the cell cycle. This work suggests that FA-SeNPs are significantly more cytotoxic in cancer cells compared to normal cells and this system may provide a suitable approach for surmounting multidrug resistant cancers.
Fig. 24.
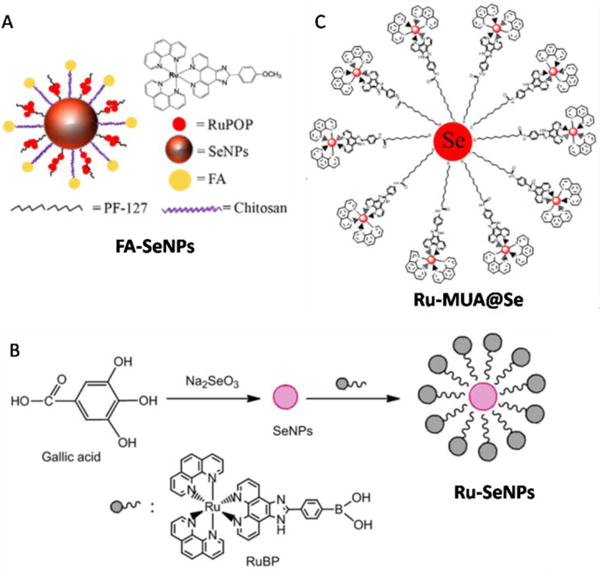
(A) The diagrammatic figure of FA-SeNPs and the structure of RuPOP. (B) The method for the synthesis of the Ru-SeNPs. (C) The structure of the Ru-MUA@Se. Reproduced with permission from ref. 274, 275 and 276, respectively. Copyright 2015, Elsevier.
A key study by Sun et al. demonstrated the anti-angiogenic effects of Ru-SeNPs, which are luminescent ruthenium selenium nanoparticles (Fig. 24B).275 The chicken chorioall-antoic membrane (CAM) assay in vivo was used to evaluate the anti-angiogenic effects of Ru-SeNPs. At 50 μg, the nanoparticles significantly reduced the angiogenesis in the chorioallantoic membrane, compared to the control. Moreover, Ru-SeNPs inhibited the proliferation of the following cancer cell lines: prostate PC-3, MCF-7, human umbilical vein endothelial cells (Huvec), SW480, and HepG2, with IC50s in the range of 3–20 μg/ml. A concentration-dependent increase in apoptosis occurred in HepG2 cells after 24 h of exposure to Ru-SeNPs. Furthermore, an inhibition of the phosphorylation of FGFR1, Erk1/2, and AKT validated the anti-angiogenic effects of Ru-SeNPs.275 Another study indicated that the anti-angiogenic effects of ruthenium-thiol selenium nanoparticles (Ru-MUA@Se) by the same group (Fig. 24C).276 Ru-MUA@Se directly suppressed HepG2 tumor growth, with no significant body weight loss, and also blocked blood vessel growth in vivo by decreasing microvessels. Ru-MUA@Se entered the cells via a clathrin-mediated endocytosis pathway. This system was shown to inhibit mitochondrial membrane function and induce ROS formation in HepG2 cells. Moreover, there was a significant reduction in tumor angiogenesis and low-systemic toxicity in HepG2 tumor.
6.2 Ru(II)-gold nanomaterials
Gold nanomaterials have been a topic of significant interest due to their interesting surface plasmon resonance (SPR) properties.277–282 The functionalized gold nanostructures have the properties of good biocompatibility, small size and shape-dependence, and they have proven to be a versatile platform for a wide range of biomedical applications. Similarly, Ru(II)-polypyridyl complexes, due to their photophysical properties, high luminescence and large two-photon absorption cross sections, have emerged as promising theranostic tools for cancer treatment.283–285 In addition, there are two excellent examples for Ru(II) complex functionalized gold nanomaterials as anticancer compounds for photothermal therapy (PTT). Zhang et al. developed the gold nanoparticles for PTT by grafting two-photon luminescent Ru(II) complexes to the surface of gold nanoparticles as antenna molecules (Fig. 25A).286 The Ru@AuNPs converted NIR (808 nm) light to heat (ΔT = 9.4–38.5 °C), with high photothermal therapy efficiency (ΔT = 38.5 °C, η = 33.3%), which was well over the required temperature increase for efficient cancer photothermal therapy. More importantly, the in vivo experiments indicated that Ru@AuNPs, as PTT compounds, had significant tumor ablation efficacy. Indeed, under diode laser (808 nm) irradiation at the power density of 0.8 W/cm2 for 5 min, tumors shrunk gradually or disappeared individually after 10 days of treatments.286 This study with Ru(II) complex modifications provides an effective solution for overcoming the typically poor NIR absorbance and low photostability (melting effect) of gold nanoparticles in PTT. Recently, the same group developed two novel gold nanostructures to get the Ru(II)-functionalized gold nanorods, (AuNRs@Ru) and nanostars (AuNTs@Ru) (see Fig. 25B).287 AuNRs@Ru and AuNTs@Ru had higher photothermal stability and photothermal efficiency compared to pure AuNRs and AuNTs (ΔTAuNRs@Ru= 41.1 °C, ΔTAuNTs@Ru =38.9 °C; ΔTAuNRs = 21.0 °C, ΔTAuNTs = 19.2 °C). AuNRs@Ru and AuNTs@Ru exhibited efficacious photothermal therapy both in vitro and in vivo under low power (808 nm laser, 0.25 W cm−2), which suggested that the two candidates have potential application as cancer therapy through photothermal destruction.
Fig. 25.
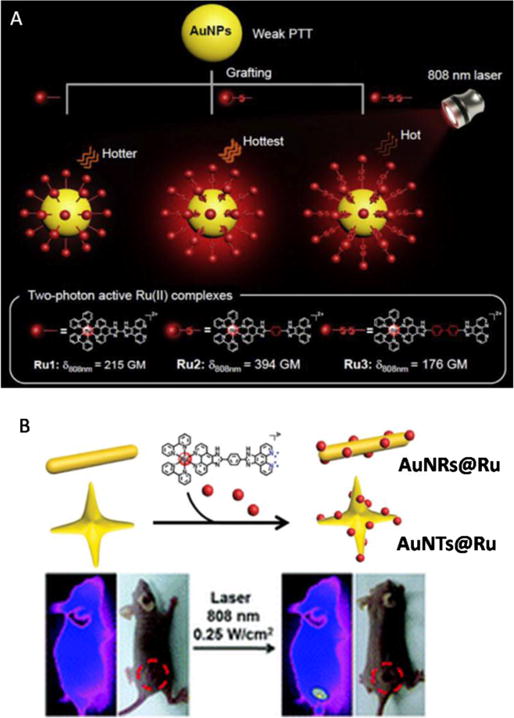
(A) Schematic illustration of the photothermal efficiency of Ru@AuNPs under two-photon luminescence. (B) The structure of AuNRs@Ru and AuNTs@Ru, and the schematic illustration of photothermal treatment on mice. Reproduced with permission from ref. 286 (Copyright 2015, Elsevier) and 287 (Copyright 2012, Royal Society of Chemistry), respectively.
6.3 Ru(II)-silica composites
Silica has long been used as a nanocarrier to deliver drugs for therapeutic uses.288,289 Silica nanoparticles are non-toxic to cells and readily undergo endocytosis in acidic liposomes. The release of these nanoparticle in specific pH environments, photon activation, redox activation, and tumor targeting, makes them an ideal nanocarrier for Ru(II) complexes and other drugs. Frasconi et al. developed ruthenium-silica nanoparticles (Fig. 26A), with enhanced cellular uptake and photoactivation.290 l The Ru(II) complex was linked covalently to the mesoporous silica nanoparticles (MSNPs) to form MSNPs2 by coordination of the monodentate ligand (3-isocyanato- propylethoxysilane with 4-(aminomethyl)-benzonitrile).290 The MSNPs2 showed rapid cellular uptake and the ruthenium complexes were rapidly released and transformed upon light irradiation into a cytotoxic aqua complex that formed monoadducts with DNA. In addition, the MSNPs2 could load paclitaxel with an 82% uptake efficiency and 35% release efficiency. Cytotoxicity studies showed that empty MSNPs2 had no significant cytotoxicity against MDAMB-231 cells. In contrast, light activation enhanced the cytotoxicity of docetaxel-loaded MSNPs2 significantly in MDA-MB-468 and MDAMB-231 breast cancer cell lines but had no effect on the cytotoxicity of free paclitaxel. Moreover, a photoactive drug delivery system, using only low light intensity, was developed by He et al. by grafting mesoporous silica coated UCNPs (lanthanide-doped upconverting nanoparticles) with Ru(II) complexes.291 The photoactive and blue light-cleavable Ru(II) complexes were grafted to the surface of the UCNPs to generate DOX-UCNP@mSiO2-Ru (Fig. 26B).291 The concept of this system is that the UCNPs can convert NIR light (∼980 nm) to UV and visible light which subsequently activates Ru(II) complexes to initiate photoreactions, releasing doxorubicin. Approximately 27% of doxorubicin and 59% Ru(II) complexes in this system can be released after 974 nm light irradiation, with 0.35 mW cm−2 for 5 hours. Using the same irradiation condition, NIR irradiation of DOX-UCNP@mSiO2-Ru for 10–30 min reduced cell viability to 40–29%. No obvious burn wound was observed when the light intensity was lower than 1 mW cm−2. The results of the above experiments indicate that the UNCPs may be an efficacious and safe therapy strategy using low intensity NIR light.
Fig. 26.
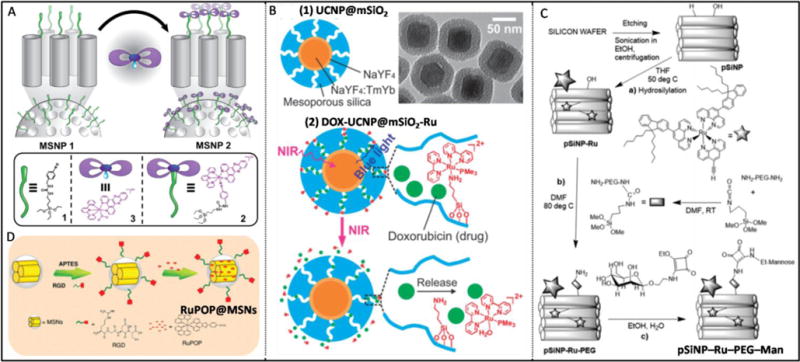
(A) Graphical representation for the assembly of mechanized MSNPs. (B) Schematic model and TEM image of UCNP@mSiO2 nanoparticles (1), the schematic illustrationof the drug release from DOX-UCNP@mSiO2-Ru nanoparticles (2). (C) Synthetic scheme for the pSiNP–Ru–PEG–Man. (D) The reaction pathways for the construction of the materials RuPOP@MSNs. Reproduced with permission from ref. 290 (Copyright 2015, American Chemical Society), 291, 292 (Copyright 2012, Royal Society of Chemistry) and 293 (Copyright 2015, Wiley-VCH), respectively.
Knežević et al. used multifunctionalized, porous silicon nanoparticles (pSiNPs) to deliver the photosensitized ruthenium complex for PDT (Fig. 26C).292 This ruthenium complex had good two photon absorption at 800 nm, yielding a ROS that killed cancer cells upon light irradiation. In addition, polyethylene glycol (PEG) was added to the surface of the nanoparticles to improve the dispersibility and biocompatibility of pSiNP-Ru-PEG-Man.292 Cytotoxicity studies indicated that 87% of MCF-7 cancer cells died at 80 mg/mL−1, following a 5 h pre-incubation upon 800 nm irradiation of pSiNP-Ru-PEG-Man. Recently, He et al. developed silica nanoparticle as a carrier for delivering anticancer Ru(II) complexes.292 The RGD (arginine-glycine-aspartic acid) peptide, which targets the ανβ3 integrin receptor, was attached to the surface of the mesoporous silica nanoparticles to improve their selectivity for cancer cells compared to and cells.293 The nanoparticle system, RuPOP@MSNs (Fig. 26D), released Ru(II) complexes much faster at pH 5.3 than at pH 7.4, with a 63.3% release for pH 5.3 and 43.1% for pH 7.4 after 12 days.293 Under a fluorescence microscope, RuPOP@MSNs was shown to enter the cells through endocytosis and release the Ru(II) complexes from lysosomes into the cytosol. The nanoparticle system had unprecedented, enhanced cytotoxicity in cancer cells overexpressing the integrin receptor and the mechanistic studies indicated that ROS overproduction induced by RuPOP@MSNs was involved in the cancer cell apoptosis through the regulation of AKT and MAPK signaling pathways.
6.4 Ru(II)-carbon nanotubes
Carbon nanotubes (CNTs) are one-dimensional, columniform structures of wrapped graphene sheets, forming tubular architectures. Single-walled nanotubes (SWNTs) and multiwalled nanotubes (MWNTs) are two main types of carbon nanotubes that can have high structural perfection.294 Single-walled nanotubes (SWNTs) are formed by a single graphite sheet seamlessly wrapped into a cylindrical tube. Multi-walled carbon nanotubes (MWCNT) contain several graphene sheets, leading to multiple concentric tubes of different diameters.295,296 Carbon nanotubes have unique properties that make them a highly promising system for biomedical application, as they can be used to deliver therapeutic drugs and diagnostic agents into cells.297,298 However, carbon nanotubes can also absorb light in the near-infrared region as photosensitizers and can kill cancer cells by localized photothermal effects.299
Wang et al. determined if nanostructured RuPOP@MWCNTs (Fig. 27A) are efficacious in surmounting multidrug resistance and radio resistance in cancer cells.300 RuPOP@MWCNTs was formed by loading a potent anticancer Ru(II) polypyridyl complex (RuPOP) in multiwalled carbon nanotubes via π−π interactions and by formation of a hydrogen bond. The RuPOP@MWCNTs entered cells via endocytosis and enhanced the selective cellular uptake of RuPOP in HepG2 and multidrug-resistant R-HepG2 cells. A concentration-dependent cell apoptosis occurred in HepG2 and R-HepG2 cells with the RuPOP@MWCNTs. The IC50 values of RuPOP@MWCNTs against R-HepG2 and HepG2 cells were 40 and 100 ng/mL, respectively. However, RuPOP@MWCNTs had significantly lower cytotoxicity towards L02 normal liver cells at the same concentration. Importantly, the nanosystem was found to significantly enhance the anticancer efficacy of clinically used X-rays in R-HepG2 cells through induction of apoptosis and G0/G1 cell cycle arrest, which involved the generation of ROS. Moreover, the nanosystem effectively reduced the toxic side effects of loaded drugs and prolonged the blood circulation of RuPOP in vivo.
Fig. 27.
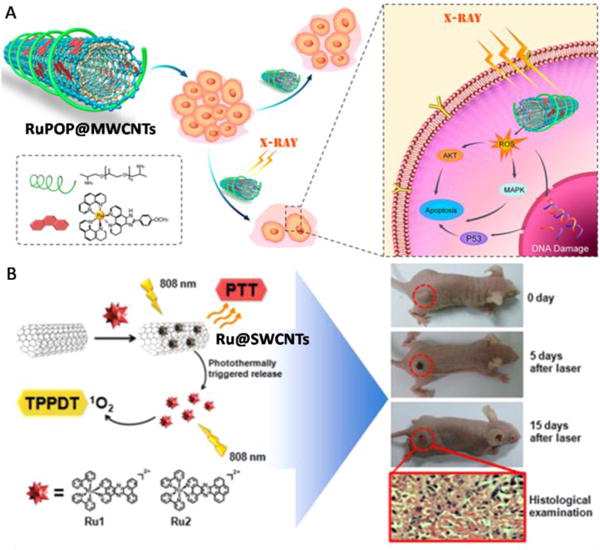
(A) The design and radiosensitization action mechanisms of the RuPOP@MWCNTs nanosystem. (B) Schematic Design of Ru@SWCNTs for bimodal photothermal therapy and two-photon photodynamic therapy with 808 nm Irradiation, and the presentative photographs of HeLa tumors in mice Ru@SWCNTs treatments. Reproduced with permission from ref. 300 and 301, respectively. Copyright 2015, American Chemical Society.
Zhang et al. used the single-walled carbon nanotube, loaded with Ru(II) complexes (Fig. 27B) for bimodal photothermal and photodynamic therapy, with near-infrared (808nm) irradiation.301 The carbon nanotube nanosystem had a high phothothermal conversion efficiency of ∼39.4% for Ru1@SWCNTs and 38.3% for Ru2@SWCNTs.300 The nanosystem can convert NIR (808 nm) light to heat (creating temperature changes from 20.3 to 58.5 °C for Ru1@SWCNTs and from 20.2 to 56.6 °C for Ru2@SWCNTs), a marked improvement from pure SWCNTs (where temperatures changed from 20.2 °C to 44.2 °C). The Ru(II) complex in the system can be released from the Ru@SWCNTs upon irradiation (808 nm, 0.25 W/cm2). Furthermore the released Ru(II) complexes produced 1O2 upon two photon laser irradiation (808 nm, 0.25 W/cm2). Cytotoxicity studies in vitro indicated that the Ru@SWCNTs (50 μg/mL) could kill all the HeLa cells under 808 nm laser irradiation at 0.25 W/cm2 for 5 min. More importantly, the tumors in nude mice shrank gradually or disappeared individually after 15 days of intratumoral injection with 100 μL of an aqueous dispersion of 1 mg/mL of Ru@SWCNTs upon the irradiation (808 nm laser).
6.5 The organic and biomaterial Ru(II) nanomaterials
Due to its biocompatibility and biodegradability, poly-(DL-lactic-co glycolic acid) (PLGA) is widely used as a nanocarrier to deliver drugs. Boeuf et al. encapsulated Ru(II) photosensitizers in PLGA-based nanosystems and determined their cytotoxicity in C6 glioma cells.302 The ruthenium-PLGA nanoparticles typically released a lower amount of the ruthenium complexes in phosphate buffer of pH 7.4 at 37 °C. In contrast, the nanoparticle systems exhibited faster release of the ruthenium complexes upon light irradiation. After exposure to a pulsed laser at 740 nm, the released Ru(II) complexes produced 1O2 and had potent cytotoxicity in the C6 glioma cells. Moreover, light exposure to PLGA-based ruthenium nanoparticles enhanced the cytotoxicity in C6 cells by ≈ 60%, supporting the notion that ruthenium-PLGA nanoparticles could be good candidates for two-photon-excited PDT applications in the future. In an effort to improve the blood circulation times and increase in the cellular uptake of polymetallodrugs, Wu et al. used the poly(ethylene glycol) (PEG) block to bind metal ruthenium to form a nanoparticle, PolyRu, via self-assembly.303 The PolyRu efficiently accumulated at the tumor sites through the enhanced permeability and retention (EPR) effect. Furthermore, PolyRu released anticancer Ru(II) complexes and generate cytotoxic 1O2 and inhibited the growth of tumors under 656 nm red light, with minimal systemic toxicity.
Zhou et al. investigated the anticancer efficacy of functionalized Ru(II) nanoparticles (RuNPs) loaded with luminescent Ru(II) complexes (RuBB) and epigallocatechingallate (EGCG).72 The nanoparticle system (RuBB-loaded EGCG-RuNPs) displayed red fluorescence and were entered SMMC-7721 liver cancer cells by 67LR-mediated endocytosis, with drug accumulation in the cytoplasm after 6 h.72 However, some of the nanoparticles were found in the nucleus after 12 h of incubation; the gradual accumulation indicated that the nucleus is a potential organelle target of the nanoparticles. Moreover, the nanoparticles significantly inhibited migration and invasion of the liver cancer cells and induced apoptosis by generating ROS and activating both the intrinsic and extrinsic apoptotic pathways. In vivo studies indicated that RuBB-loaded EGCG-RuNPs (50 μg/mouse) decreased the average tumor volume to 23% of the control level after 15 days of treatment.
In addition, a 3P-Ru/PbPS nanocapsule system for delivering the tris (1,10-phenanthroline) Ru(II) complex (3P-Ru) to tumor cells based on a pH-sensitive Poly (2-diisopropyl- aminoethyl methacrylate)-blockpoly (2-aminoethyl methacryl-ate hydrochloride) was designed by Chen et al.304 This system released 35% of the 3P-Ru in the nanocapsule in a pH 7.4 solution after 48 h of dialysis. However, the release rate reached 65% in a solution at pH 6.5, indicating a selective and rapid release in acidic environments. The 3P-Ru/PbPS system delivered the 3P-Ru into gliomas cells with high efficiency and inhibited U251 cell proliferation in a concentration-dependent manner via an apoptosis pathway. More importantly, the 3P-Ru/PbPS system significantly decreased tumor growth in tumor-bearing mice, resulting in smaller tumor volumes (5 mm−3), compared to mice treated for 8 days with PBS (25 mm−3), PbPS-NC (26 mm−3) and PEG-Ru-NC (20 mm−3) mice.
Recently, Chakrabortty et al. used the TPP-functionalized bloodplasma protein serum albumin (HSA) as the targeting peptide to combine with a Ru(II) complex to serve as a new photosentizer (cHSA-PEO-TPP-Ru) (Fig. 28).305 cHSA-PEO-TPP-Ru produced an ∼8-fold improvement in 1O2 quantum yields and a five times higher TP action cross section compared to the single Ru(II) complex. cHSA-PEO-TPP-Ru was highly localized in mitochondria. cHSA-PEO-TPP-Ru had potent phototoxicity, with an IC50 value of 34.9 nM against HeLa cells after light irradiation for 5 min (470 nm, ∼20 mW/cm2) and had a significantly high PI of 250. Further study indicated that cHSA-PEO-TPP-Ru had effective antileukemic activity, which occurred by decreasing cell proliferation and clonogenic property of the myeloid leukemic cell line OCI-AML3. However, cHSA-PEO-TPP-Ru was less toxicity to normal bone marrow cells compared to leukemic cells.
Fig. 28.
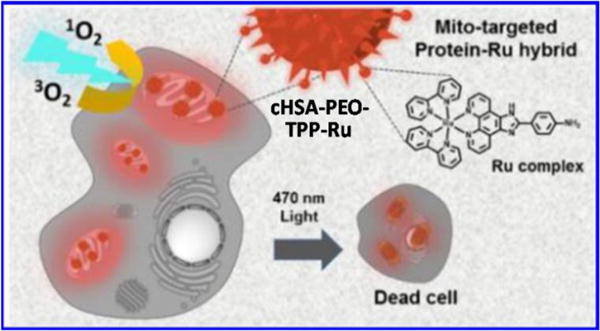
Schematic designof cHSA-PEO-TPP-Ru for PDT. Reproduced with permission from ref. 305. Copyright 2015, American Chemical Society.
In addition, there are other nanomaterials functionalized with Ru(II) complexes for cancer treatments, such as nanographene oxide,306 polymers,307 DNA origami and liposomes.308–311 These Ru(II) complex, functionalized-nanomaterials were safety and efficacious. Although further research, including clinical studies, is needed to verify these results, these examples serve as the basis of new developments for treating cancer.
7. Ru(II) complexes for bioorthogonal catalysis
The sections above presented ruthenium complexes as anticancer drugs or drug delivery systems, which play the role of reagents and direct targeting of tumor cells. Furthermore, ruthenium complexes could also be used as catalysis in biological system. The field, called bioorthogonal catalysis, has extended our understanding and is useful in imaging and drug development. Bioorthogonal chemistry allows for the occurrence of chemical reactions inside living cells without interfering with native biochemical processes. Thus, for this reason, bioorthogonal catalysts should selectively recognize specific functional, unnatural groups and catalyze the chemical reaction, especially in living systems. The catalyst needs to balance reactivity and stability. As a result, there is a lack of efficient bioorthogonal catalysts available today that match these crucial criteria.312 The design of bioorthogonal synthetic catalyst/substratepairs, which can passively diffuse into cells for use as tools in chemical biology studies, is a highly formidable challenge that has only recently started to be investigated.313 Recently, unique opportunities, arising from the catalysis of transition metals,314,315 have been explored.
Metal ruthenium complexes represent a powerful toolkit for selective synthesis and lysis of chemical bonds, thus offering plentiful physicochemical properties. It is possible that Ru(II) complexes may serve as catalysts in bioorthogonal chemistry. Based on how they enter into cells, Ru(II) complexes could also be cataloged into direct catalysis and nano-systems.
7.1 Ru(II) complexes as catalytic agents
Within the last decade, significant attention has been centered around improving the biocompatibility of a Cu(I)-catalyzed bioorthogonal reaction in living cells. Meanwhile, additional transition metals, such as palladium or ruthenium, have been examined as alternative sources to facilitate a bioorthogonal conjugation reaction in living cells.316 Current concepts of bioorthogonal chemistry have largely centered on ‘bond formation’ reactions between two mutually reactive bioorthogonal handles (Fig. 29). Recently, in a reverse strategy, a collection of ‘bond cleavage’ reactions has emerged with excellent biocompatibility.317 In 2006, Meggers et al. reported allylcarbamate cleavage in living cells using Ru(II) complexes (Ru1).318 In addition, researchers in the above group made progress towards synthesizing organometallic Ru(II) complexes for the catalytic uncaging of allyloxycarbonyl-protected amines under biologically relevant conditions and within living mammalian cells (Ru2).312,313 To find a catalyst with an improved activity, they screened a set of ruthenium complex and found that catalytic efficiency is fine-tuned by ligand- modifications (Ru3-Ru5).319 Using those Ru(II) complexes catalysts, the fluorescence development was more pronounced in the presence of thiophenol, yielding a 10-fold increase within 15 min in the cytoplasm of HeLa cells.313
Fig. 29.
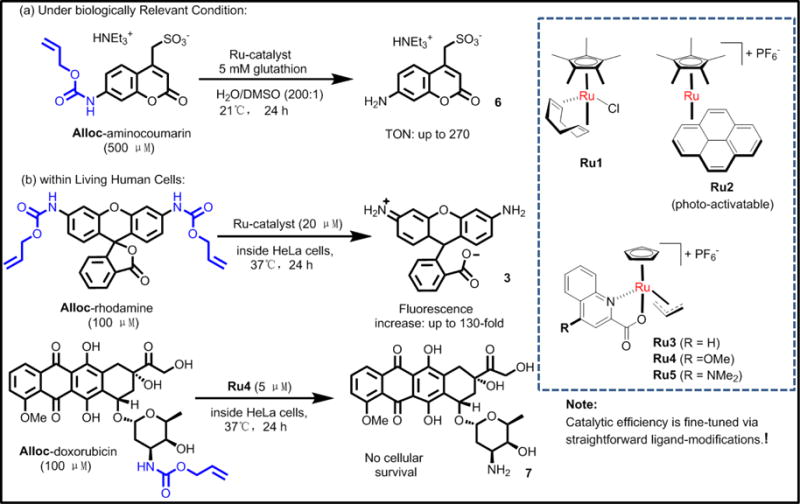
Uncaging reactions of allyloxycarbonyl (alloc) protected amines under (a) biologically relevant conditions and (b) within living human cells with ruthenium(II) complexes reported by Meggers et al. Reproduced with permission from ref. 319. Copyright 2015, Elsevier.
Sadhu et al. developed a luminescent Ru(II) complex that labeled proteins, enabling the direct visualization and photocatalytic reduction of arylazide in live cells.320 Hsu et al. reported that a bioorthogonal precatalyst Ru(II) complex cleaves a novel caged bioluminescence probe in luciferase- transfected 4T1 cells. The rate of the probe release and enzymatic turnover could be evaluated in 4T1 cells using a luciferase reporter system.321 With this method, researchers could measure the catalytic cleavage of a pro-probe and intracellular enzyme-mediated turnover of the released probe. This approach provides a set of critical metrics to observe the performance of biological catalysts and caging strategies for analogously cleavable pro-drugs.321 Similarly, Mascarenas et al. developed a Ru(II) catalyst in the mitochondria of living cells by integrating the phosphonium-targeting moieties to Ru(II) complexes.322 This metal catalysts was had significant catalytic efficacy and produced a smooth and rapid ruthenium-dependent depolarization of the mitochondria.
Sadler and colleagues summarized the design approaches of catalytic metallodrugs. Of special interest was the development of redox-modulating drugs, including the thiol oxidation and transfer hydrogenation reactions.323 The complexes [(η6-arene)Ru(azpy)I] (where arene = biphenyl, and azpy = N,N-dimethylphenyl- or hydroxyphenyl-azopyridine) were highly cytotoxic to A2780 and A549 cell lines, with IC50 values from 2–6 μM.324 The replacement of iodide by chloride dramatically decreased the cytotoxicity of the arene Ru(II) complexes.325 Intriguingly, the iodide Ru(II) complexes were catalysts in reactions with the tripeptide glutathione (γ-L-Glu-L-Cys-Gly).324 In addition, millimolar concentrations of glutathione were oxidized to glutathione disulfide in the presence of micromolar concentrations of Ru(II) complexes (Fig. 30A), significantly influencing intracellular redox processes. In addition, the same group also reported that another class of Ru(II) complexes with a chelated sulfonylethylamine ligand can convert coenzyme NAD+ into NADH in the presence of formate, thereby modulating the NAD+/NADH redox couple, as depicted in Fig. 30B.326 The efficacy of the arene Ru(II) sulfonamidoethyleneamine complexes toward ovarian cancer cells was enhanced by up to 50-fold in the presence of low, non-toxic concentrations of formate. The IC50 of this Ru(II) complex towards A2780 cells decreased from 13.6 μM (in the absence of formate), to 1.0 μM, in the presence of 2 mM formate, making the complex equipotent to cisplatin.326 Catalytic reactions in cancer cells offer a new strategy for the design of safe, Ru-based anticancer drugs that may contribute to further insight into the mechanism of cell death. The catalytic metallodrugs may offer the prospect of low-dose therapy and a challenging new design strategy for future exploration.323
Fig. 30.
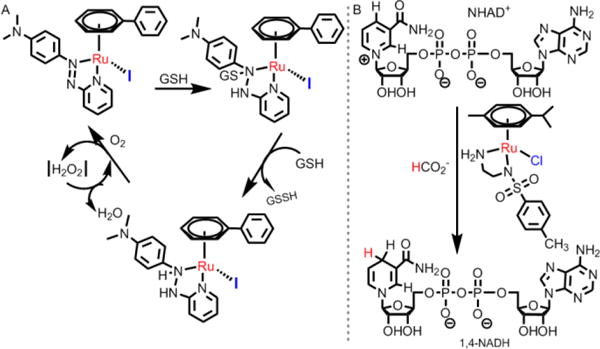
The Ru(II) catalytic reactions reported by Sadler group. Reproduced with permission from ref. 324 (Copyright 2008, National Academy of Sciences.) and 326 (Copyright 2015, Nature Publishing Group), respectively.
Currently, the catalytic efficiency of most precious-metal organometallic catalysis within living cells or in the presence of cell lysates has not been maximized. There are two major restrictions for organometallic catalysis in a cellular environment: 1) the catalyst and enzymes would affect each other and 2) even millimolar concentration of glutathione in cells under aerobic conditions would inhibit the precious metals catalytic activity.327 Enzymes have evolved to be efficient biocatalysts, and with the development of protein engineering, researchers have the potential obtain powerful tools to exploit more matallo-enzymes combined with natural proteins.328 The undeniable advantage of signal amplification through catalytic turnover has been successfully exploited in the area of enzyme-based bio-imaging and sensing.329
7.2 Ru(II) complexes as nano-catalytic system
In the sections above, results indicated that by combining ruthenium complexes and Au nanoparticles, the metallodrug could enter into living cells more easily and effectively. Au nanoparticles (AuNPs) provide non-toxic carriers for drug and gene delivery applications. In these systems, the gold core imparts stability to the assembly, while the monolayer allows tuning of surface properties, such as charge and hydrophobicity.279 The adaptable functionalization of both selective and specific recognition elements and environmentally responsive optoelectronic properties of AuNPs can be utilized to accomplish the transduction of the binding event with appropriate affinity and selectivity toward target analytes. Functionalized AuNPs may be both molecular receptor and signal transducer in a single sensing motif, thereby simplifying the sensor design while improving the sensitivity.282 All of the characteristics are present in bioorthogonal catalysis. Utilizing the well known property of thiols to bind to gold nanoparticles, researcher could produce facile access to multivalent, functional systems anchored on support. In addition, this complex would be soluble, with limited mobility and conformational restraint, thereby being suitable to act cooperatively in a catalytic process.281 Thus, various research groups have reported that Ru(II) form complexes with traditional gold nanoparticles, which are so-called nanozymes (see Fig. 31).
Fig. 31.
Spatiotemporal resolution of the sequential actions performed by Rotello’s nanobots. Reproduced with permission from ref. 333. Copyright 2015, Nature Publishing Group.
Recently, researchers have altered the surface of AuNPs with a variety of unnatural molecules such as sialic acids with azide groups, acylhydrazide, amine, or azide moieties.330,331 Supramolecules were also introduced onto gold nanoparticles to target different sites in cells and control the size of NPs.332 The new in vivo targeting strategy of nanoparticles, based on bioorthogonal, copper-free click chemistry, greatly broadens nano applications.333 The catalytic efficiencies of ruthenium complex nanoparticles in solutions and in cells have been measured and compared by Rotello et al.334 This bioorthogonal catalysis can be used not only in therapeutic applications, but also in treating noncancerous, chronic diseases. The system introduces biomimetics into bioorthogonal chemistry, providing a new platform for imaging and therapeutic applications, as well as combining pharmacological treatments with human made synthetic tools.333
8. Conclusions and future perspectives
Ru(II) compounds have highly promising anticancer activity in in vitro and in vivo models. Compared to platinum(II) compounds, ruthenium can be coordinated at two additional axial sits and it tends to form octahedral compounds. In general, the ligand combination and coordination geometry between ruthenium and its ligands mainly determine the activity of ruthenium compounds, mostly in their reactivity, hydrophobicity, binding, cellular uptake and intracellular distribution. In this regard, several different Ru(II) compounds in this review have been reported to have high selectivity and targeting, ultimately improving efficiency in cancer cells and minimizing toxicity in normal cells. Further studies of ruthenium compounds should investigate structure-activity relationships (SARs) to determine how modifying different functional groups on the ligands affect the anticancer efficacy of the complexes. As expected, most of the ruthenium complexes are lipophilic and hold positive charge, which facilitates their diffusion across the cell membrane, which is composed of negatively-charged and similarly lipophilic phospholipids. In addition, DNA, proteins and mitochondria often contain negative charges, allowing ruthenium complexes to selectively target these biomolecules or organelles and have significant efficacy. However, the ruthenium complexes must be non-toxic or relatively less toxic in normal tissue before use in patients. Simply modulating the ligands or increasing the lipophilicity and the charge of ruthenium complexes does not decrease their adverse effects, limiting their clinical application.
The introduction of PDT allows for the design of additional Ru(II) complexes with enhanced anticancer efficacy and higher selectivity. Ruthenium complexes have been proven to be effective photosensitizers for PDT due to the relatively long lifetimes of their excited states and efficient, low-energy visible-infrared light absorption. PDT has been shown to have high efficacy and minimal adverse effects, and has also been used to overcome resistance in tumor cells. Ru(II) complexes have long 3MLCT-based, excited states, with a luminescent lifetime of 1.1 μs and 3IL-based excited states, with remarkably long and tunable lifetimes (from 22 to 270 μs).335 These lifetimes is sufficient to produce ROS, which kill cancer cells upon light irradiation. Moreover, the use of O2-independent, Ru(II) complex photosensitizers may kill hypoxic tumors with enhanced cytotoxicity. Many Ru(II) complexes have efficient two-photon absorption in the NIR or IR region. These Ru(II) complexes were developed into the photosensitizers for two-photon absorption PDT, with less photodamage and a maximum tissue penetration depth. These Ru(II) complexes have the potential to be the next-generation photosensitizer compounds for PDT. Therefore, the characterization of Ru(II) complexes, with excellent physicochemical properties, remains urgent. In parallel with these advances in chemistry, improvement in the methods of irradiation should further improve the efficacy of Ru(II) complexes for PDT.
In this review, we discussed typical nanostructured Ru(II) complexes. The application of nanostructures improves the delivery and penetration of Ru(II) complexes,336,337 thus increasing the concentration in cells. For example, the combination of nanomedicine and Ru(II) complexes yields significant anticancer efficacy in drug resistant cancer cells as they are not substrates for MDR transporters. The encapsulation and delivery of Ru(II) complexes with nanomaterials may also improve certain pharmacological barriers relevant to drugs such as bioavailability, targeting ability, solubility, degradation and adverse effects. In addition, the Ru(II) complexes are designed to allow the nanomaterial systems to control the release of drugs and maintain efficacy within an acidic tumor environment. Despite the major benefits of the nano-functionalization of Ru(II) complexes in cancer therapy, nanomaterials also produce a certain level of toxicity in normal cells. The physical properties of nanomaterials affect the efficacy and toxicity of the nanomaterials. Therefore, further research should investigate the structure-activity relationship in nanostructured Ru(II) complexes. Finally, new ideas and breakthroughs in nano-functionalized complexes are expected to produce safer and more efficacious anticancer drugs.
Finally, we briefly discussed the role of Ru(II) complexes as bioorthogonal catalysts. With the formation and especially lysis of chemical bond, Ru(II) complexes accelerate biochemical reactions in living systems without interfering with normal physiological processes. The metallo-drugs could directly function at the specific groups, and also be delivered to cellular targets in encapsulated nanoparticles. With the development of protein engineering, it is essential that we integrate more transitional metal-based biological catalysts with natural biomolecules. By combining fluorescence with Ru complex moieties, we hypothesize that we can realize a real-time tracking of catalysts in cells, which would greatly improve our understanding of biological processes.
The complete mechanisms of action of the Ru(II) complexes are diverse and still poorly understood. However, this review delineates a number of different mechanisms where Ru(II) complexes have efficacy in certain cancers, with the ultimate goal of obtaining clinically acceptable candidates in the near future. We also believe that the design of metallo-drugs based on nanomaterials have potential as anticancer treatments.338 Also, we anticipate that metallodrugs will foster interdisciplinary research among organometallics, oncology, photochemistry, biology and nanomedicine.
Acknowledgments
We thank Dr. Charles R. Ashby, Jr. (St. John’s University, New York) for reviewing and editing the article. We thank Juanjuan Huang (Sun Yat-Sen University), for making the graphic image. We thank the support from Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John’s University and International Program for Ph.D. Candidates, Sun Yat-Sen University. This work was also supported by National Institute of Health-USA (Nos. 1R15CA143701 and 1R15GM116043-01), the National Science Foundation of China (Nos. 21471164 and 21525105), the 973 program (Nos. 2014CB845604 and 2015CB856301), and the Fundamental Research Funds for the Central Universities.
Biographies

Enju Wang receivedher PhDdegree from ETH-Zurichin 1989under the guidance ofthe later Prof. Simon in Analytical Chemistry. She joined Kagoshima University as a Research associate before her Post-doctoral research in the University of Michigan with Dr. Meyerhoff in 1992. She then joined thefaculty atSt. John’s University in 1993 as an assistant professor and become full professor in 2005. She is the author of over 30 peer reviewed publications and 3 bookchapters. Hercurrent research interest is focused ondeveloping Os/Ru-complex based optical sensors for Heparin and DNA polyanion sensing and detection.

Hui Chao received his PhD degree from Sun Yat-Sen University in 2000 under the guidance of Prof. Liangnian Ji. Subsequently, he joined the faculty at Sun Yat-Sen University. During 2000-2003, he attended Hong Kong University of Science and Technology every year as a short-term visiting scholar. In 2004-2005, he conducted postdoctoral work with Prof. F. A. Cotton at Texas A&M University. He was promoted to full professor in Sun Yat-Sen University in 2007. He is the author of over 180 peer reviewed publications and 4 book chapters. His current research interest is focused on metal-based anticancer complexes and bioimaging agents.

Leli Zeng received his MS degree in 2013 at XiangTan University and he worked under the supervision of Prof. Hui Chao from 2013 to 2017 at Sun Yat-Sen University, where he received his PhD degree in June, 2017. During 2016-2017, he had a one-year’s study on the multidrug resistance under the guidance of Prof. Zhe-Sheng Chen at St. John’s University. His primary work is focused on the design of ruthenium-based cancer drugs and nanodrugs, as well as the study of drug resistance.

Liangnian Ji was elected as a member of the Chinese Academy of Sciences in 2003. Prof. Ji is actively involved in bioinorganic chemistry research. He has designed and synthesized over 600 new coordination compounds. The results have been published in over 700 research papers, 3 monographs and 15 Chinese invention patents. Prof. Ji has been invited to be an organizing committee or advisory committee member in more than 10 international conferences or symposia on bioinorganic chemistry. He is also on the editorial board or an advisory member for 8 international or national chemical journals.

Pranav Gupta received his Bachelor’s degree in Pharmacy from Gurukul University and Master's degree in Pharmacology from St. John's University. He has worked as a graduate student researcher at St. Johns' University and has been a STEM mentor with the New York Academy of Sciences. He has published original research articles and invited reviews and book chapters in peer reviewed journals. Most recently, he is working on developing novel inhibitors to overcome chemotherapeutic multidrug resistance under Dr. Zhe-Sheng Chen.

Yanglu Chen graduated from Princeton University in 2017 with a Bachelor’s degree in Chemistry. She has also conducted research at Fox Chase Cancer Center, St. John’s University, the National Institutes of Health, University of Melbourne, and Rockefeller University, with interests ranging from immunology to genomics to nanomedicine. Most recently, she uses ultrafast spectroscopy to study photo-induced nanotheranostics under Professor Gregory Scholes.

Zhe-Sheng Chen received MS (Sun-Yat-Sen-University), PhD (Kagoshima-University). He had postdoctoral training at Fox-Chase-Cancer-Center. He joined St. John’s University in 2004 as an assistant professor. He is a full professor and director of Institute for Biotechnology. He is an editor-in-chief of Journal of Cancer Research Updates, and New Developments on Chemistry, an editor of African Journal of Pharmacy and Pharmacology, an editorial board member of 26 journals and a reviewer of~190 journals. He is an Ad-hoc reviewer of grants including NIH, New Zealand, Poland, China, Hungary, Netherlands and Canada etc. He established research projects to study ABC transporters and MDR.
References
- 1.Shi G, Monro S, Hennigar R, Colpitts J, Fong J, Kasimova K, Yin H, DeCoste R, Spencer C, Chamberlain L. Coord Chem Rev. 2015;282:127–138. [Google Scholar]
- 2.Garbutcheon-Singh KB, Grant MP, Harper BW, Krause-Heuer AM, Manohar M, Orkey N, Aldrich-Wright JR. Curr Top Med Chem. 2011;11:521–542. doi: 10.2174/156802611794785226. [DOI] [PubMed] [Google Scholar]
- 3.Tao Y, Li M, Ren J, Qu X. Chem Soc Rev. 2015;44:8636–8663. doi: 10.1039/c5cs00607d. [DOI] [PubMed] [Google Scholar]
- 4.Li F, Collins JG, Keene FR. Chem Soc Rev. 2015;44:2529–2542. doi: 10.1039/c4cs00343h. [DOI] [PubMed] [Google Scholar]
- 5.Muhammad N, Guo Z. Curr Opin Chem Biol. 2014;19:144–153. doi: 10.1016/j.cbpa.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Johnstone TC, Suntharalingam K, Lippard SJ. Chemi Rev. 2016;116:3436–3486. doi: 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Z, Sadler PJ. Acc Chem Res. 2014;47:1174–1185. doi: 10.1021/ar400266c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 9.Chu G. J Biol Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- 10.Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Pérez JM. Anti-Cancer Agents Med Chem. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 11.Sledge G, Loehrer PJ, Roth BJ, Einhorn LH. J Clin Oncol. 1988;6:1811–1814. doi: 10.1200/JCO.1988.6.12.1811. [DOI] [PubMed] [Google Scholar]
- 12.Arany I, Safirstein RL. Semin Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 13.Florea A-M, Büsselberg D. Cancers. 2011;3:1351–1371. doi: 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 15.Zheng YR, Suntharalingam K, Johnstone TC, Yoo H, Lin W, Brooks JG, Lippard SJ. J Am Chem Soc. 2014;136:8790–8798. doi: 10.1021/ja5038269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suntharalingam K, Song Y, Lippard SJ. Chem Commun. 2014;50:2465–2468. doi: 10.1039/c3cc48740g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graf N, Bielenberg DR, Kolishetti N, Muus C, Banyard J, Farokhzad OC, Lippard SJ. ACS nano. 2012;6:4530–4539. doi: 10.1021/nn301148e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Natl Acad Sci. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhar S, Liu Z, Thomale J, Dai H, Lippard SJ. J Am Chem Soc. 2008;130:11467–11476. doi: 10.1021/ja803036e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang XJ, Meng H, Wang Y, He H, Meng J, Lu J, Wang PC, Zhao Y, Gao X, Sun B. Proc Natl Acad Sci. 2010;107:7449–7454. doi: 10.1073/pnas.0909707107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fong TTH, Lok CN, Chung CYS, Fung YME, Chow PK, Wan PK, Che CM. Angew Chem, Int Ed. 2016;55:11935–11939. doi: 10.1002/anie.201602814. [DOI] [PubMed] [Google Scholar]
- 22.Cao ZT, Chen ZY, Sun CY, Li HJ, Wang HX, Cheng QQ, Zuo ZQ, Wang JL, Liu YZ, Wang YC, Wang J. Biomaterials. 2016;94:9–19. doi: 10.1016/j.biomaterials.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Abid M, Shamsi F, Azam A. Mini-Rev Med Chem. 2016;16:772–786. doi: 10.2174/1389557515666151001142012. [DOI] [PubMed] [Google Scholar]
- 24.Schmid WF, John RO, Arion VB, Jakupec MA, Keppler BK. Organometallics. 2007;26:6643–6652. [Google Scholar]
- 25.Hearn JM, Romero-Canelón I, Qamar B, Liu Z, Hands-Portman I, Sadler PJ. ACS Chem Biol. 2013;8:1335–1343. doi: 10.1021/cb400070a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romero-Canelón I, Salassa L, Sadler PJ. J Med Chem. 2013;56:1291–1300. doi: 10.1021/jm3017442. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Z, Wang X, Li T, Aime S, Sadler PJ, Guo Z. Angew Chem, Int Ed. 2014;53:13225–13228. doi: 10.1002/anie.201407406. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Romero-Canelon I, Qamar B, Hearn JM, Habtemariam A, Barry NPE, Pizarro AM, Clarkson GJ, Sadler PJ. Angew Chem, Int Ed. 2014;53:3941–3946. doi: 10.1002/anie.201311161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye RR, Tan CP, He L, Chen MH, Ji LN, Mao ZW. Chem Commun. 2014;50:10945–10948. doi: 10.1039/c4cc05215c. [DOI] [PubMed] [Google Scholar]
- 30.Zhang CX, Lippard SJ. Curr Opin Chem Biol. 2003;7:481–489. doi: 10.1016/s1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 31.Kostova I. Curr Med Chem. 2006;13:1085–1107. doi: 10.2174/092986706776360941. [DOI] [PubMed] [Google Scholar]
- 32.Smith GS, Therrien B. Dalton Trans. 2011;40:10793–10800. doi: 10.1039/c1dt11007a. [DOI] [PubMed] [Google Scholar]
- 33.Liu P, Jia J, Zhao Y, Wang K. Mini-Rev Med Chem. 2015;16:272–289. doi: 10.2174/1389557516666151120120524. [DOI] [PubMed] [Google Scholar]
- 34.Furrer J, Süss-Fink G. Coord Chem Rev. 2016;309:36–50. [Google Scholar]
- 35.Levina A, Mitra A, Lay PA. Metallomics. 2009;1:458–470. doi: 10.1039/b904071d. [DOI] [PubMed] [Google Scholar]
- 36.Hartinger CG, Jakupec MA, Zorbas-Seifried S, Groessl M, Egger A, Berger W, Zorbas H, Dyson PJ, Keppler BK. Chem Biodiversity. 2008;5:2140–2155. doi: 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- 37.Duan L, Fischer A, Xu Y, Sun L. J Am Chem Soc. 2009;131:10397–10399. doi: 10.1021/ja9034686. [DOI] [PubMed] [Google Scholar]
- 38.Abid M, Shamsi F, Azam A. Mini-Rev Med Chem. 2016;10:772–786. doi: 10.2174/1389557515666151001142012. [DOI] [PubMed] [Google Scholar]
- 39.Dragutan I, Dragutan V, Demonceau A. Molecules. 2015;20:17244–17274. doi: 10.3390/molecules200917244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartinger CG, Groessl M, Meier SM, Casini A, Dyson PJ. Chemical Society Reviews. 2013;42:6186–6199. doi: 10.1039/c3cs35532b. [DOI] [PubMed] [Google Scholar]
- 41.Antonarakis ES, Emadi A. Cancer Chemother Pharmacol. 2010;66:1–9. doi: 10.1007/s00280-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minchinton AI, Tannock IF. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 43.Bergamo A, Zorzet S, Gava B, Sorc A, Alessio E, Iengo E, Sava G. Anti-cancer drugs. 2000;11:665–672. doi: 10.1097/00001813-200009000-00012. [DOI] [PubMed] [Google Scholar]
- 44.Jones SL. Arch Psychiat Nurs. 1999;13:1–2. doi: 10.1016/s0883-9417(99)80011-9. [DOI] [PubMed] [Google Scholar]
- 45.Sava G, Gagliardi R, Cocchietto M, Clerici K, Capozzi I, Marrella M, Alessio E, Mestroni G, Milanino R. Pathol, Oncol Res. 1998;4:30–36. doi: 10.1007/BF02904692. [DOI] [PubMed] [Google Scholar]
- 46.Bacac M, Vadori M, Sava G, Pacor S. Cancer Immunol, Immun. 2004;53:1101–1110. doi: 10.1007/s00262-004-0566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergamo A, Gava B, Alessio E, Mestroni G, Serli B, Cocchietto M, Zorzet S, Sava G. Int J Oncol. 2002;21:1331–1338. [PubMed] [Google Scholar]
- 48.Debidda M, Sanna B, Cossu A, Posadino AM, Tadolini B, Ventura C, Pintus G. Int J Oncol. 2003;23:477–482. [PubMed] [Google Scholar]
- 49.Pacor S, Zorzet S, Cocchietto M, Bacac M, Vadori M, Turrin C, Gava B, Castellarin A, Sava G. J, Pharmacol Exp Ther. 2004;310:737–744. doi: 10.1124/jpet.104.066175. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Chen L, Liao S, Zheng K, Ji L. J, phys, chem, B. 2007;111:7862–7869. doi: 10.1021/jp0711794. [DOI] [PubMed] [Google Scholar]
- 51.Bacac M, Hotze AC, van der Schilden K, Haasnoot JG, Pacor S, Alessio E, Sava G, Reedijk J. J Inorg Biochem. 2004;98:402–412. doi: 10.1016/j.jinorgbio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Rademaker-Lakhai JM, van den Bongard D, Pluim D, Beijnen JH, Schellens JH. Clin Cancer res. 2004;10:3717–3727. doi: 10.1158/1078-0432.CCR-03-0746. [DOI] [PubMed] [Google Scholar]
- 53.Alessio E. Eur J Inorg Chem. 2017;2017:1549–1560. [Google Scholar]
- 54.Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK. J Inorg Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 55.Hartinger CG, Jakupec MA, Zorbas-Seifried S, Groessl M, Egger A, Berger W, Zorbas H, Dyson PJ, Keppler BK. Chem Biodiversity. 2008;5:2140–2155. doi: 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- 56.Bytzek AK, Koellensperger G, Keppler BK, Hartinger CG. J Inorg Biochem. 2016;160:250–255. doi: 10.1016/j.jinorgbio.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 57.Wang F, Habtemariam A, van der Geer EPL, Fernández R, Melchart M, Deeth RJ, Aird R, Guichard S, Fabbiani FPA, Lozano-Casal P, Oswald IDH, Jodrell DI, Parsons S, Sadler PJ. Proc Natl Acad Sci. 2005;102:18269–18274. doi: 10.1073/pnas.0505798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris RE, Aird RE, del Socorro Murdoch P, Chen H, Cummings J, Hughes ND, Parsons S, Parkin A, Boyd G, Jodrell DI. J Med Chem. 2001;44:3616–3621. doi: 10.1021/jm010051m. [DOI] [PubMed] [Google Scholar]
- 59.Rodrigues FP, Carneiro ZA, Mascharak P, Curti C, da Silva RS. Coord Chem Rev. 2016;306:701–707. [Google Scholar]
- 60.Kwong WL, Lam KY, Lok CN, Lai YT, Lee PY, Che CM. Angew Chem. 2016;128:13722–13726. doi: 10.1002/anie.201608094. [DOI] [PubMed] [Google Scholar]
- 61.Montani M, Pazmay GVB, Hysi A, Lupidi G, Pettinari R, Gambini V, Tilio M, Marchetti F, Pettinari C, Ferraro S. Pharmacol Res. 2016;107:282–290. doi: 10.1016/j.phrs.2016.03.032. [DOI] [PubMed] [Google Scholar]
- 62.Fan W, Huang P, Chen X. Chem Soc Rev. 2016;45:6488–6519. doi: 10.1039/c6cs00616g. [DOI] [PubMed] [Google Scholar]
- 63.Mari C, Pierroz V, Ferrari S, Gasser G. Chem Sci. 2015;6:2660–2686. doi: 10.1039/c4sc03759f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dickerson M, Howerton B, Bae Y, Glazer EC. J Mater Chem B. 2016;4:394–408. doi: 10.1039/C5TB01613D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glazer EC. Isr J Chem. 2013;53:391–400. [Google Scholar]
- 66.Shen Y, Shuhendler AJ, Ye D, Xu JJ, Chen HY. Chem Soc Rev. 2016;45:6725–6741. doi: 10.1039/c6cs00442c. [DOI] [PubMed] [Google Scholar]
- 67.Vajpayee V, Yang YJ, Kang SC, Kim H, Kim IS, Wang M, Stang PJ, Chi KW. Chem Commun. 2011;47:5184–5186. doi: 10.1039/c1cc10167f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y, Guan R, Zhang C, Huang J, Ji L, Chao H. Coord Chem Rev. 2016;310:16–40. [Google Scholar]
- 69.Barry NPE, Sadler PJ. ACS nano. 2013;7:5654–5659. doi: 10.1021/nn403220e. [DOI] [PubMed] [Google Scholar]
- 70.http://theralase.com/pressrelease/health-canada-approvesclinical-trial-application-anti-cancer-drug/.
- 71.Gill MR, Thomas JA. Chem Soc Rev. 2012;41:3179–3192. doi: 10.1039/c2cs15299a. [DOI] [PubMed] [Google Scholar]
- 72.Zhou Y, Yu Q, Qin X, Bhavsar D, Yang L, Chen Q, Zheng W, Chen L, Liu J. ACS Appl Mater Interfaces. 2016;8:15000–15012. doi: 10.1021/acsami.5b02261. [DOI] [PubMed] [Google Scholar]
- 73.Liang R, Wei M, Evans DG, Duan X. Chem Commun. 2014;50:14071–14081. doi: 10.1039/c4cc03118k. [DOI] [PubMed] [Google Scholar]
- 74.Puckett CA, Barton JK. J Am Chem Soc. 2007;129:46–47. doi: 10.1021/ja0677564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Puckett CA, Ernst RJ, Barton JK. Dalton Trans. 2010;39:1159–1170. doi: 10.1039/b922209j. 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Komor AC, Barton JK. Chem Commun. 2013;49:3617–3630. doi: 10.1039/c3cc00177f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Puckett CA, Barton JK. J Am Chem Soc. 2009;131:8738–8739. doi: 10.1021/ja9025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Puckett CA, Barton JK. Bioorg Med Chem. 2010;18:3564–3569. doi: 10.1016/j.bmc.2010.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan C, Lai S, Wu S, Hu S, Zhou L, Chen Y, Wang M, Zhu Y, Lian W, Peng W. J Med Chem. 2010;53:7613–7624. doi: 10.1021/jm1009296. [DOI] [PubMed] [Google Scholar]
- 80.Huang H, Zhang P, Yu B, Chen Y, Wang J, Ji L, Chao H. J Med Chem. 2014;57:8971–8983. doi: 10.1021/jm501095r. [DOI] [PubMed] [Google Scholar]
- 81.Groessl M, Zava O, Dyson PJ. Metallomics. 2011;3:591–599. doi: 10.1039/c0mt00101e. [DOI] [PubMed] [Google Scholar]
- 82.Green DR, Reed JC. Science. 1998;281:1309. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 83.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Acc Chem Res. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 84.Yousif LF, Stewart KM, Kelley SO. Chembiochem. 2009;10:1939–1950. doi: 10.1002/cbic.200900185. [DOI] [PubMed] [Google Scholar]
- 85.Bhat TA, Kumar S, Chaudhary AK, Yadav N, Chandra D. Drug Discov Today. 2015;20:635–643. doi: 10.1016/j.drudis.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pierroz V, Joshi T, Leonidova A, Mari C, Schur J, Ott I, Spiccia L, Ferrari S, Gasser G. J Am Chem Soc. 2012;134:20376–20387. doi: 10.1021/ja307288s. [DOI] [PubMed] [Google Scholar]
- 87.Joshi T, Pierroz V, Mari C, Gemperle L, Ferrari S, Gasser G. Angew Chem, Int Ed. 2014;53:2960–2963. doi: 10.1002/anie.201309576. [DOI] [PubMed] [Google Scholar]
- 88.Dickerson M, Sun Y, Howerton B, Glazer EC. Inorg Chem. 2014;53:10370–10377. doi: 10.1021/ic5013796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qian C, Wang JQ, Song CL, Wang LL, Ji LN, Chao H. Metallomics. 2013;5:844–854. doi: 10.1039/c3mt20270d. [DOI] [PubMed] [Google Scholar]
- 90.Wang JQ, Zhang PY, Qian C, Hou XJ, Ji LN, Chao H. J Biol Inorg Chem. 2014;19:335–348. doi: 10.1007/s00775-013-1069-2. [DOI] [PubMed] [Google Scholar]
- 91.Liu J, Chen Y, Li G, Zhang P, Jin C, Zeng L, Ji L, Chao H. Biomaterials. 2015;56:140–153. doi: 10.1016/j.biomaterials.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 92.Luzio JP, Pryor PR, Bright NA. Nat Rev Mol Cell Biol. 2007;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 93.Cho K, Wang X, Nie S, Shin DM. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 94.He L, Tan CP, Ye RR, Zhao YZ, Liu YH, Zhao Q, Ji LN, Mao ZW. Angew Chem, Int Ed. 2014;53:12137–12141. doi: 10.1002/anie.201407468. [DOI] [PubMed] [Google Scholar]
- 95.He L, Li Y, Tan CP, Ye RR, Chen MH, Cao JJ, Ji LN, Mao ZW. Chem Sci. 2015;6:5409–5418. doi: 10.1039/c5sc01955a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qiu K, Liu Y, Huang H, Liu C, Zhu H, Chen Y, Ji L, Chao H. Dalton Trans. 2016;45:16144–16147. doi: 10.1039/c6dt03328h. [DOI] [PubMed] [Google Scholar]
- 97.Qiu K, Huang H, Liu B, Liu Y, Huang Z, Chen Y, Ji LN, Chao H. ACS Appl Mater Interfaces. 2016;8:12702. doi: 10.1021/acsami.6b03422. [DOI] [PubMed] [Google Scholar]
- 98.Huang H, Yu B, Zhang P, Huang J, Chen Y, Gasser G, Ji L, Chao H. Angew Chem. 2015;127:14255–14258. doi: 10.1002/anie.201507800. [DOI] [PubMed] [Google Scholar]
- 99.Kroemer G, Jäättelä M. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 100.Casini A, Mastrobuoni G, Ang WH, Gabbiani C, Pieraccini G, Moneti G, Dyson PJ, Messori L. Chemmedchem. 2007;2:631–635. doi: 10.1002/cmdc.200600258. [DOI] [PubMed] [Google Scholar]
- 101.Zhao R, Hammitt R, Thummel RP, Liu Y, Turro C, Snapka RM. Dalton Trans. 2009:10926–10931. doi: 10.1039/b913959a. [DOI] [PubMed] [Google Scholar]
- 102.Zeng L, Xiao Y, Liu J, Tan L. J Mol Struct. 2012;1019:183–190. [Google Scholar]
- 103.Aird RE, Cummings J, Ritchie AA, Muir M, Morris RE, Chen H, Sadler PJ, Jodrell DI. Br J Cancer. 2002;86:1652–1657. doi: 10.1038/sj.bjc.6600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pascu GI, Hotze AC, Sanchez-Cano C, Kariuki BM, Hannon MJ. Angew Chem. 2007;119:4452–4456. doi: 10.1002/anie.200700656. [DOI] [PubMed] [Google Scholar]
- 105.Wu K, Hu W, Luo Q, Li X, Xiong S, Sadler PJ, Wang F. J Am Soc Mass Spectrom. 2013;24:410–420. doi: 10.1007/s13361-012-0539-z. [DOI] [PubMed] [Google Scholar]
- 106.Chen H, Parkinson JA, Morris RE, Sadler PJ. J Am Chem Soc. 2003;125:173–186. doi: 10.1021/ja027719m. [DOI] [PubMed] [Google Scholar]
- 107.Turro C. Proc Natl Acad Sci. 2011;108:17573–17574. doi: 10.1073/pnas.1114519108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Friedman AE, Chambron JC, Sauvage JP, Turro NJ, Barton JK. J Am Chem Soc. 1990;112:4960–4962. [Google Scholar]
- 109.Gill MR, Harun SN, Halder S, Boghozian RA, Ramadan K, Ahmad H, Vallis KA. Sci Rep. 2016;6:31973. doi: 10.1038/srep31973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Önfelt B, Göstring L, Lincoln P, Nordén B, Önfelt A. Mutagenesis. 2002;17:317–320. doi: 10.1093/mutage/17.4.317. [DOI] [PubMed] [Google Scholar]
- 111.Gill MR, Garcia-Lara J, Foster SJ, Smythe C, Battaglia G, Thomas JA. Nat Chem. 2009;1:662–667. doi: 10.1038/nchem.406. [DOI] [PubMed] [Google Scholar]
- 112.Baggaley E, Gill MR, Green NH, Turton D, Sazanovich IV, Botchway SW, Smythe C, Haycock JW, Weinstein JA, Thomas JA. Angew Chem. 2014;126:3435–3439. doi: 10.1002/anie.201309427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang L, Carroll P, Meggers E. Org Lett. 2004;6:521–523. doi: 10.1021/ol036283s. [DOI] [PubMed] [Google Scholar]
- 114.Meggers E. Chem Commun. 2009:1001–1010. doi: 10.1039/b813568a. [DOI] [PubMed] [Google Scholar]
- 115.Aman F, Hanif M, Kubanik M, Ashraf A, Soehnel T, Jamieson S, Siddiqui W, Hartinger C. Chem – Eur J. 2017;23:4893–4902. doi: 10.1002/chem.201700263. [DOI] [PubMed] [Google Scholar]
- 116.Babak MV, Meier SM, Huber KVM, Reynisson J, Legin AA, Jakupec MA, Roller A, Stukalov A, Gridling M, Bennett KL, Colinge J, Berger W, Dyson PJ, Superti-Furga G, Keppler BK, Hartinger CG. Chem Sci. 2015;6:2449–2456. doi: 10.1039/c4sc03905j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kilpin KJ, Dyson PJ. Chem Sci. 2013;4:1410–1419. [Google Scholar]
- 118.Blanck S, Maksimoska J, Baumeister J, Harms K, Marmorstein R, Meggers E. Angew Chem, Int Ed. 2012;51:5244–5246. doi: 10.1002/anie.201108865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Meggers E. Curr Opin Chem Biol. 2007;11:287–292. doi: 10.1016/j.cbpa.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 120.Dwyer FP, Gyarfas EC, Rogers WP, Koch JH. Nature. 1952;170:190. doi: 10.1038/170190a0. [DOI] [PubMed] [Google Scholar]
- 121.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 122.Rüegg UT, Gillian B. Trends Pharmacol Sci. 1989;10:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- 123.Bregman H, Carroll PJ, Meggers E. J Am Chem Soc. 2006;128:877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 124.Feng L, Geisselbrecht Y, Blanck S, Wilbuer A, Atilla-Gokcumen GE, Filippakopoulos P, Kräling K, Celik MA, Harms K, Maksimoska J. J Am Chem Soc. 2011;133:5976–5986. doi: 10.1021/ja1112996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bregman H, Meggers E. Org Lett. 2006;8:5465–5468. doi: 10.1021/ol0620646. [DOI] [PubMed] [Google Scholar]
- 126.Meggers E, Atilla-Gokcumen GE, Gruendler K, Frias C, Prokop A. Dalton Trans. 2009:10882–10888. doi: 10.1039/b917792b. [DOI] [PubMed] [Google Scholar]
- 127.Pagano N, Wong EY, Breiding T, Liu H, Wilbuer A, Bregman H, Shen Q, Diamond SL, Meggers E. J Org Chem. 2009;74:8997–9009. doi: 10.1021/jo901641k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qin J, Rajaratnam R, Feng L, Salami J, Barber-Rotenberg JS, Domsic J, Reyes-Uribe P, Liu H, Dang W, Berger SL, Villanueva J, Meggers E, Marmorstein R. J Med Chem. 2015;58:305–314. doi: 10.1021/jm5011868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vock CA, Ang WH, Scolaro C, Phillips AD, Lagopoulos L, Juillerat-Jeanneret L, Sava G, Scopelliti R, Dyson PJ. J Med Chem. 2007;50:2166–2175. doi: 10.1021/jm070039f. [DOI] [PubMed] [Google Scholar]
- 130.Ang WH, De Luca A, Chapuis-Bernasconi C, Juillerat-Jeanneret L, LoBello M, Dyson PJ. Chemmedchem. 2007;2:1799–1806. doi: 10.1002/cmdc.200700209. [DOI] [PubMed] [Google Scholar]
- 131.Respondek T, Garner RN, Herroon MK, Podgorski I, Turro C, Kodanko JJ. J Am Chem Soc. 2011;133:17164–17167. doi: 10.1021/ja208084s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tan CP, Lu YY, Ji LN, Mao ZW. Metallomics. 2014;6:978–995. doi: 10.1039/c3mt00225j. [DOI] [PubMed] [Google Scholar]
- 133.Kangdi Z, Qiong W, Chengxi W, Weijun T, Wenjie M. Anti-Cancer Agent. 2017;17:29–39. [Google Scholar]
- 134.Cao W, Zheng W, Chen T. Sci Rep. 2015;5:9157. doi: 10.1038/srep09157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu Y, Chen T, Liu J, Wong YS. Medchemcomm. 2013;4:865–869. [Google Scholar]
- 136.Chen L, Li G, Peng F, Jie X, Dongye G, Cai K, Feng R, Li B, Zeng Q, Lun K, Chen J, Xu B. Oncotarget. 2016;7:80716–80734. doi: 10.18632/oncotarget.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Majno G, Joris I. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 138.Castonguay A, Doucet C, Juhas M, Maysinger D. J Med Chem. 2012;55:8799–8806. doi: 10.1021/jm301103y. [DOI] [PubMed] [Google Scholar]
- 139.Yuan J, Lei Z, Wang X, Zhu F, Chen D. Metallomics. 2015;7:896–907. doi: 10.1039/c5mt00010f. [DOI] [PubMed] [Google Scholar]
- 140.Mühlgassner G, Bartel C, Schmid WF, Jakupec MA, Arion VB, Keppler BK. J Inorg Biochem. 2012;116:180–187. doi: 10.1016/j.jinorgbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Suss-Fink G. Dalton Trans. 2010;39:1673–1688. doi: 10.1039/b916860p. [DOI] [PubMed] [Google Scholar]
- 142.Murray BS, Babak MV, Hartinger CG, Dyson PJ. Coord Chem Rev. 2016;306:86–114. Part 1. [Google Scholar]
- 143.Dougan SJ, Sadler PJ. Chimia. 2007;61:704–715. [Google Scholar]
- 144.Wang F, Chen H, Parsons S, Oswald ID, Davidson JE, Sadler PJ. Chem – Eur J. 2003;9:5810–5820. doi: 10.1002/chem.200304724. [DOI] [PubMed] [Google Scholar]
- 145.Romero-Canelon I, Pizarro AM, Habtemariam A, Sadler PJ. Metallomics. 2012;4:1271–1279. doi: 10.1039/c2mt20189e. [DOI] [PubMed] [Google Scholar]
- 146.Montani M, Pazmay GVB, Hysi A, Lupidi G, Pettinari R, Gambini V, Tilio M, Marchetti F, Pettinari C, Ferraro S, Iezzi M, Marchini C, Amici A. Pharm Res. 2016;107:282–290. doi: 10.1016/j.phrs.2016.03.032. [DOI] [PubMed] [Google Scholar]
- 147.Chow MJ, Licona C, Yuan Qiang Wong D, Pastorin G, Gaiddon C, Ang WH. J Med Chem. 2014;57:6043–6059. doi: 10.1021/jm500455p. [DOI] [PubMed] [Google Scholar]
- 148.Chow MJ, Licona C, Pastorin G, Mellitzer G, Ang WH, Gaiddon C. Chem Sci. 2016;7:4117–4124. doi: 10.1039/c6sc00268d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chow MJ, Babak MV, Wong DYQ, Pastorin G, Gaiddon C, Ang WH. Mol Pharm. 2016;13:2543–2554. doi: 10.1021/acs.molpharmaceut.6b00348. [DOI] [PubMed] [Google Scholar]
- 150.Grozav A, Balacescu O, Balacescu L, Cheminel T, Berindan-Neagoe I, Therrien B. J Med Chem. 2015;58:8475–8490. doi: 10.1021/acs.jmedchem.5b00855. [DOI] [PubMed] [Google Scholar]
- 151.Betanzos-Lara S, Salassa L, Habtemariam A, Sadler PJ. Chem Commun. 2009:6622–6624. doi: 10.1039/b914153g. [DOI] [PubMed] [Google Scholar]
- 152.Betanzos-Lara S, Salassa L, Habtemariam A, Novakova O, Pizarro AM, Clarkson GJ, Liskova B, Brabec V, Sadler PJ. Organometallics. 2012;31:3466–3479. [Google Scholar]
- 153.Wang T, Zhou Q, Zhang Y, Zheng Y, Wang W, Hou Y, Jiang G, Cheng X, Wang X. RSC Adv. 2016;6:45652–45659. [Google Scholar]
- 154.Chen Y, Lei W, Hou Y, Li C, Jiang G, Zhang B, Zhou Q, Wang X. Dalton Trans. 2015;44:7347–7354. doi: 10.1039/c5dt00939a. [DOI] [PubMed] [Google Scholar]
- 155.Motswainyana WM, Ajibade PA. Adv Chem. 2015;2015:859730. [Google Scholar]
- 156.Chelopo MP, Pawar SA, Sokhela MK, Govender T, Kruger HG, Maguire GE. Eur J Med Chem. 2013;66:407–414. doi: 10.1016/j.ejmech.2013.05.048. [DOI] [PubMed] [Google Scholar]
- 157.Habtemariam A, Melchart M, Fernández R, Parsons S, Oswald ID, Parkin A, Fabbiani FP, Davidson JE, Dawson A, Aird RE. J Med Chem. 2006;49:6858–6868. doi: 10.1021/jm060596m. [DOI] [PubMed] [Google Scholar]
- 158.Malgorzata Frik AM, Elie Benelita T, Gonzalo Oscar, Ramírez de Mingo Daniel, Sanaú Mercedes, Sánchez-Delgado roberto, Sadhukha Tanmoy, Prabha Swayam, Ramos Joe W, Marzo Isabel, Contel María. J Med Chem. 2014;57:9995–10012. doi: 10.1021/jm5012337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lord RM, Hebden AJ, Pask CM, Henderson IR, Allison SJ, Shepherd SL, Phillips RM, McGowan PC. J Med Chem. 2015;58:4940–4953. doi: 10.1021/acs.jmedchem.5b00455. [DOI] [PubMed] [Google Scholar]
- 160.Clavel CM, Paunescu E, Nowak-Sliwinska P, Griffioen AW, Scopelliti R, Dyson PJ. J Med Chem. 2015;58:3356–3365. doi: 10.1021/jm501655t. [DOI] [PubMed] [Google Scholar]
- 161.Seršen S, Kljun J, Kryeziu K, Panchuk R, Alte B, Körner W, Heffeter P, Berger W, Turel I. J Med Chem. 2015;58:3984–3996. doi: 10.1021/acs.jmedchem.5b00288. [DOI] [PubMed] [Google Scholar]
- 162.Pettinari R, Pettinari C, Marchetti F, Skelton BW, White AH, Bonfili L, Cuccioloni M, Mozzicafreddo M, Cecarini V, Angeletti M, Nabissi M, Eleuteri AM. J Med Chem. 2014;57:4532–4542. doi: 10.1021/jm500458c. [DOI] [PubMed] [Google Scholar]
- 163.Yellol GS, Donaire A, Yellol JG, Vasylyeva V, Janiak C, Ruiz J. Chem Commun. 2013;49:11533–11535. doi: 10.1039/c3cc46239k. [DOI] [PubMed] [Google Scholar]
- 164.Yellol J, Pérez SA, Buceta A, Yellol G, Donaire A, Szumlas P, Bednarski PJ, Makhloufi G, Janiak C, Espinosa A. J Med Chem. 2015;58:7310–7327. doi: 10.1021/acs.jmedchem.5b01194. [DOI] [PubMed] [Google Scholar]
- 165.Bratsos I, Gianferrara T, Alessio E, Hartinger CG, Jakupec MA, Keppler BK. In: Bioinorganic Medicinal Chemistry. Alessio E, editor. Wiley-VCH; Weinheim: 2011. pp. 151–174. [Google Scholar]
- 166.Scolaro C, Bergamo A, Brescacin L, Delfino R, Cocchietto M, Laurenczy G, Geldbach TJ, Sava G, Dyson PJ. J Med Chem. 2005;48:4161–4171. doi: 10.1021/jm050015d. [DOI] [PubMed] [Google Scholar]
- 167.Bergamo A, Masi A, Dyson PJ, Sava G. Int J Oncol. 2008;33:1281. [PubMed] [Google Scholar]
- 168.Weiss A, Berndsen RH, Dubois M, Muller C, Schibli R, Griffioen AW, Dyson PJ, Nowak-Sliwinska P. Chem Sci. 2014;5:4742–4748. [Google Scholar]
- 169.Pettinari R, Marchetti F, Condello F, Pettinari C, Lupidi G, Scopelliti R, Mukhopadhyay S, Riedel T, Dyson PJ. Organometallics. 2014;33:3709–3715. [Google Scholar]
- 170.Allardyce CS, Dyson PJ, Ellis DJ, Heath SL. Chem Commun. 2001:1396–1397. [Google Scholar]
- 171.Adhireksan Z, Davey GE, Campomanes P, Groessl M, Clavel CM, Yu H, Nazarov AA, Yeo CHF, Ang WH, Dröge P, Rothlisberger U, Dyson PJ, Davey CA. Nat Commun. 2014;5:3462. doi: 10.1038/ncomms4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Adhireksan Z, Palermo G, Riedel T, Ma Z, Muhammad R, Rothlisberger U, Dyson PJ, Davey CA. Nat Commun. 2017;8:14860. doi: 10.1038/ncomms14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Casini A, Gabbiani C, Sorrentino F, Rigobello MP, Bindoli A, Geldbach TJ, Marrone A, Re N, Hartinger CG, Dyson PJ. J Med Chem. 2008;51:6773–6781. doi: 10.1021/jm8006678. [DOI] [PubMed] [Google Scholar]
- 174.Linares F, Galindo MA, Galli S, Romero MA, Navarro JA, Barea E. Inorg Chem. 2009;48:7413–7420. doi: 10.1021/ic900980y. [DOI] [PubMed] [Google Scholar]
- 175.Mishra A, Kang SC, Chi KW. Eur J Inorg Chem. 2013;2013:5222–5232. [Google Scholar]
- 176.Singh AK, Pandey DS, Xu Q, Braunstein P. Coord Chem Rev. 2014;270:31–56. [Google Scholar]
- 177.Mendoza-Ferri MG, Hartinger CG, Mendoza MA, Groessl M, Egger AE, Eichinger RE, Mangrum JB, Farrell NP, Maruszak M, Bednarski PJ. J Med Chem. 2009;52:916–925. doi: 10.1021/jm8013234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mendoza-Ferri MG, Hartinger CG, Eichinger RE, Stolyarova N, Severin K, Jakupec MA, Nazarov AA, Keppler BK. Organometallics. 2008;27:2405–2407. [Google Scholar]
- 179.Nováková O, Nazarov AA, Hartinger CG, Keppler BK, Brabec V. Biochem Pharmacol. 2009;77:364–374. doi: 10.1016/j.bcp.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 180.Gras M, Therrien B, Süss-Fink G, Zava O, Dyson PJ. Dalton Trans. 2010;39:10305–10313. doi: 10.1039/c0dt00887g. [DOI] [PubMed] [Google Scholar]
- 181.Therrien B. CrystEngComm. 2015;17:484–491. [Google Scholar]
- 182.Schmitt Fdr, Freudenreich J, Barry NP, Juillerat-Jeanneret L, Süss-Fink G, Therrien B. J Am Chem Soc. 2011;134:754–757. doi: 10.1021/ja207784t. [DOI] [PubMed] [Google Scholar]
- 183.Mattsson J, Govindaswamy P, Renfrew AK, Dyson PJ, Štěpnička P, Süss-Fink G, Therrien B. Organometallics. 2009;28:4350–4357. [Google Scholar]
- 184.Therrien B. Chem – Eur J. 2013;19:8378–8386. doi: 10.1002/chem.201301348. [DOI] [PubMed] [Google Scholar]
- 185.Therrien B, Süss-Fink G, Govindaswamy P, Renfrew AK, Dyson PJ. Angew Chem. 2008;120:3833–3836. doi: 10.1002/anie.200800186. [DOI] [PubMed] [Google Scholar]
- 186.Mattsson J, Zava O, Renfrew AK, Sei Y, Yamaguchi K, Dyson PJ, Therrien B. Dalton Trans. 2010;39:8248–8255. doi: 10.1039/c0dt00436g. [DOI] [PubMed] [Google Scholar]
- 187.Medlycott EA, Hanan GS. Chem Soc Rev. 2005;34:133–142. doi: 10.1039/b316486c. [DOI] [PubMed] [Google Scholar]
- 188.Gill MR, Thomas JA. Chem Soc Rev. 2012;41:3179–3192. doi: 10.1039/c2cs15299a. [DOI] [PubMed] [Google Scholar]
- 189.Salassa L. Eur J Inorg Chem. 2011;2011:4931–4947. [Google Scholar]
- 190.Li G, Sun L, Ji L, Chao H. Dalton Trans. 2016;45:13261–13276. doi: 10.1039/c6dt01624c. [DOI] [PubMed] [Google Scholar]
- 191.Dolmans DE, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 192.Kilah NL, Meggers E. Aust J Chem. 2012;65:1325–1332. [Google Scholar]
- 193.Tan C, Wu S, Lai S, Wang M, Chen Y, Zhou L, Zhu Y, Lian W, Peng W, Ji L. Dalton Trans. 2011;40:8611–8621. doi: 10.1039/c1dt10084j. [DOI] [PubMed] [Google Scholar]
- 194.Song H, Kaiser JT, Barton JK. Nat chem. 2012;4:615–620. doi: 10.1038/nchem.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhu BZ, Chao XJ, Huang CH, Li Y. Chem Sci. 2016;7:4016–4023. doi: 10.1039/c5sc03796d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Puckett CA, Barton JK. Biochemistry. 2008;47:11711–11716. doi: 10.1021/bi800856t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gill MR, Cecchin D, Walker MG, Mulla RS, Battaglia G, Smythe C, Thomas JA. Chem Sci. 2013;4:4512–4519. doi: 10.1039/c3sc51725j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zeng ZP, Wu Q, Sun FY, Zheng KD, Mei WJ. Inorg Chem. 2016;55:5710–5718. doi: 10.1021/acs.inorgchem.6b00824. [DOI] [PubMed] [Google Scholar]
- 199.Yu Q, Liu Y, Xu L, Zheng C, Le F, Qin X, Liu Y, Liu J. Eur J Med Chem. 2014;82:82–95. doi: 10.1016/j.ejmech.2014.05.040. [DOI] [PubMed] [Google Scholar]
- 200.Li L, Wong YS, Chen T, Fan C, Zheng W. Dalton Trans. 2012;41:1138–1141. doi: 10.1039/c1dt11950h. [DOI] [PubMed] [Google Scholar]
- 201.Chen T, Mei WJ, Wong YS, Liu J, Liu Y, Xie HS, Zheng WJ. Medchemcomm. 2010;1:73–75. doi: 10.1039/c8md90010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhao Z, Luo Z, Wu Q, Zheng W, Feng Y, Chen T. Dalton Trans. 2014;43:17017–17028. doi: 10.1039/c4dt01392a. [DOI] [PubMed] [Google Scholar]
- 203.Du Y, Fu X, Li H, Chen B, Guo Y, Su G, Zhang H, Ning F, Lin Y, Mei W, Chen T. ChemMedChem. 2014;9:714–718. doi: 10.1002/cmdc.201300379. [DOI] [PubMed] [Google Scholar]
- 204.Chen T, Liu Y, Zheng WJ, Liu J, Wong YS. Inorg Chem. 2010;49:6366–6368. doi: 10.1021/ic100277w. [DOI] [PubMed] [Google Scholar]
- 205.Deng Z, Gao P, Yu L, Ma B, You Y, Chan L, Mei C, Chen T. Biomaterials. 2017;129:111–126. doi: 10.1016/j.biomaterials.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 206.Cardoso CR, Lima MVS, Cheleski J, Peterson EJ, Venâncio T, Farrell NP, Carlos RM. J Med Chem. 2014;57:4906–4915. doi: 10.1021/jm5005946. [DOI] [PubMed] [Google Scholar]
- 207.Griffith C, Dayoub AS, Jaranatne T, Alatrash N, Mohamedi A, Abayan K, Breitbach ZS, Armstrong DW, MacDonnell FM. Chem Sci. 2017 doi: 10.1039/C6SC04094B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang YC, Qian C, Peng ZL, Hou XJ, Wang LL, Chao H, Ji LN. J Inorg Biochem. 2014;130:15–27. doi: 10.1016/j.jinorgbio.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 209.Kou JF, Qian C, Wang JQ, Chen X, Wang LL, Chao H, Ji LN. J Biol Inorg Chem. 2012;17:81–96. doi: 10.1007/s00775-011-0831-6. [DOI] [PubMed] [Google Scholar]
- 210.He X, Zeng L, Yang G, Xie L, Sun X, Tan L. Inorg Chimi Acta. 2013;408:9–17. [Google Scholar]
- 211.Shi S, Xu JH, Gao X, Huang HL, Yao TM. Chem – Eur J. 2015;21:11435–11445. doi: 10.1002/chem.201501093. [DOI] [PubMed] [Google Scholar]
- 212.Zhang Z, Wu Q, Wu XH, Sun FY, Chen LM, Chen JC, Yang SL, Mei WJ. Eur J Med Chem. 2014;80:316–324. doi: 10.1016/j.ejmech.2014.04.070. [DOI] [PubMed] [Google Scholar]
- 213.Fan C, Wu Q, Chen T, Zhang Y, Zheng W, Wang Q, Mei W. Medchemcomm. 2014;5:597–602. [Google Scholar]
- 214.Chen X, Wu JH, Lai YW, Zhao R, Chao H, Ji LN. Dalton Trans. 2013;42:4386–4397. doi: 10.1039/c3dt32921f. [DOI] [PubMed] [Google Scholar]
- 215.Liao G, Chen X, Wu J, Qian C, Wang Y, Ji L, Chao H. Dalton Trans. 2015;44:15145–15156. doi: 10.1039/c4dt03585b. [DOI] [PubMed] [Google Scholar]
- 216.Chen ZF, Qin QP, Qin JL, Zhou J, Li YL, Li N, Liu YC, Liang H. J Med Chem. 2015;58:4771–4789. doi: 10.1021/acs.jmedchem.5b00444. [DOI] [PubMed] [Google Scholar]
- 217.Ye RR, Ke ZF, Tan CP, He L, Ji LN, Mao ZW. Chem – Eur J. 2013;19:10061–10061. doi: 10.1002/chem.201300814. [DOI] [PubMed] [Google Scholar]
- 218.Luo Z, Yu L, Yang F, Zhao Z, Yu B, Lai H, Wong KH, Ngai SM, Zheng W, Chen T. Metallomics. 2014;6:1480–1490. doi: 10.1039/c4mt00044g. [DOI] [PubMed] [Google Scholar]
- 219.Ferstl W, Sakodinskaya IK, Beydoun-Sutter N, Le Borgne G, Pfeffer M, Ryabov AD. Organometallics. 1997;16:411–418. [Google Scholar]
- 220.Dehand J, Pfeffer M. Coord Chem Rev. 1976;18:327–352. [Google Scholar]
- 221.Sortais JB, Pannetier N, Holuigue A, Barloy L, Sirlin C, Pfeffer M, Kyritsakas N. Organometallics. 2007;26:1856–1867. [Google Scholar]
- 222.Djukic JP, Sortais JB, Barloy L, Pfeffer M. Eur J Inorg Chem. 2009;2009:817–853. [Google Scholar]
- 223.Gaiddon C, Jeannequin P, Bischoff P, Pfeffer M, Sirlin C, Loeffler JP. J. P. E. T. 2005;315:1403–1411. doi: 10.1124/jpet.105.089342. [DOI] [PubMed] [Google Scholar]
- 224.Meng X, Leyva ML, Jenny M, Gross I, Benosman S, Fricker B, Harlepp S, Hébraud P, Boos A, Wlosik P. Cancer Res. 2009;69:5458–5466. doi: 10.1158/0008-5472.CAN-08-4408. [DOI] [PubMed] [Google Scholar]
- 225.Licona C, Spaety ME, Capuozzo A, Ali M, Santamaria R, Armant O, Delalande F, Dorsselaer AV, Cianferani S, Spencer J, Pfeffer M, Mellitzer G, Gaiddon C. Oncotarget. 2017;8:2568–2584. doi: 10.18632/oncotarget.13711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Boff B, Ali M, Alexandrova L, Espinosa-Jalapa NÁ, Saavedra-Díaz RO, Le Lagadec R, Pfeffer M. Organometallics. 2013;32:5092–5097. [Google Scholar]
- 227.Leyva L, Sirlin C, Rubio L, Franco C, Le Lagadec R, Spencer J, Bischoff P, Gaiddon C, Loeffler JP, Pfeffer M. Eur J Inorg Chem. 2007;2007:3055–3066. [Google Scholar]
- 228.Fetzer L, Boff B, Ali M, Meng X, Collin JP, Sirlin C, Gaiddon C, Pfeffer M. Dalton Trans. 2011;40:8869–8878. doi: 10.1039/c1dt10322a. [DOI] [PubMed] [Google Scholar]
- 229.Pena B, David A, Pavani C, Baptista MS, Pellois JP, Turro C, Dunbar KR. Organometallics. 2014;33:1100–1103. [Google Scholar]
- 230.Albani BA, Pena B, Dunbar KR, Turro C. Photochem Photobiol Sci. 2014;13:272–280. doi: 10.1039/c3pp50327e. [DOI] [PubMed] [Google Scholar]
- 231.Huang H, Zhang P, Chen H, Ji L, Chao H. Chem – Eur J. 2015;21:715–725. doi: 10.1002/chem.201404922. [DOI] [PubMed] [Google Scholar]
- 232.Zeng L, Chen Y, Huang H, Wang J, Zhao D, Ji L, Chao H. Chem – Eur J. 2015;21:15308–15319. doi: 10.1002/chem.201502154. [DOI] [PubMed] [Google Scholar]
- 233.Zeng L, Chen Y, Liu J, Huang H, Guan R, Ji L, Chao H. Sci Rep. 2016;6:19449. doi: 10.1038/srep19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Klajner M, Licona C, Fetzer L, Hebraud P, Mellitzer G, Pfeffer M, Harlepp S, Gaiddon C. Inorg Chem. 2014;53:5150–5158. doi: 10.1021/ic500250e. [DOI] [PubMed] [Google Scholar]
- 235.Colasson B, Credi A, Ragazzon G. Coord Chem Rev. 2016;325:125–134. [Google Scholar]
- 236.Smith NA, Zhang P, Greenough SE, Horbury MD, Clarkson GJ, McFeely D, Habtemariam A, Salassa L, Stavros VG, Dowson CG, Sadler PJ. Chemical Science. 2017;8:395–404. doi: 10.1039/c6sc03028a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Karaoun N, Renfrew AK. Chem Commun. 2015;51:14038–14041. doi: 10.1039/c5cc05172j. [DOI] [PubMed] [Google Scholar]
- 238.Knoll JD, Turro C. Coord Chem Rev. 2015;282-283:110–126. doi: 10.1016/j.ccr.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Singh TN, Turro C. Inorg Chem. 2004;43:7260–7262. doi: 10.1021/ic049075k. [DOI] [PubMed] [Google Scholar]
- 240.Salassa L, Ruiu T, Garino C, Pizarro AM, Bardelli F, Gianolio D, Westendorf A, Bednarski PJ, Lamberti C, Gobetto R, Sadler PJ. Organometallics. 2010;29:6703–6710. [Google Scholar]
- 241.Palmer AM, Peña B, Sears RB, Chen O, El Ojaimi M, Thummel RP, Dunbar KR, Turro C. Philos Trans R Soc, A. 2013;371:20120135. doi: 10.1098/rsta.2012.0135. [DOI] [PubMed] [Google Scholar]
- 242.Sgambellone MA, David A, Garner RN, Dunbar KR, Turro C. J Am Chem Soc. 2013;135:11274–11282. doi: 10.1021/ja4045604. [DOI] [PubMed] [Google Scholar]
- 243.Albani BA, Peña B, Leed NA, de Paula NABG, Pavani C, Baptista MS, Dunbar KR, Turro C. J Am Chem Soc. 2014;136:17095–17101. doi: 10.1021/ja508272h. [DOI] [PubMed] [Google Scholar]
- 244.van Rixel VHS, Siewert B, Hopkins SL, Askes SHC, Busemann A, Siegler MA, Bonnet S. Chem Sci. 2016;7:4922–4929. doi: 10.1039/c6sc00167j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Siewert B, van Rixel VHS, van Rooden EJ, Hopkins SL, Moester MJB, Ariese F, Siegler MA, Bonnet S. Chem – Eur J. 2016;22:10960–10968. doi: 10.1002/chem.201600927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Howerton BS, Heidary DK, Glazer EC. J Am Chem Soc. 2012;134:8324–8327. doi: 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]
- 247.Wachter E, Heidary DK, Howerton BS, Parkin S, Glazer EC. Chem Commun. 2012;48:9649–9651. doi: 10.1039/c2cc33359g. [DOI] [PubMed] [Google Scholar]
- 248.Hidayatullah AN, Wachter E, Heidary DK, Parkin S, Glazer EC. Inorg Chem. 2014;53:10030–10032. doi: 10.1021/ic5017164. [DOI] [PubMed] [Google Scholar]
- 249.Wachter E, Zamora A, Heidary DK, Ruiz J, Glazer EC. Chem Commun. 2016;52:10121–10124. doi: 10.1039/c6cc04813g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Davia K, King D, Hong Y, Swavey S. Inorg Chem Commun. 2008;11:584–586. [Google Scholar]
- 251.Rani-Beeram S, Meyer K, McCrate A, Hong Y, Nielsen M, Swavey S. Inorg Chem. 2008;47:11278–11283. doi: 10.1021/ic8015589. [DOI] [PubMed] [Google Scholar]
- 252.Ke H, Wang H, Wong WK, Mak NK, Kwong DWJ, Wong KL, Tam HL. Chem Commun. 2010;46:6678–6680. doi: 10.1039/c0cc01848a. [DOI] [PubMed] [Google Scholar]
- 253.Frei A, Rubbiani R, Tubafard S, Blacque O, Anstaett P, Felgentraeger A, Maisch T, Spiccia L, Gasser G. J Med Chem. 2014;57:7280–7292. doi: 10.1021/jm500566f. [DOI] [PubMed] [Google Scholar]
- 254.Pierroz V, Rubbiani R, Gentili C, Patra M, Mari C, Gasser G, Ferrari S. Chem Sci. 2016;7:6115–6124. doi: 10.1039/c6sc00387g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Cloonan SM, Elmes RBP, Erby M, Bright SA, Poynton FE, Nolan DE, Quinn SJ, Gunnlaugsson T, Williams DC. J Med Chem. 2015;58:4494–4505. doi: 10.1021/acs.jmedchem.5b00451. [DOI] [PubMed] [Google Scholar]
- 256.Lameijer LN, Hopkins SL, Brevé TG, Askes SHC, Bonnet S. Chem – Eur J. 2016;22:18484–18491. doi: 10.1002/chem.201603066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Monro S, Scott J, Chouai A, Lincoln R, Zong R, Thummel RP, McFarland SA. Inorg Chem. 2010;49:2889–2900. doi: 10.1021/ic902427r. [DOI] [PubMed] [Google Scholar]
- 258.Lincoln R, Kohler L, Monro S, Yin Hm, Stephenson M, Zong Rf, Chouai A, Dorsey C, Hennigar R, Thummel RP, McFarland SA. J Am Chem Soc. 2013;135:17161–17175. doi: 10.1021/ja408426z. [DOI] [PubMed] [Google Scholar]
- 259.Stephenson M, Reichardt C, Pinto M, Wächtler M, Sainuddin T, Shi G, Yin H, Monro S, Sampson E, Dietzek B. J Phys Chem A. 2014;118:10507–10521. doi: 10.1021/jp504330s. [DOI] [PubMed] [Google Scholar]
- 260.Arenas Y, Monro S, Shi G, Mandel A, McFarland S, Lilge L. Photodiagn Photodyn Ther. 2013;10:615–625. doi: 10.1016/j.pdpdt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 261.Yin H, Stephenson M, Gibson J, Sampson E, Shi G, Sainuddin T, Monro S, McFarland SA. Inorg Chem. 2014;53:4548–4559. doi: 10.1021/ic5002368. [DOI] [PubMed] [Google Scholar]
- 262.Sainuddin T, McCain J, Pinto M, Yin H, Gibson J, Hetu M, McFarland SA. Inorg Chem. 2015;55:83–95. doi: 10.1021/acs.inorgchem.5b01838. [DOI] [PubMed] [Google Scholar]
- 263.Tabrizi L, Chiniforoshan H. Dalton Trans. 2016;45:18333–18345. doi: 10.1039/c6dt03502g. [DOI] [PubMed] [Google Scholar]
- 264.Liu J, Chen Y, Li G, Zhang P, Jin C, Zeng L, Ji L, Chao H. Biomaterials. 2015;56:140–153. doi: 10.1016/j.biomaterials.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 265.Zeng L, Kuang S, Li G, Jin C, Ji L, Chao H. Chem Commun. 2017;53:1977–1980. doi: 10.1039/c6cc10330h. [DOI] [PubMed] [Google Scholar]
- 266.Zhuang X, Ma X, Xue X, Jiang Q, Song L, Dai L, Zhang C, Jin S, Yang K, Ding B. ACS Nano. 2016;10:3486–3495. doi: 10.1021/acsnano.5b07671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Jiang Q, Song C, Nangreave J, Liu X, Lin L, Qiu D, Wang ZG, Zou G, Liang X, Yan H. J Am Chem Soc. 2012;134:13396–13403. doi: 10.1021/ja304263n. [DOI] [PubMed] [Google Scholar]
- 268.Huang K, Ma H, Liu J, Huo S, Kumar A, Wei T, Zhang X, Jin S, Gan Y, Wang PC. ACS nano. 2012;6:4483–4493. doi: 10.1021/nn301282m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lee CC, MacKay JA, Fréchet JM, Szoka FC. Nat Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 270.Ahmed F, Pakunlu RI, Brannan A, Bates F, Minko T, Discher DE. J Controlled Release. 2006;116:150–158. doi: 10.1016/j.jconrel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 271.Tran PA, Webster TJ. Int J Nanomed. 2011;6:1553–1558. doi: 10.2147/IJN.S21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ramamurthy C, Sampath K, Arunkumar P, Kumar MS, Sujatha V, Premkumar K, Thirunavukkarasu C. Bioprocess Biosyst Eng. 2013;36:1131–1139. doi: 10.1007/s00449-012-0867-1. [DOI] [PubMed] [Google Scholar]
- 273.Liu W, Li X, Wong YS, Zheng W, Zhang Y, Cao W, Chen T. ACS nano. 2012;6:6578–6591. doi: 10.1021/nn202452c. [DOI] [PubMed] [Google Scholar]
- 274.Liu T, Zeng L, Jiang W, Fu Y, Zheng W, Chen T. Nanomedicine. 2015;11:947–958. doi: 10.1016/j.nano.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 275.Sun D, Liu Y, Yu Q, Zhou Y, Zhang R, Chen X, Hong A, Liu J. Biomaterials. 2013;34:171–180. doi: 10.1016/j.biomaterials.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 276.Sun D, Liu Y, Yu Q, Qin X, Yang L, Zhou Y, Chen L, Liu J. Biomaterials. 2014;35:1572–1583. doi: 10.1016/j.biomaterials.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 277.Cobley CM, Chen J, Cho EC, Wang LV, Xia Y. Chem Soc Rev. 2011;40:44–56. doi: 10.1039/b821763g. [DOI] [PubMed] [Google Scholar]
- 278.Huang X, Jain PK, El-Sayed IH, El-Sayed MA. Lasers Med Sci. 2008;23:217–228. doi: 10.1007/s10103-007-0470-x. [DOI] [PubMed] [Google Scholar]
- 279.Ghosh P, Han G, De M, Kim CK, Rotello VM. Adv Drug Deliver Rev. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 280.De M, Ghosh PS, Rotello VM. Adv Mater. 2008;20:4225–4241. [Google Scholar]
- 281.Manea F, Houillon FB, Pasquato L, Scrimin P. Angew Chem Int Edit. 2004;43:6165–6169. doi: 10.1002/anie.200460649. [DOI] [PubMed] [Google Scholar]
- 282.Saha K, Agasti SS, Kim C, Li X, Rotello VM. Chem Rev. 2012;112:2739–2779. doi: 10.1021/cr2001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Elmes RBP, Orange KN, Cloonan SM, Williams DC, Gunnlaugsson T. J Am Chem Soc. 2011;133:15862–15865. doi: 10.1021/ja2061159. [DOI] [PubMed] [Google Scholar]
- 284.Rogers NJ, Claire S, Harris RM, Farabi S, Zikeli G, Styles IB, Hodges NJ, Pikramenou Z. Chem Commun. 2014;50:617–619. doi: 10.1039/c3cc47606e. [DOI] [PubMed] [Google Scholar]
- 285.Zhang P, Wang J, Huang H, Chen H, Guan R, Chen Y, Ji L, Chao H. Biomaterials. 2014;35:9003–9011. doi: 10.1016/j.biomaterials.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 286.Zhang P, Wang J, Huang H, Yu B, Qiu K, Huang J, Wang S, Jiang L, Gasser G, Ji L, Chao H. Biomaterials. 2015;63:102–114. doi: 10.1016/j.biomaterials.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 287.Zhang P, Wang J, Huang H, Qiu K, Huang J, Ji L, Chao H. J Mater Chem B. 2017;5:671–678. doi: 10.1039/c6tb01991a. [DOI] [PubMed] [Google Scholar]
- 288.Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ. Acc Chem Res. 2013;46:792–801. doi: 10.1021/ar3000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Knežević NŽ, Trewyn BG, Lin VSY. Chem–Eur J. 2011;17:3338–3342. doi: 10.1002/chem.201002960. [DOI] [PubMed] [Google Scholar]
- 290.Frasconi M, Liu Z, Lei J, Wu Y, Strekalova E, Malin D, Ambrogio MW, Chen X, Botros YY, Cryns VL. J Am Chem Soc. 2013;135:11603–11613. doi: 10.1021/ja405058y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.He S, Krippes K, Ritz S, Chen Z, Best A, Butt H-J, Mailänder V, Wu S. Chem Commun. 2015;51:431–434. doi: 10.1039/c4cc07489k. [DOI] [PubMed] [Google Scholar]
- 292.Knežević NŽ, Stojanovic V, Chaix A, Bouffard E, Cheikh KEl, Morère A, Maynadier M, Lemercier G, Garcia M, Gary-Bobo M. J Mater Chem B. 2016;4:1337–1342. doi: 10.1039/c5tb02726h. [DOI] [PubMed] [Google Scholar]
- 293.He, Huang Y, Zhu H, Pang G, Zheng W, Wong YS, Chen T. Adv Funct Mater. 2014;24:2754–2763. [Google Scholar]
- 294.Baughman RH, Zakhidov AA, de Heer WA. Science. 2002;297:787–792. doi: 10.1126/science.1060928. [DOI] [PubMed] [Google Scholar]
- 295.Huang K, Saha A, Dirian K, Jiang C, Chu PLE, Tour JM, Guldi DM, Marti AA. Nanoscale. 2016;8:13488–13497. doi: 10.1039/c6nr02577c. [DOI] [PubMed] [Google Scholar]
- 296.Bianco A, Kostarelos K, Prato M. Curr Opin Chem Biol. 2005;9:674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 297.Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Xue X, Yang JY, He Y, Wang LR, Liu P, Yu LS, Bi GH, Zhu MM, Liu YY, Xiang RW. Nat Nanotechnol. 2016;11:613–620. doi: 10.1038/nnano.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kam NWS, O’Connell M, Wisdom JA, Dai H. Proc Natl Acad Sci. 2005;102:11600–11605. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wang N, Feng Y, Zeng L, Zhao Z, Chen T. ACS Appl Mater Interfaces. 2015;7:14933–14945. doi: 10.1021/acsami.5b03739. [DOI] [PubMed] [Google Scholar]
- 301.Zhang P, Huang H, Huang J, Chen H, Wang J, Qiu K, Zhao D, Ji L, Chao H. ACS Appl Mater Interfaces. 2015;7:23278–23290. doi: 10.1021/acsami.5b07510. [DOI] [PubMed] [Google Scholar]
- 302.Bœuf G, Roullin GV, Moreau J, Van Gulick L, Terryn C, Ploton D, Andry MC, Chuburu F, Dukic S, Molinari M. ChemPlusChem. 2014;79:171–180. doi: 10.1002/cplu.201300242. [DOI] [PubMed] [Google Scholar]
- 303.Sun W, Li S, Häupler B, Liu J, Jin S, Steffen W, Schubert US, Butt H-J, Liang X-J, Wu S. Adv Mater. 2017;29:1603702. doi: 10.1002/adma.201603702. [DOI] [PubMed] [Google Scholar]
- 304.Chen L, Fu C, Deng Y, Wu W, Fu A. Pharm Res. 2016;33:2989–2998. doi: 10.1007/s11095-016-2021-2. [DOI] [PubMed] [Google Scholar]
- 305.Chakrabortty S, Agrawalla BK, Stumper A, Vegi NM, Fischer S, Reichardt C, Kögler M, Dietzek B, Feuring-Buske M, Buske C, Rau S, Weil T. J Am Chem Soc. 2017;139:2512–2519. doi: 10.1021/jacs.6b13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Zhang DY, Zheng Y, Tan CP, Sun JH, Zhang W, Ji LN, Mao ZW. ACS Appl Mater Interfaces. 2017;9:6761–6771. doi: 10.1021/acsami.6b13808. [DOI] [PubMed] [Google Scholar]
- 307.Valente A, Garcia MH, Marques F, Miao Y, Rousseau C, Zinck P. J Inorg Biochem. 2013;127:79–81. doi: 10.1016/j.jinorgbio.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 308.Huang Y, Huang W, Chan L, Zhou B, Chen T. Biomaterials. 2016;103:183–196. doi: 10.1016/j.biomaterials.2016.06.053. [DOI] [PubMed] [Google Scholar]
- 309.Süss-Fink G, Khan F-A, Juillerat-Jeanneret L, Dyson PJ, Renfrew AK. J Cluster Sci. 2010;21:313–324. [Google Scholar]
- 310.Zhang G, Wu C, Ye H, Yan H, Wang X. J Nanobiotechnol. 2011;9:6. doi: 10.1186/1477-3155-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Romero-Canelón I, Phoenix B, Pitto-Barry A, Tran J, Soldevila-Barreda JJ, Kirby N, Green S, Sadler PJ, Barry NP. J Organomet Chem. 2015;796:17–25. [Google Scholar]
- 312.Völker T, Dempwolff F, Graumann PL, Meggers E. Angew Chem Int Ed. 2014;53:10536–10540. doi: 10.1002/anie.201404547. [DOI] [PubMed] [Google Scholar]
- 313.Sasmal PK, Streu CN, Meggers E. Chem Commun. 2013;49:1581–1587. doi: 10.1039/c2cc37832a. [DOI] [PubMed] [Google Scholar]
- 314.Zheng Y, Tan Y, Harms K, Marsch M, Riedel R, Zhang L, Meggers E. J Am Chem Soc. 2017;139:4322–4325. doi: 10.1021/jacs.7b01098. [DOI] [PubMed] [Google Scholar]
- 315.Zhang L, Meggers E. Acc Chem Res. 2017;50:320–330. doi: 10.1021/acs.accounts.6b00586. [DOI] [PubMed] [Google Scholar]
- 316.Yang M, Li J, Chen PR. Chem Soc Rev. 2014;43:6511–6526. doi: 10.1039/c4cs00117f. [DOI] [PubMed] [Google Scholar]
- 317.Li J, Chen PR. Nat Chem Biol. 2016;12:129–137. doi: 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- 318.Streu C, Meggers E. Angew Chem, Int Ed. 2006;45:5645–5648. doi: 10.1002/anie.200601752. [DOI] [PubMed] [Google Scholar]
- 319.Völker T, Meggers E. Curr Opin Chem Biol. 2015;25:48–54. doi: 10.1016/j.cbpa.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 320.Sadhu KK, Lindberg E, Winssinger N. Chem Commun. 2015;51:16664–16666. doi: 10.1039/c5cc05405b. [DOI] [PubMed] [Google Scholar]
- 321.Hsu HT, Trantow BM, Waymouth RM, Wender PA. Bioconjugate Chem. 2016;27:376–382. doi: 10.1021/acs.bioconjchem.5b00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Tomas-Gamasa M, Martinez-Calvo M, Couceiro JR, Mascarenas JL. Nat Commun. 2016;7:12538. doi: 10.1038/ncomms12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Soldevila-Barreda JJ, Sadler PJ. Curr Opin Chem Biol. 2015;25:172–183. doi: 10.1016/j.cbpa.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 324.Dougan SJ, Habtemariam A, McHale SE, Parsons S, Sadler PJ. Proc Natl Acad Sci. 2008;105:11628–11633. doi: 10.1073/pnas.0800076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Dougan SJ, Melchart M, Habtemariam A, Parsons S, Sadler PJ. Inorg Chem. 2006;45:10882–10894. doi: 10.1021/ic061460h. [DOI] [PubMed] [Google Scholar]
- 326.Soldevila-Barreda JJ, Romero-Canelón I, Habtemariam A, Sadler PJ. Nat Commun. 2015;6:6582. doi: 10.1038/ncomms7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wilson YM, Dürrenberger M, Nogueira ES, Ward TR. J Am Chem Soc. 2014;136:8928–8932. doi: 10.1021/ja500613n. [DOI] [PubMed] [Google Scholar]
- 328.Ritter C, Nett N, Acevedo-Rocha CG, Lonsdale R, Kräling K, Dempwolff F, Hoebenreich S, Graumann PL, Reetz MT, Meggers E. Angew Chem, Int Ed. 2015;54:13440–13443. doi: 10.1002/anie.201506739. [DOI] [PubMed] [Google Scholar]
- 329.Sanchez MI, Penas C, Vazquez ME, Mascarenas JL. Chem Sci. 2014;5:1901–1907. [PMC free article] [PubMed] [Google Scholar]
- 330.Li X, Guo J, Asong J, Wolfert MA, Boons GJ. J Am Chem Soc. 2011;133:11147–11153. doi: 10.1021/ja2012164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Koo H, Lee S, Na JH, Kim SH, Hahn SK, Choi K, Kwon IC, Jeong SY, Kim K. Angew Chem, Int Ed. 2012;51:11836–11840. doi: 10.1002/anie.201206703. [DOI] [PubMed] [Google Scholar]
- 332.Han Y, Yang X, Liu Y, Ai Q, Liu S, Sun C, Liang F. Sci Rep. 2016;6:22239. doi: 10.1038/srep22239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Unciti-Broceta A. Nat Chem. 2015;7:538–539. doi: 10.1038/nchem.2291. [DOI] [PubMed] [Google Scholar]
- 334.Tonga GY, Jeong Y, Duncan B, Mizuhara T, Mout R, Das R, Kim ST, Yeh YC, Yan B, Hou S, Rotello VM. Nat Chem. 2015;7:597–603. doi: 10.1038/nchem.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Ji S, Guo H, Yuan X, Li X, Ding H, Gao P, Zhao C, Wu W, Wu W, Zhao J. Org Lett. 2010;12:2876–2879. doi: 10.1021/ol100999j. [DOI] [PubMed] [Google Scholar]
- 336.Li G, Sasaki T, Asahina S, Roy MC, Mochizuki T, Koizumi K, Zhang Y. Chem. 2017;2:283–298. [Google Scholar]
- 337.Shen J, Kim HC, Wolfram J, Mu C, Zhang W, Liu H, Xie Y, Mai J, Zhang H, Li Z, Guevara M, Mao ZW, Shen H. Nano Lett. 2017;17:2913–2920. doi: 10.1021/acs.nanolett.7b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Wang YM, Liu W, Yin XB. Chem Sci. 2017;8:3891–3897. doi: 10.1039/c6sc05616d. [DOI] [PMC free article] [PubMed] [Google Scholar]